

Abstract
Prostate cancer (PCa) metabolism appears to be unique in comparison with other type of solid cancers. Normal prostate cells mainly rely on glucose oxidation to provide precursors for the synthesis and secretion of citrate, resulting in an incomplete Krebs cycle and minimal oxidative phosphorylation for energy production. In contrast, during transformation, PCa cells no longer secrete citrate and they reactivate the Krebs cycle as energy source. Moreover, primary PCas do not show increased aerobic glycolysis and therefore they are not efficiently detectable with 18F-FDG-PET. However, increased de novo lipid synthesis, strictly intertwined with deregulation in classical oncogenes and oncosuppressors, is an early event of the disease. Up-regulation and increased activity of lipogenic enzymes (including fatty acid synthase and choline kinase) occurs throughout PCa carcinogenesis and correlates with worse prognosis and poor survival. Thus, lipid precursors such as acetate and choline have been successfully used as alternative tracers for PET imaging. Lipid synthesis intermediates and FA catabolism also emerged as important players in PCa maintenance. Finally, epidemiologic studies suggested that systemic metabolic disorders including obesity, metabolic syndrome, and diabetes as well as hypercaloric and fat-rich diets might increase the risk of PCa. However, how metabolic disorders contribute to PCa development and whether dietary lipids and de novo lipids synthesized intra-tumor are differentially metabolized still remains unclear. In this review, we examine the switch in lipid metabolism supporting the development and progression of PCa and we discuss how we can exploit its lipogenic nature for therapeutic and diagnostic purposes.
Keywords: prostate cancer, lipid metabolism, fatty acids, imaging, metabolic diseases
Introduction
Prostate cancer (PCa) is the most commonly diagnosed malignancy in men and the second leading cause of cancer-related deaths in the USA [1]. Although the advent of PSA screening has led to an increase in the number of diagnoses for early intervention, PSA testing does not appear to decrease PCa mortality and some patients still experience disease progression after receiving primary treatment [2]. Currently, the standard systemic treatment for advanced PCa is based on androgen deprivation (LHRH agonists, often in combination with oral anti-androgens) to which tumors initially respond but eventually become castrate resistant by acquiring changes that include androgen-receptor overexpression and up-regulation of enzymes involved in androgen biosynthesis, which result in reactivation of the receptor. Based on the new understanding of these resistance mechanisms and androgen biosynthesis pathways, new anti-androgens and androgen-depleting agents have been developed (including enzalutamide and abiraterone acetate), hopefully increasing efficacy in advanced cancers [3]. Men with metastatic castration-resistant PCa (CRPC) have a median survival of less than 2 years [4]. Currently, there is no standard therapeutic modality for this fatal stage of PCa. Broad-band chemotherapeutic drugs are commonly used as a last resort; the taxane-derivative docetaxel is the only US Food and Drug administration (FDA)-approved treatment for use, showing a survival benefit, although several others are approved for palliative indications [5, 6]. Unfortunately, tumors often develop resistance to taxanes [6]. Thus, alternative therapeutic strategies are urgently sought.
In the 1920s, the German biochemist Otto Warburg made the first observation that cancer cells undergo a metabolic reprogramming by increasing the uptake of glucose and converting it to lactate using glycolysis, even in the presence of normoxic conditions (a phenomenon called Warburg effect or aerobic glycolysis) [7]. Despite being a less efficient mechanism of ATP production, aerobic glycolysis proved to be the core cellular metabolism to provide cancer cells with building blocks for macromolecule synthesis, such as carbohydrates, proteins, lipids, and nucleic acids to support their accelerated proliferative rate [8]. After one century, the importance of metabolic reprogramming in cancer has been re-discovered and in the last decade the use of new molecular tools allowed unraveling the numerous connections between oncogenic pathways and metabolic activities. Indeed, the identification of metabolic weaknesses of cancer cells has led to new strategies for treating cancer. PCa presents very unique metabolic features, since primary tumors do not show the classical “glycolytic switch” as the majority of other solid tumors, and so are not efficiently detectable with FDG-PET, a technique successfully used in oncology for non-invasive diagnosis, staging and treatment follow-up. However, an aberrant increase in de novo lipogenesis, directly coupled with glucose and glutamine metabolisms, is observed at early stages of the disease, significantly associated with tumor progression, worse prognosis, and shorter survival [9–12]. Moreover, lipid deregulation, due to systemic metabolic diseases (including obesity, metabolic syndrome, diabetes) or specific diet behaviors has been linked to an increased risk of prostate cancer, but the mechanism for this relationship is still not fully understood [13]. Although the production of amino acids and nucleic acids is certainly an integral characteristic of the cancer anabolic reprogramming as well, here, we discuss the alterations of lipid metabolism in PCa, how oncogenic signaling, diet, and lifestyle regulate it, and finally how we can take advantage of this understanding to develop novel diagnostic tools and therapeutic strategies.
Role of lipids in cancer
Lipids contribute to several aspects of tumor biology due to the diversity of their biological roles (Figure 1). First, they function as building blocks for biological membranes to support the high proliferative rate of cancer cells. Several endogenously synthesized fatty acids (FAs) are esterified to phospholipids, which provide pivotal structural lipids, facilitate the formation of detergent-resistant membrane microdomains for signal transduction, intracellular trafficking, polarization, and migration required for cancer cells [9, 14]. The importance of membrane synthesis in cancer cells has been underscored by the observation that the expression and activity of choline kinase, an enzyme required for the synthesis of phosphatidylcholine and phosphatidylethanolamine (the major phospholipids found in cellular membranes) is increased in tumors from several tissues, including PCa, and correlates with poor prognosis [15, 16]. Choline kinase has oncogenic activity when overexpressed, suggesting that the synthesis of phospholipids is rate limiting for transformation [17, 18].
Figure 1. The role of lipids in PCa cells.
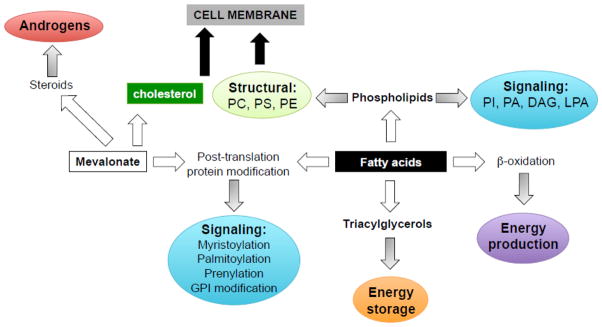
Lipids exert several biological functions in cancer cells, including energy supply and storage, membrane building blocks, signaling molecules, protein post-translation modifications, substrates for steroids to support PCa cells survival, proliferation, migration, and invasion. PI, phosphatidylinositol, PA, phopshatidic acid; DAG, diacylglycerol; LPA, lysophosphatidic acid; PC, phosphocholine; PS, Phosphatidylserine; PE, Phosphatidylethanolamine; GPI, Glycosylphosphatidylinositol.
Second, intermediates of de novo lipogenesis, such as diacylglycerol (DAG), phosphatidic acid (PA), lysophosphatidic acid (LPA), and sphingosine 1-phosphate (S1P) also function as second messengers in signal transduction pathways and are involved in crucial aspects of cell to cell communication. Once generated, these lipids can be released and act extracellularly in an autocrine/paracrine manner, controlling a variety of cellular functions such as cell proliferation, survival, and migration/invasion by either activating other signaling proteins, or by engaging a series of G protein-coupled receptors (GPCRs) on the cell surfaces [19]. Third, lipid-mediated post-translational modification of proteins is also a vital process in regulating expression, localization and function of various signaling proteins [20, 21]. Phosphatidylinositol (PI)-associated modification through a carbohydrate linker to the proteins (GPI-anchored proteins) directs them towards the cell surface from the endoplasmic reticulum (ER) [22]. Some GPI-anchored proteins, such as urokinase-type plasminogen activator (uPA)-receptor (uPAR) and membrane anchored serine protease matriptase (also known as MT-SP1 and epithin), have strong association with cancer [23, 24]. Covalent attachment of FA palmitic acid to a serine residue (also called palmitoylation) of Wnt, an important signal molecule, regulates its signaling capacity and secretion [25, 26]. Cytoplasmic stabilization of β-catenin through palmitoylation of Wnt-1 and subsequent activation of the pathway has been shown as a potential mechanism of fatty acid synthase (FASN)-mediated oncogenicity in PCa [27]. Moreover, the irreversible attachment of a myristoyl group (derived from FA myristic acid) to N-terminal amino acid of Akt has been shown to activate the kinase and to induce cell transformation [28, 29]. Prenylation (the transfer of either a farnesyl or a geranyl-geranyl moiety to C-terminal cysteine(s) of the target protein) is also involved in the regulation of signaling outputs and controls the trafficking of proteins among ER, Golgi and plasma membranes, including the Ras small GTPase family members [30]. Finally, in response to glucose limitation, FAs can also be consumed through β-oxidation to provide key substitute energy for cancer cell survival. It is reported that increased FA oxidation is sufficient for cell survival and to protect cells from glucose withdrawal-induced death in Akt-overexpressing glioblastoma [31]. In PCa, FA oxidation has been observed to be a dominant bioenergetic pathway, as discussed below [32].
Alteration of lipid metabolism in prostate cancer
Dysregulation of FA biosynthesis and its pleiotropic role in cancer cells
Altered lipid metabolism has been increasingly recognized as a classical hallmark of cancer cells and a growing body of literature has been focused on the relationship between up-regulated lipogenesis (the so called “lipogenic phenotype”) and PCa pathogenesis [10, 11, 27, 33, 34]. A number of lipogenic enzymes utilize reduced nicotinamide adenine dinucleotide phosphate (NADPH) and acetyl-CoA generated from glucose and glutamine oxidation to synthesize FAs and their derivatives (Figure 2). Therefore, the exacerbated lipogenesis in cancer cells is not only caused by the up-regulation of lipid metabolizing enzymes, but is also directly coupled to other common metabolic pathways and associated cell signaling pathways. FA synthesis occurs in the cytosol and begins with the conversion of citrate [derived from the tricarboxylic acid (TCA) cycle in the mitochondria] to acetyl-CoA and oxaloacetate by the enzyme ATP citrate lyase (ACLY). Acetyl-CoA is then converted to malonyl-CoA by the rate-limiting enzyme acetyl-CoA carboxylase (ACACA, commonly referred as ACC), whereas oxaloacetate can be converted into pyruvate by the malic enzyme. This reaction generates NADPH and, along with the NADPH-producing reactions in the pentose phosphate pathway, provides the reducing power for lipid synthesis. The multi-enzymatic complex FASN then processes one acetyl-CoA and seven malonyl-CoA molecules through a series of catalytic domains to produce palmitate (16:0), a saturated 16-carbon FA that represents about 80–90% of total FAs produced in a cell, and the less abundant FAs myristate (14:0) and stearate (18:0) [9]. Saturated FAs may then undergo further modifications at the cytoplasmatic face of the ER membrane by desaturases and/ or elongases to insert double bonds or increase the carbon chain length, respectively, before FAs are ultimately utilized for energy, protein modification, or incorporation into complex lipid structures for cellular signaling and membrane integrity [35] (Figure 2). Desaturation is catalyzed by fatty acyl-CoA desaturases, including the family of stearoyl-CoA desaturases (SCDs) also known as fatty acyl-CoA delta-9 desaturases, that catalyze the introduction of the first double bond in the cis-delta-9 position of several saturated fatty acyl-CoAs principally palmitoyl-CoA and stearoyl-CoA, to yield palmitoleoyl (16:1)- and oleoyl-CoA (18:1), respectively [36]. The best-characterized desaturase is SCD1, which is highly expressed in oncogene-transformed lung fibroblasts and in several cancers [37–40]. Recently, SCD1 has been found overexpressed both at the mRNA and protein level in androgen-sensitive and resistant PCa cell lines as well as human tumor tissues. In the latter, its overexpression correlates with a higher gleason grade, suggesting a role of this desaturase in PCa progression [41]. Elongation of FAs to produce FAs with longer chains such as stearic acid is obtained by a family of elongases that add two carbons to the end of the chain in each cycle of reactions. This family comprises seven members (ELOVL1–7) with different chain lengths and saturation specificity. Tamura and coworkers have recently shown that ELOVL7 is overexpressed in PCa and is involved in PCa growth and survival through the metabolism of very-long-chain saturated FAs (SVLFAs) and their derivatives, as discussed in the following paragraph [42]. Normal adult human tissues preferentially use dietary (exogenous) lipids for their metabolic demand, whereas de novo (endogenous) FA synthesis is usually suppressed (except for some activity in the liver, adipose tissue, and lactating breast) and the expression of lipogenic enzymes is maintained at low levels [43]. By contrast, about 60 years ago, Medes et al., showed that cancer cells synthesize large amounts of de novo FAs and cholesterol irrespective of the circulating lipid levels [44] and they shunt glucose-derived carbons from the mitochondrial TCA cycle to the cytosol to feed FA synthesis [8]. In line with this observation, the expression and activity of many enzymes involved in the FA synthesis pathway such as ACLY, ACC, FASN are up-regulated in many cancers [as reviewed in 12, 14, 45, 46], including PCa [10, 33]. Overexpression of FASN is an early event in PCa pathogenesis, associated with PCa progression and metastases to the bone [11, 33]. Elevated expression of FASN has been linked with poor prognosis and reduced disease-free survival in PCa [47]. Moreover, forced ectopic expression of FASN in immortalized prostate epithelial cells transforms them, inducing invasive tumors when injected in immunodeficient mice whereas transgenic mice expressing FASN in the prostate develop prostatic intra-epithelial neoplasia [34]. The proposed mechanisms of FASN oncogenicity include cytoplasmic stabilization of β-catenin through acylation of Wnt-1, inhibition of the intrinsic/mitochondrial pathway of apoptosis, protection from ER stress, and the structural need to synthesize lipids [27, 34, 48]. The major transcriptional regulator of the lipogenic enzymes ACLY, ACC, FASN is the sterol response element binding protein-1 (SREBP-1). Expression of SREBP-1 can be stimulated by androgens and epidermal growth factor (EGF) in PCa [49, 50], and in turn, expression of the androgen receptor is regulated by SREBP-1, creating a positive feedback loop for the expression of lipogenic enzymes [51]. Moreover, overexpression of SREBP-1 is sufficient to increase tumorigenicity and invasion of PCa [51]. Pharmacological or small-interfering RNA (siRNA)-mediated inhibition of enzymes in the de novo fatty acid synthesis pathway, namely ACLY, ACC, FASN, or SCD1 results in either the inhibition of PCa growth or cell death [52–56], demonstrating the requirement for de novo fatty acids for PCa growth and survival. FA synthesis is an extremely demanding process: seven molecules of ATP and 14 molecules of NADPH are required to synthesize one molecule of palmitate. Thus, PCa cancer cells have adapted the use of alternative metabolic pathways and enzymes to provide the necessary substrates for lipogenesis and prevent its inhibition by upstream signaling mediators. These alternative pathways include isoform switching, truncation of the TCA cycle, the use of TCA cycle intermediates for FA synthesis, and the increased use of other non-canonical metabolic enzymes such as hexokinase 2 in response to androgen, the embryonic form of pyruvate kinase (PKM2), the pentose phosphate pathway (PPP), and cytosolic TCA cycle enzymes [8, 57] (Figure 3).
Figure 2. Increased de novo lipogenesis in PCa.

Citrate generate in TCA cycle is exported to the cytosol to fuel the mevalonate and fatty acid (FA) synthesis pathways by conversion to acetyl-CoA by ATP citrate lyase (ACLY). Acetyl-CoA acetyltransferases (ACAT) is the first enzyme of cholesterol synthesis pathway that converts acetyl-CoA into acetoacetyl-CoA, which functions as a substrate for the synthesis of mevalonate by HMG-CoA synthase (HMGCS), followed by the reductase (HMGCR). The latter is the rate-limiting step for the production of cholesterol, which is the precursor to androgens. Acetyl-CoA carboxylase 1 (ACC) initiates the first committed step to FA synthesis to produce malonyl-CoA. Seven malonyl-CoA molecules are added to acetyl-CoA by fatty acid synthase (FASN) to produce palmitic acid, a 16-carbon saturated FA (SFA). Palmitic acid can be further elongated to form long SFAs and/or desaturated by stearoyl-CoA desaturase 1 (SCD1) and by other desaturases to produce monounsaturated FA (MUFAs). The reducing factor nicotinamide adenine dinucleotide phosphate (NADPH), which is essential for FA synthesis, is provided by pentose phosphate pathway (PPP), by malic enzyme (ME1), which produces pyruvate from malate (shunt malate-pyruvate), and by cytosolic isocitrate dehydrogenase (IDH1) to produce α-ketoglutarate (αKG) from isocitrate (shunt isocitrate αKG). TAG, triacylglycerols; PL, phospholipids; CYP11A1, p450, family 11, subfamily A, polypeptide 1; CYP17A1; p450, family 11, subfamily A, polypeptide 1; 3β-HSD1, 2, 3-β-hydroxysteroid dehydrogenase/Δ-5-4 isomerase 1, 2; 17β-HSD, 17β-Hydroxysteroid dehydrogenase; SRD5A 1, 2, steroid-5α-reductase 1, 2.
Figure 3. Regulation of lipid metabolism by oncogenes and tumor suppressors.

PCa cancer cells show high rates of de novo lipid synthesis and the enzymes belonging to the pathway are regulated by oncogenic signals. Growth factor-activated PI3K-Akt or hypoxia-induced HIF stimulates glucose uptake and hexokinases to promote glycolysis, providing more synthetic precursors for FA synthesis. Akt also activates the lipogenic enzyme activity and expression through direct phosphorylation or SREBP-mediated transcription of lipogenic genes. Activation of E2F following loss of the retinoblastoma (Rb) protein increases expression of SREBPs and their target genes. The tumor suppressor p53 plays a role in glucose uptake, pentose phosphate pathway, transcription of lipogenic genes, and anaplerosis of citrate. Mutant p53 (p53mut) also increases the expression of genes within the cholesterol biosynthesis (mevalonate). The tumor suppressor LKB1-AMPK axis, activated in response to low cellular energy levels, inhibits de novo lipogenesis through direct inhibition of ACC and SREBP-1. Myc promotes citrate anaplerosis through increasing glutamine transporters and glutaminase 2 expression. The activity and expression of lipogenic enzyme, ACLY, ACC, and FASN are also regulated at multiple levels through mTORC1 and mitogen-activated protein kinases (MAPKs). IDH1, isocitrate dehydrogenase 1; pRB, retinoblastoma 1; Glut, Glucose transporter; FBP, Fructose-1,6-bisphosphate; PEP, phosphoenolpyruvate; HK, Hexokinase; G6PDH, Glucose-6-phosphate dehydrogenase; PFK, Phospho-fructokinase; PKM2, Tumor-specific pyruvate kinase M2; LDHA, Lactate dehydrogenase A; HIF, Hypoxia-inducible factor; AMPK, AMP-activated protein kinase; PI3K, Phosphatidylinositol 3-kinase; MAPK, Mitogen activated protein kinase; mTOR, Mammalian target of rapamycin; E2F, DNA-binding transcription factor; SREBP-1, Sterol-regulatory element-binding protein-1; TCA cycle, tricarboxylic acid cycle; OAA, Oxaloacetate; Mal, Malate; ME, Malic enzyme; αKG, α-ketoglutarate; ACLY, ATP citrate lyase; ACC, Acetyl-CoA carboxylase; FASN, Fatty acid synthase; HMGCR, HMG-CoA reductase; NADPH, Nicotinamide adenine dinucleotide phosphate; Glt, glutamate.
Although de novo synthesized FAs are typically derived from glucose, some cancer cells rely on glutamine metabolism (Figure 3). Because glucose carbons are shunted from the mitochondria for FA synthesis, PCa cells convert glutamine to α-ketoglutarate and shuttle it into the mitochondria to replenish the TCA cycle intermediates for anaplerotic reactions [58, 59]. Additionally, cancer cells experiencing selective pressure by hypoxia or a defective electron transport chain use glutamine, rather than glucose, as the major lipid precursor. In such a scenario, stabilized hypoxia inducible factor 1α (HIF-1α) inhibits the conversion of pyruvate to acetyl-CoA and isocitrate dehydrogenase (IDH1) works in reverse to convert α-ketoglutarate to citrate, which is then metabolized by ACLY for lipid synthesis [60, 61] (Figure 3). One major purpose for increased FA synthesis is to supply the heavy demand for new membrane biogenesis. However, Swinnen and coworkers showed that de novo fatty acids used for membrane biogenesis also preferentially localize in the membrane’s signaling raft domains involved in signal transduction pathways [62]. Thus, de novo FAs may play a significant role in propagating oncogenic signals from the plasma membrane, such as Akt activation in PCa [63]. Overexpression of FASN in PCa also supports palmitoylation and activation of oncogenic β-catenin [27]. While the enzymes FASN and ACC are also involved in the control of cell cycle progression, the role of de novo synthesized FAs in this context remains to be determined [56, 64]. The role of de novo FA synthesis goes beyond merely facilitating proliferation and survival. Indeed, FA synthesis appears to play a role in energy homeostasis, resistance to oxidative stress, and cell polarity.
a. Energy homeostasis
As aforementioned, cancer cells use large amounts of glucose for energetic and biosynthetic purposes [8] resulting in a high rate of lactate production and secretion that lead to the acidification of the tumor microenvironment [65]. Thus, it is possible that lipid synthesis may function as a carbon sink to sequester excess pyruvate and avoid lactate production to equilibrate the intracellular pH. Furthermore, it may also contribute to redox balance. Indeed, it has been shown that in PCa cells, the oxidizing power of the FA synthesis pathway is so large that redox is stabilized more favorably (more oxidized) than in normal prostate cells. This FASN-facilitated redox improvement occurs despite the fact that malignant cells are more O2 limited and therefore express more HIF-1α and hypoxia-regulated genes [66]. Additionally, in hypoxic cells, lipid synthesis-derived NADP+ could also increase the availability of cytoplasmic NAD+ required to maintain glycolysis, thanks to a recently proposed mitochondria-cytosolic NADPH shuttle [67]. Lipid synthesis would thus harness its oxidizing power to improve redox balance between the cytoplasm and the mitochondria and contributes to maximize glycolysis.
b. Resistance to oxidative stress
A recent mass spectrometry-based phospholipid analysis revealed that prostate tumors displaying the lipogenic phenotype (due to increased FASN expression) show a consistent increase in saturated and monounsaturated FAs (SFAs and MUFAs) and a decrease in polyunsaturated species compared to normal tissues [68]. Reversal of the lipogenic phenotype by the small molecule inhibitor soraphen A or siRNAs markedly decreases the saturated and monounsaturated phospholipid species and increases the relative degree of polyunsaturation. This lipogenic switch that favors SFAs appears to protect cancer cells from oxidative stress and from doxorubicin-induced cell death [68]. Furthermore, the tendency of phospholipids containing mainly SFAs to segregate into lipid rafts/detergent-resistant membranes would markedly alter signal transduction cascades, vesicular trafficking and cell migration [69].
c. Cell polarity
Recent evidence has shown that de novo lipogenesis and particularly the activation of SREBP-1c distorts cell polarization and suppresses the formation of the primary cilium [70]. This microtubular-based sensory organelle is expressed on the surface of nearly every cell type and it is lost in many cancers, including PCa [71]. Since the primary cilium concentrates and regulates several signaling pathways, SREBP-induced repression of the primary cilium may render cancer cells less dependent on external cues and anti-growth signals. This may also contribute to the constitutive activation of cancer-associated signaling pathways, such as Wnt signaling, which, further has been reported to be activated by FASN-induced palmitoylation in PCa cells [27]. Suppression of the primary cilium by SREBP-1 and by de novo lipogenesis may also release a direct physical restraint on the cell cycle as the primary cilium is composed of the same structural elements that are used for mitotic spindle formation [72]. These findings, not only provide new insights into the role of the de novo lipogenesis in PCa cells but they also provide a rationale for the use of lipogenesis inhibitors as antineoplastic agents and as chemotherapeutic sensitizers.
Dysregulation of FA remodeling
Despite early experimental and epidemiological observations reported elevated levels of MUFA in cancer cells and tissues, the precise mechanism of action of SCDs in carcinogenesis remains elusive. SCD1 expression promotes transformation, accelerated rate of cell proliferation, increased invasiveness, enhanced survival and, ultimately, greater tumorigenic capacity [reviewed in 73]. Constitutively active SCD1 promotes this metabolic balance that favors cell proliferation and survival by at least three potential mechanisms (i) stimulation of ACC activity by the suppression of allosteric feedback inhibition by SFAs and the inactivation of AMPK phosphorylation, described in the following paragraphs; (ii) activation of the Akt pathway, with potential positive effects on the transcription of lipogenic enzymes and downstream mitogenic signaling proteins and (iii) stimulation of the lipid biosynthetic machinery by continuous supply with its preferred substrates, the MUFAs [73]. Furthermore, the overexpression of SCD1 in cancer cells ensures that harmful accumulation of SFAs does not occur, hence preventing the cytotoxic effects of these FAs [74]. In keeping with these findings, Fritz et al. found that lipids composition in human PCa is characterized by an increased ratio of MUFAs to SFAs, compared to normal prostate, and confirmed the overexpression of SCD1 in human PCa [41]. They also showed that pharmacological inhibition of SCD1 activity impairs lipid synthesis and results in decreased proliferation of both androgen-sensitive and resistant PCa cells, abrogates the growth of prostate tumor xenografts in nude mice, and confers a survival benefit [41]. At a molecular level, SCD1 inhibition results in the suppression of the Akt pathway and reduced concentration of phosphatidyl inositol tri-phosphate (PI (3,4,5) P3), in the promotion of AMPK and glycogen synthase kinase α/β (GSK3α/β) activation. Finally, SCD1 activity appears to be required for cell transformation by Ras oncogene [41]. Changes in the elongation of FA have also been reported in PCa cells [42]. ELOVLs are human homologues of yeast ELOs and catalyze the elongation reaction of very-long-chain FA [75, 76]. The elongation system, which is responsible for the addition of two carbon units to the carboxyl end of a FA chain, is composed of four enzymes: a condensing enzyme (elongase, β-ketoacyl CoA synthase), β-ketoacyl CoA reductase, β-hydroxyacyl CoA dehydrase, and trans-2, 3-enoyl-CoA reductase [75]. Seven distinct FA elongase subtypes (ELOVL1-7) have been reported in mammals thus far, and each of these multiple elongation enzymes is thought to work specifically for different chain length saturated or unsaturated FAs [77]. The metabolic pathways of long-chain FAs play an important role in the maintenance of membrane lipid composition as well as in the generation of precursors for cell signaling molecules, such as eicosanoids and sphingosine-1 phosphate involved in cell physiology and cancer pathology [76, 77]. In this regard, ELOVL7 have been shown to be overexpressed in high grade PCas and its expression to be regulated by the androgen pathway through SREBP-1. Knockdown of ELOVL7 resulted in drastic attenuation of PCa cell growth, whereas high-fat diet promoted in vivo tumor growth of ELOVL7-overexpressing PCas [42]. FA elongation assay and composition analyses indicated that ELOVL7 is preferentially involved in FA elongation of saturated very-long-chain fatty acids (SVLFA, C20:0). Moreover, de novo androgen synthesis is significantly affected by EVOLV7 inhibition [42]. While further characterization of SCD1 and EVOLV7 biochemical and biological functions is still required, these enzymes represent valid targets for novel pharmacological approaches in PCa. Indeed, potent and specific small molecule inhibitors of SCD1 activity have recently been discovered.
Alterations in FA catabolism
a) Increased FA breakdown
The majority of research on cancer lipid metabolism has focused on the contribution to tumor growth by increased of FA synthesis. However, cancer cells also contain increased numbers of lipid droplets, highly ordered intracellular structures derived from the ER where triacylglycerols and cholesterol esters are stored [78]. Thus, Nomura and coworkers reasoned that cancer cells should possess a complementary lipolytic pathway to liberate fatty acyl moieties from those reservoirs for signaling purpose [81]. Using an elegant proteomic approach, the activity of monoacylglycerol lipase (MAGL), an enzyme that has long been known for its participation in the breakdown of adipose triacyglycerol (TAG) stores, was unexpectedly found to be highly elevated in aggressive cancer cells from multiple tissues of origin [79]. Furthermore, increased free FA (FFA) levels were measured in the more aggressive cancer cell lines and high-grade primary tumors. Genetic or pharmacological blockade of MAGL rendered these tumors FFA-depleted and less malignant, whereas ectopic overexpression of MAGL in non-aggressive cells rendered them FFA-enriched and more malignant [79]. Whereas MAGL-regulated lipid hydrolysis appears to be important for the transformed properties of tumor cells, its inhibition suppresses migration, invasion and survival of cancer cells in vitro and xenograft tumor growth in mice. Strikingly, the functional defects of MAGL inhibition were reversed by exogenous addition of saturated FAs in vitro or feeding mice a high-fat diet [79]. Thus, MAGL supports the malignant properties of cancer cells by maintain elevated levels of FFAs, which, in turn become mostly channeled to the production of various oncogenic lipid messengers, such as PA, LPA, and prostaglandin E rather than to fuel β–oxidation, sustain glycolysis, or generate membrane lipids [79]. It was also shown that MAGL-silenced cancer cells display enhanced sensitivity to the anti-migratory and anti-survival effects of the EGF receptor inhibitor tyrphostin. Thus, understanding whether MAGL inhibitors render human tumors more responsive to the antineoplastic action of tyrosine kinase-targeted chemotherapeutic drugs is worth f urther investigation. Although acute/pharmacological and sustained/genetic loss of MAGL function generated somewhat distinct metabolomics profiles in the cancer cells studied, the work of Nomura and coworkers (2010) turns MAGL into an unexpected and promising new candidate for cancer therapy [81]. The study raised also the exciting question why both lipogenesis and lipolysis are increased in cancer cells. Further studies need to be performed to test whether FAs derived from de novo synthesis versus lipolytic release differ in both compositions and functions.
b) Increased β-oxidation
While most tumors exhibit a high rate of glucose uptake, which contributes to support both their energetic and biosynthetic requirements [8], PCas generally display a low rate of glucose utilization [80, 81], increased uptake of FAs, such as palmitate [82], and increased FA oxidation [32] with overexpression and increased activity of enzymes of the peroxisomal branched chain fatty acid β-oxidation, including the D-bifunctional protein (DBP) and the α-Methylacyl-CoA racemase AMACR [83–85]. This increase in β-oxidation may be caused by the specialized metabolism of normal prostate epithelial cells. Normal epithelial prostate cells inhibit the enzyme aconitase, responsible for the conversion of citrate in isocitrate, and secrete large amounts of citrate into the prostatic fluid (40–150 mM citrate in the prostatic fluid in comparison with 0.2 mM in blood plasma) [86]. Citrate is likely to act as a seminal buffering agent, a chelator of free Ca++ and Zn++, and possibly a scavenger of free radicals [87–90]. In contrast, during transformation, PCa cells no longer secrete citrate and reactivate instead the TCA cycle, increasing the oxidation of citrate [91] as energy source. The transformation of a citrate-producing normal epithelial cell to a malignant citrate-oxidating cell thus results in a more efficient energy-generating system. Indeed, the metabolic switch towards citrate oxidation is an early event in PCa pathogenesis [92]. Activation of β-oxidation may also be crucial to support cancer-cell viability during periods of energy stress. Constitutive activation of the PI3K/Akt pathway sensitizes haematopoietic cells to withdrawal of glucose or growth factors and activation of β-oxidation is sufficient to maintain cell viability under these conditions [31, 93]. Finally, increase β-oxidation has recently been shown to contribute to resistance to oxidative stress by providing substrates for NAPDH and glutathione production, allowing cells to remove reactive oxygen species [94]. FA oxidation, representing as a dominant bioenergetic pathway in PCa, could therefore be exploited for drug targeting and diagnostic imaging [95].
Increased cholesterol and endogenous steroid synthesis in PCa
Increased accumulation of cholesterol in PCa cells
Another important biosynthetic process that is highly up-regulated in PCa is the mevalonate pathway, which facilitates the accumulation of cholesterol [96] (Figure 2). A large number of epidemiological studies have been conducted to address the association between cholesterol levels and PCa, reporting that men with hypercholesterolemia are at increased risk for aggressive PCa [reviewed in 96]. Moreover, pre-clinical animal models showed that hypercholesterolemia is associated with increased growth of PCa, progression to metastasis, Akt activation, and increased levels of cyclin D1 [97, 98]. The first steps of cholesterol biosynthesis involve the condensation of acetyl-CoA with acetoacetyl-CoA to form 3-hydroxy-3-methylglutaryl (HMG)-CoA. The reduction of HMG-CoA to mevalonate by HMG-CoA reductase (HMGCR) represents the rate-limiting reaction of the cholesterol synthesis pathway and is highly regulated at the transcriptional and post-transcriptional levels through phosphosphorylation by the energy sensor AMPK [99]. Transcriptional regulation of the enzymes in this pathway occurs through activation of SREBP-2 [100]. SREBP-2, a regulator of androgen synthesis, it is also itself regulated by androgens, demonstrating a direct feedback circuit for regulation of androgen production [100]. Interestingly, SREBP-2 expression increases during PCa progression, it is significantly higher after castration and lacks its feedback inhibition in PCa cells [101, 102]. Cholesterol is an important component of biological membranes as it modulates the fluidity of the lipid bilayer and also forms lipid rafts, including caveolae that coordinate the activation of some oncogenic transduction signaling, including the PI3K/Akt pathway [69]. The cholesterol binding protein caveolin-1 (Cav-1), the major structural protein within caveolae, has been implicated as a promoter of PCa. Cav-1 expression correlates positively with aggressive PCa, metastatic potential, and its serum levels are elevated in patients with PCa [103]. Moreover, several studies showed that Cav-1 is capable of actively promoting the metastatic and castrate-resistant phenotypes [103] and its deletion in the PCa mouse model TRAMP decreases tumor growth and delays advanced PCa development [104]. During the synthesis of cholesterol, several metabolic intermediates are also generated, such as isoprenoids, geranylgeranyl pyrophosphate, and farnesyl pyrophosphate [105] (Figure 2). These protein modifiers can prenylate oncogenic proteins such as Ras and Rho, which regulate proliferation, migration, and invasion of PCa cells [106]. Finally, sterols have an important role in PCa development as they form the structural backbone for the synthesis of the steroid hormones androgens on which PCa cells depends for their growth and proliferation.
Statin drugs were designed to inhibit hepatic HMGCR, the rate-limiting enzyme of cholesterol synthesis, and were originally indicated for the treatment of cardiovascular disease; however, long term use of statin drugs has shown beneficial effects in reducing the risk of advanced PCa [reviewed in 96]. Therefore, significant efforts have been devoted to the understanding of the mechanisms of action of statins [reviewed in 106–108].
Increased intra-tumoral steroidogenesis
PCa cells respond to androgens through the action of the androgen receptor (AR), a nuclear receptor that controls PCa cell proliferation at all stages of disease, including late-stage, and castration resistant disease. As a result, therapy for advanced PCa almost invariably involves deprivation of androgens, resulting in decreased circulating androgens and PCa shrinkage. However, despite these initial responses, essentially all patients experience relapse and progression to CRPC and metastasis. It is now well recognized that CRPCs, including bone metastases, still express AR and maintain an active androgen-signaling pathway [109]. In addition intra-prostatic and/or tumor cell androgen synthesis occur despite androgen ablation therapy, and this is sufficient to activate AR and to partly explain the development of castration-resistance. Interestingly, increased cholesterol levels were identified in PCa bone metastases, along with increased expression of enzymes involved in steroidogenesis [110–112]. Substantial up-regulation was noted for CYP17A (which converts progestins to androgens), for 17-keto-reductase (which converts androstenedione to testosterone), and for 5α-reductase 1 (which converts testosterone into the more potent androgen dihydrotestosterone) (Figure 2). Recent studies have also suggest these intra-tumoral androgens may be derived from de novo synthesized cholesterol of the mevalonic acid pathway [110], as well as circulating cholesterol from the diet [113].
Novel drugs aimed at blocking de novo steroidogenesis in advanced PCa have been recently developed with promising results [3]. In 2011, Abiraterone acetate, an inhibitor of CYP17A, was approved by the FDA for the treatment of men with CRPC who received prior docetaxel. On December 10, 2012 the FDA extended its use in combination with prednisone for the treatment of men with CRPC before chemotherapy [http://www.fda.gov].
Oncogenes and tumor suppressors tightly regulate de novo lipogenesis
In addition to regulating cell proliferation and survival, it is now well established that oncogenic/tumor suppressor signaling pathways control the expression and activity of enzymes involved in lipid metabolism [114] (Figure 3). The major positive regulators of the lipogenic enzymes ACLY, FASN, and ACC are the PI3K/Akt and the mitogen-activated protein kinase (MAPK) pathways [115, 116]. Around 42% of primary and over 70% of metastastatic PCas show constitutive activation of PTEN/PI3K/Akt/mTORC1 pathway resulting predominantly, but not exclusively, from PTEN loss [117]. PTEN deletion in mice results in invasive prostatic adenocarcinoma [118]. PTEN knockdown in PCa cells induces the overexpression of FASN, whereas, treatment with a PI3K inhibitor or reintroduction of PTEN in PTEN-null PCa cells reduces FASN expression [119]. In contrast, inhibition of FASN reduces expression and activation of Akt [120, 121]. Additionally, cell growth suppression by FASN inhibition can be overcome by hyperactivation of Akt [121]. Increased FASN expression has also been shown to correlate with activation and nuclear localization of Akt in PCa tissues, suggesting a coordinate feedback between lipogenesis and PTEN/PI3K/Akt oncogenic signaling to promote PCa growth and tumor progression [63]. As previously mentioned, the synthesis of most enzymes involved in FA and cholesterol synthesis is regulated by SREBPs [100, 122]. Recently, the activity of Akt and its downstream effector mTORC1 (mainly involved in the regulation of protein synthesis) has been shown to be required for the nuclear accumulation of mature SREBP-1 [123]. Moreover, transcriptional profiling of cells deficient for the tuberous sclerosis complex 1 and 2 (TSC1/2), two negative regulators of mTORC1, revealed that SREBP-1 is an important component of the metabolic regulatory network downstream of this signaling axis [124]. mTORC1 also regulates the expression of SREBP-1 and it is required for the stimulation of lipogenesis in the liver [125]. Akt itself can also directly phosphorylate and activate lipogenic enzymes such as ACLY [126] and activate the expression of several genes involved in cholesterol and FA biosynthesis [127] (Figure 3). SREBP-1 function is also essential for Akt-dependent regulation of cell size, suggesting that Akt/mTORC1 signaling axis regulates protein and lipid synthesis in concert to fulfill increased request of building blocks by cancer cells [123]. The overexpression of the oncogene HER2, (found in approximately 20% of PCas), associated with high gleason grade, proliferation, tumor recurrence, and progression to androgen independence [128], also activates FA synthesis via PI3K/Akt signaling pathway [129]. This activation, however, does not occur transcriptionally via SREBP-1c, but rather, translationally through activation of mTORC1 and increased FASN protein [130]. Conversely, the inhibition of FASN attenuates HER2 oncogenic signaling and down-regulates activation of Akt and ERK 1/2 [131]. The oncogene c-Myc (Myc), involved in the early stages of PCa development [132], has been shown to enhance mitochondrial synthesis of Acetyl-CoA, which, in turn, contributes to significant increases in histone acetylation and FA biosynthesis in rapidly dividing cells. Using 13C-glucose as tracer, Morrish and coworkers have recently shown that Myc mobilizes glucose-derived acetyl groups for synthesis of the FA palmitate during cell cycle entry and it increases SFA levels [133]. Same results were obtained when cells were provided with glutamine, acetate, or acetoacetate as a sole carbon source [133]. The mechanisms responsible for Myc-induced enhanced contribution of glucose to cytosolic acetyl-CoA likely include Myc activation of mitochondrial biogenesis, increased glycolytic flux and entry of glucose carbons into the TCA cycle during cell cycle [133]. Myc-induced temporal activation of lipogenic genes, including acetoacetyl-CoA synthetase (AACS) and ACLY was also observed [134].
AMPK is a master energy sensor that responds to increased AMP/ATP ratio by turning on ATP-generating pathways, while switching off ATP-consuming ones, including mTORC1 and lipogenesis. The tumor-suppressor LKB1 negatively regulates the lipogenic enzymes by activating the downstream target AMPK. AMPK inhibits the enzyme FASN, ACLY, ACC, SCD1, and HMGCR both at the transcriptional and posttranslational level [135, 136]. The latter occurs by a direct inhibitory phosphorylation of ACC and HMGCR. AMPK-mediated reduction of the transcription of lipogenic enzymes is obtained by a direct phosphorylation of SREBP-1, thus preventing its proteolytic activation or by a reduction of its expression [137, 138] (Figure 3).
Patients with the metabolic syndrome (MS), a metabolic disease associated with increased risk of high grade PCa [139, 140], show deregulation of the LKB1/AMPK pathway [141], thus allowing enhanced lipogenesis to occur. Other tumor-suppressors such as Rb and p53 are also involved in the regulation of lipogenesis. Indeed, inactivation of the tumor suppressor Rb (reported in 5% of primary PCa and 37% of advanced tumors) enhances isoprenylation and activation of oncogenic N-Ras, through induction of SREBP-1 and 2 [142]. Moreover, genome-wide expression analysis identified the mevalonate pathway as significantly up-regulated in p53 mutant cells (which occurs in approximately 3–20% of PCas) [143] (Figure 3).
Interfering with lipid metabolism as a therapeutic approach
Activation of the FA and mevalonate synthesis pathways appears to be essential in both prostatic tumorigenesis and progression. Furthermore, lipid synthesis is especially active in metastatic, castration-resistant disease. Thus, significant effort has been made to target lipogenic enzymes and their key upstream regulators. Two attractive targets include the key enzyme for FA synthesis FASN and the master metabolic sensor AMPK (Figure 4).
Figure 4. Drug-mediated targeting of lipogenic enzymes.

A lot of efforts have been spent to identify inhibitors of de novo FA and cholesterol synthesis as well as inhibitors of intra-tumor de novo steroidogenesis to use in cancer therapy. In this diagram, inhibitors of FA and cholesterol synthesis are depicted in red and blue squares, respectively. The enzymes belonging to the two pathways are highlighted in corresponding red and blue colors. Novel small molecules targeting fatty acid synthase (FASN), AMP-activated protein kinase (AMPK), and intra-tumor synthesis of androgens have been recently developed, as discussed in the text. ACLY, ATP citrate lyase; ACC, Acetyl-CoA carboxylase; ACAT, Acetyl-CoA acetyltransferases; HMGCS, HMG-CoA synthase; HMGCR, HMG-CoA reductase; SCD1, stearoyl-CoA desaturase 1; CYP11A1, p450, family 11, subfamily A, polypeptide 1; CYP17A1; p450, family 11, subfamily A, polypeptide 1; 3β-HSD1, 2, 3-β-hydroxysteroid dehydrogenase/Δ-5-4 isomerase 1, 2; 17β-HSD, 17β-Hydroxysteroid dehydrogenase; SRD5A 1, 2, steroid-5α-reductase 1, 2.
Inhibitors of FASN
Several small molecule inhibitors of FASN have been described so far, including Orlistat, Cerulenin, C75 and C93. Orlistat, which was originally developed as an anti-obesity drug, is a potent irreversible inhibitor of FASN; its mechanism of action entails the formation of a covalent adduct with the thioesterase domain of the molecule. In 2004, Kridel and coworkers identified orlistat as an inhibitor of FASN with anti-tumor activity in PCa, following a proteomic screen for PCa specific enzymes [144]. Treatment with Orlistat resulted in the reduction of PC3 cell proliferation, induction of apoptosis, and inhibition of the growth of PC3 xenografts [reviewed in 145]. Several analogs have been recently developed to improve its pharmacological limitations, which include poor oral bioavailability and poor metabolic stability, low solubility, low cell permeability and lack of selectivity [145]. Early small molecule FASN inhibitors such as Cerulenin (inhibitor of the ketoacyl synthase domain) and C75 (inhibitor of the β-ketoacyl synthase, enoyl reductase and thioesterase domains) demonstrated significant antitumor activity. However, both of them induce dramatic effects on food intake and body weight in mice that limited their development for cancer therapy [reviewed in 146]. In order to overcome the lack of potency and the side effects of C75, naturally occurring thiolactomycins have been used as a template to develop a new class of type I FASN inhibitors (C93, C24, FAS31). C93 has been shown to effectively inhibit FASN without stimulating FA oxidation in lung cancer cells and to significantly inhibit the growth of non small cell lung and ovarian cancer xenograft tumors without causing anorexia and weight loss in the treated animals. Similar effects were seen for C247 and FAS31 in a transgenic model of breast cancer and in ovarian cancer xenograft models, respectively [146].
Currently, newer more potent FASN inhibitors have been identified through medicinal chemistry programs and high-throughput screening: Astra Zeneca developed a series of bisamide derivatives, Merck a series of 3-aryl-4-hydroxyquinolin-2(1H)-one derivatives, and GlaxoSmithKline produced GSK837149, a FASN inhibitor with nanomolar potency [reviewed in 146]. Scientists at GlaxoSmithKline have also recently patented the molecular structure of novel triazolone derivatives as FASN inhibitors (US20120316151 A1, www.google.com/patents). However, to the best of our knowledge, none of the FASN inhibitors available is currently in cancer clinical trials. One of the main reasons for the clinical failure of FASN inhibitors so far has been the anorexia and sustained body weight loss associated with FASN inhibition. Moreover, some available FASN inhibitors, such as Orlista, are poorly absorbed by the gastrointestinal tract. The other issue is the unanswered question as to whether exogenous lipids can rescue the effects of lipid synthesis inhibition.
Other inhibitors of lipogenesis
In addition to FASN, other lipogenic enzymes represent promising targets for cancer therapy. ACLY inhibition by RNA interference (RNAi) or with the chemical inhibitor SB-204990 results in decreased proliferation and survival of tumor cells displaying aerobic glycolysis, and in reduced tumor growth in vivo [55, 147]. In PCa cells, short hairpin RNA-mediated ACLY inhibition induces cell cycle arrest and apoptosis. However, these effects were only observed when cells were cultured under lipid-reduced growth conditions. Proliferation arrest was partially rescued by supplementing the media with FAs and/or cholesterol, indicating that the ACLY knockdown-mediated growth arrest is the result of either FAs or cholesterol starvation or both [148]. Potent and specific small molecule inhibitors of SCD1 activity, such as BZ36, showed anticancer activity both in vitro and in vivo models [73, 149, 150]. Knockdown of the FA synthesis rate-limiting enzyme ACCα by RNAi leads to inhibition of cell proliferation and induction of caspase-mediated apoptosis in highly lipogenic LNCaP cells. Moreover, treatment of PC3-M PCa cells with Soraphen A, a potent inhibitor of ACCα resulted in significant cell death [145]. Finally, the use of HMGCR inhibitors statins (in particular simvastatin) in PC3-M cells induced cell death, which was rescued by incubation of cells with cholesterol [148]. Treatment of both androgen-dependent and independent PCa cells (LNCaP and PC3 cells) with simvastatin significantly inhibited serum-induced cell migration, invasion, colony formation, and proliferation, which were rescued by adenovirus-mediated expression of constitutively active Akt (myristoylated Akt). Moreover, treatment of PC3 xenografts with simvastatin resulted in reduced tumor growth associated with decreased Akt activity and reduced PSA levels [151]. Finally, long-term use of statin drugs has shown beneficial effects in reducing the risk of advanced PCa [reviewed in 96], suggesting a positive relationship between cholesterol lowering levels and protection to PCa risk. At present, two clinical trials are ongoing to investigate the effect of statin therapy prior to prostatectomy (NCT00572468) or during external beam radiation therapy (NCT00580970).
AMPK activators
Since AMPK constitutes a hub for cellular metabolic and growth control, it represents an ideal therapeutic target for metabolic diseases as well as cancer. Several drugs which are largely used in the treatment of diabetes and other metabolic diseases, including metformin, have been shown to reduce cancer risk [152] and to reduce tumor cells proliferation and tumor growth in animal models mainly through AMPK activation [reviewed in 146]. AMPK activators fall in two classes: a) indirect activators including biguanides and thiazolidinediones b) direct activators, including AICAR and the last generation of small molecules.
Indirect AMPK activators
Metformin
Metformin is a derivative of guanidine and it is a widely prescribed oral drug used as first-line therapy for type 2 diabetes. The primary actions of metformin are inhibition of hepatic glucose production and reduction of insulin resistance in peripheral tissue, resulting in lower levels of circulating glucose and decreased plasma insulin. Recent evidence indicates that: a) diabetics under treatment with metformin show a reduced cancer incidence [152] and cancer-related mortality compared to patients exposed to sulfonylureas or insulin [153]; b) metformin use is associated with a 44% risk reduction in PCa cases compared to controls in Caucasian men [154]; c) breast cancer patients treated with neoadjuvant chemotherapy and metformin have significantly higher pathologic complete responses than patients not taking metformin (retrospective study) [155]. Thus, metformin’s potential antineoplastic activity is currently under investigation. Early promise has been reinforced by in vitro and xenograft mouse model studies highlighting metformin’s antitumor activity in PCa cells [156–158]. The mechanism of action for metformin’s anti-tumor effect is not completely clear and has been ascribed to both direct and indirect effect. At millimolar concentrations, metformin inhibits complex I of the respiratory chain resulting in increased AMP/ATP ratio and secondary activation of the AMPK pathway [159]. This results in inhibition of mTORC1 and p70S6kinase 1 (S6K1) activity and decreased translational efficiency in PCa cell lines [157]. However, several reports have recently shown that the inhibition of AMPK using siRNA does not prevent the anti-proliferative effect of metformin in PCa cells, and several other mechanisms involved in the anti-cancer effect of metformin have been proposed, suggesting that AMPK activation is only one of the pleiotropic effects of metformin [reviewed in 146, 158].
Thiazolidinediones
Like metformin, thiazolidinediones (TZD) are used clinically to treat type 2 diabetes. They activate AMPK likely via inhibition of complex I of the respiratory chain [160]. Currently, their activity as anticancer agents is under evaluation. Indeed, the TZD derivative CGP 52608 has been shown to have a strong cytostatic activity in LNCaP cells by reducing cell proliferation and by affecting cell cycle distribution through the modulation of the expression of cell cycle-related genes [161].
Direct AMPK activators
AICAR
The nucleoside 5-Aminoimidazole-4carboxamide-1-β-ribofuranoside (AICAR) was the first compound reported to activate AMPK in intact cells and in vivo, and has been widely used to investigate the downstream effects of AMPK activation in animals. AICAR is taken up into cells by adenosine transporters and is then converted by adenosine kinase to the monophosphorylated derivative ZMP, which is an analogue of 5′-MP and thus mimics several of its cellular effects. AICAR inhibits the growth of tumor cells in vitro and in vivo. In particular, it has been shown to inhibit PCa cell proliferation and tumor growth in PCa xenograft models [157, 162]. However, AICAR is not entirely specific for AMPK, exerting AMPK-independent effects mainly on AMP-regulated enzymes and mitochondrial oxidative phosphorylation. Moreover, it has limited oral bioavailability making it a poor candidate for long-term use in human [163]. Hence, design of novel small molecules AMPK activators with reduced off target effects is actively sought and was greatly enhanced by the publication of the crystal structure of AMPK subunits [164].
Small molecules AMPK activators
Abbott Laboratories have pioneered the area of developing subunit-specific AMPK activators and identified thienopyridone A-769662 in a screening of a chemical library of over 700,000 compounds, using partially purified AMPK αβγ complex [165]. As AMP, A-769662 (EC50 = 0.8 uM) activates AMPK via an allosteric mechanism and by inhibiting dephosphorylation at Thr172 [166, 167]. However, A-769662 does not involve the AMP binding sites on the γ subunit, since mutations that abolish AMP activation do not block A-769662. Instead, truncation mutagenesis of the carbohydrate-binding domain (CBD) as well as mutation of Ser108 to Ala in the β subunit abolishes A-769662 activation [167], suggesting A-769662 requirement for β subunits. Specifically, A-769662 is selective for the β1 isoform and does not activate AMPK heterotrimers containing β2 nor AMPK in tissues from β1 null mice [reviewed in 168]. In terms of upstream kinase requirements, A-769662 activates AMPK in HeLa cells, lacking LKB1 as well as in TAK1 null MEFs; thus the effects of A-769662 are independent of the AMPK upstream kinase utilized [reviewed in 168]. A-769662 has been shown to delay tumor development, including PCa, and decrease tumor incidence in PTEN+/– mice with a hypomorphic LKB1 allele [169], suggesting its potential anticancer properties. Unfortunately, although it has beneficial effects in vivo, the promise of A-769662 as a lead compound for drug development is tempered by its poor oral absorption and some recently reported AMPK-independent and non-specific effects [170, 171]. Thus, intense efforts have been directed to develop more specific small molecules AMPK activators. In 2008, Pang and colleagues identified PT1 as a novel direct activator of AMPK [172]. PT1 (EC50= 8–12 uM for AMPK α1 and α2, respectively) was identified in a targeted chemical library screening of 3,600 compounds, using the auto-inhibited α-subunit fragment [α1 (1–394)]. Since the constitutively active AMPK fragment α1 (1–312) is not activated by the PT1 compound, it is proposed that PT1 interacts with the auto-inhibitory domain and relieves inhibition of the kinase activity by this domain [reviewed in 168]. However, the effects of PT1 in vivo and its potential use as anticancer agent are still to be investigated. In 2011, Lee and coworkers proposed the novel compound OSU-53, which showed promising results in triple negative breast cancer [173]. OSU-53 directly stimulated recombinant AMPK activity (EC50 = 0.3 μM) and inhibited the viability and growth of breast cancer MDA-MB-231 and MDA-MB-468 cells, despite lack of LKB1 expression. Beyond AMPK-mediated effects on mTORC1 and lipogenesis, OSU-53 targeted multiple AMPK downstream pathways, including the protein phosphatase 2A-dependent dephosphorylation of Akt and it modulated energy homeostasis by suppressing FA biosynthesis and shifting the metabolism to oxidation (by the up-regulation of key regulators of mitochondrial biogenesis). Moreover, OSU-53 suppressed interleukin-6 (IL-6) production, thereby blocking subsequent Stat3 activation and inhibited hypoxia-induced epithelial-mesenchymal transition. In MDA-MB-231 tumor-bearing mice, daily oral administration of OSU-53 (50 and 100 mg/kg) suppressed tumor growth by 47–49% [173]. However, compound-induced protective autophagy reduced its anti-proliferative potency and further development. Thus, the design of novel more potent and specific AMPK direct activators is strongly sought.
Exploiting lipid deregulation to develop novel imaging techniques for diagnosis and therapy of PCa
Molecular imaging of tumor metabolism has gained considerable interest since preclinical studies indicated that alterations in the classical oncogenes and tumor suppressors are strictly intertwined with dysregulated cellular metabolism. A body of literature shows that deregulation of PI3K/Akt pathway, a very common feature of PCa, affects both glucose and lipid metabolism [reviewed in 46 and 174]. In addition, the tumor environment causes specific adaptations of cellular metabolism that increase the uptake of metabolic substrates [175]. Several clinical trials have now demonstrated that metabolic imaging can significantly impact patient management by improving tumor staging, restaging, treatment planning, and monitoring of tumor response to therapy [176]. However, the use of imaging to assess PCa remains a challenge, owing to the inherent heterogeneity of the disease. PET and PET-computed tomography (PET-CT) with the glucose analog 18F-FDG has become a routine clinical test for staging and restaging of most solid tumors [177]. However, PCa is not as glycolytic as the majority of other solid cancers and increased glycolysis is found mainly in the advanced stages of the disease. In contrast, an increase in FA synthesis seems to be both an early event in PCa tumorigenesis and correlated with the progression of the disease. Thus, metabolic imaging using lipid precursor tracers such as 11C-acetate, 18F-fluoro-acetate, 11C-choline, 18F-fluoro-choline appear to be a promising alternative. Both 11C-acetate and 11C-choline have shown increased sensitivity in the detection of primary PCa, metastasis, and recurrent disease compared with 18F-FDG PET [178]. In particular, a growing body of literature is pointing at the preferential use of 11C-choline for detecting primary PCa and restaging recurrence after treatment, whereas 11C-acetate holds promise for accurate detection of advanced PCa [179, 180]. However, both of the tracers fail to detect small local and metastatic sites [180] and 11C-acetate presents low sensitivity for disease detection at PSA levels less than 3ng/mL [178]. On a therapeutic perspective, 11C-acetate PET also proved to be a successful technique for monitoring FASN activity and PCa treatment with FASN inhibitors [181].
Whole body lipid metabolism and prostate cancer
Obesity, Metabolic syndrome and PCa
The increasing incidence of obesity and other metabolic conditions, including metabolic syndrome, poses a great challenge to global health. Metabolic syndrome (MS), is a metabolic disorder characterized by insulin resistance, hyperglycemia, dyslipidemia, hypertension, and predisposition to type 2 diabetes. MS is a frequent consequence of the metabolic dysregulation observed in PCa patients on androgen deprivation therapy [reviewed in 182]. In addition to cardiovascular disease and diabetes, a close relationship between obesity and risk for multiple types of cancer, including PCa, has emerged [183]. Indeed, a large body of epidemiologic evidence supports aa relationship between obesity and PCa progression, and suggests obesity as an adverse prognostic factor [184]. Obesity disrupts the dynamic role of the adipocyte in energy homeostasis, resulting in: a) an altered secretion of pro-inflammatory molecules [such as tumor necrosis factor alpha (TNFα, IL-6, and plasminogen activator inhibitor-1 (PAI1)] that contributes to insulin resistance, b) an altered production of the adipocytes-secreted cytokines (adipokines) leptin and adiponectin. Indeed, decreased serum levels of adiponectin and increased leptin are found to be associated with obesity [reviewed in 183]. Adiponectin and leptin seem to have opposing roles in PCa development, similar to their function in metabolic diseases [185, 186]. In fact, there is an inverse correlation between adiponectin levels and risk of PCa [187, 188] and in vitro studies confirmed adiponectin’s inhibitory effect on PCa cell growth through activation of AMPK [156, 189], although additional mechanisms to explain adiponectin’s anti-cancer effect have been recently proposed [reviewed in 183]. Additionally, obesity causes secondary changes that are related to insulin signaling such as insulin resistance and insulin-mediated increase of IGF-1 levels and subsequent activation of PI3K/mTORC1 pathway), as well as alterations in lipid metabolism that are also pro-tumorigenic [reviewed in 183]. While some studies have shown association of MS with a higher risk of PCa, the results are still inconclusive [190]. However, there is clear evidence of its relationship with increased risk of more aggressive PCa [139, 191]. The molecular mechanisms underpinning this positive association between MS and aggressive PCa are still not well defined but the involvement of AMPK inactivation is probably a central element [141]. In this regard, an association between polymorphisms in the PRKAA2 gene (encoding the α2 subunit of AMPK, which is responsible for the MS phenotype) and susceptibility to insulin resistance and diabetes has been found in the Japanese population [192, 193]. Interestingly, the same locus correlates with PCa risk [193], suggesting that AMPK dysregulation may provide a mechanistic link between MS and PCa. Finally, AMPK activating drugs that ameliorate MS, including metformin, have been associated with a reduced risk of PCa [154]. The role of intra-tumor de novo lipogenesis in the context of MS is still an unexplored area of investigation. In particular, there is no a clear understanding on whether increased circulating lipids can compensated for intra-tumor de novo lipids. However, our group has recently observed a positive association between FASN overexpression in PCa and higher body mass index (BMI), which is a component of MS, suggesting that in a metabolically permissive environment characterized by higher BMI, more abundant glucose, higher insulin/C-peptide levels, and low adiponectin, FASN may have an increased ability to exert its influence on either tumorigenesis or tumor maintenance [194].
Dietary fat and prostate cancer
The incidence and disease-specific mortality of PCa show marked geographic variation, being greatest in North America and Western Europe, and lowest in Asia [195]. These differences undoubtedly have a genetic component, but the contribution of the diet and the “western lifestyle” is nowadays gaining recognition. Indeed, diets around the globe vary in their total fat content, as well as their enrichment for specific lipids, which may contain varying quantities of SFAs, MUFAs, and PUFAs. Studies regarding the association between fat diet and risk of PCa are very controversial. This may be due to the fact that the majority of epidemiology studies rely on patient questionnaires to assess the implications of diet in PCa, and these can often inaccurately reflect the patient’s true diet [196]. In addition, it is often difficult in human studies to separate and distinguish the role of high caloric versus high fat intake. Many preclinical studies have tried to compensate this inaccuracy and to identify the dietary components affecting PCa development and progression by formulating controlled diets in mouse models. However, even preclinical studies remain controversial. A confounding factor between many preclinical studies is the variance in dietary formulation, specifically the source of dietary fat and the quantity of specific types of SFAs, MUFAs, and PUFAs, and especially their ratio to the respective control diet. Several epidemiological studies suggested that increased consumption of SFAs (palmitic acid, stearic acid), commonly included in meat, milk, eggs, palm oil, and coconut oil, correlates with increased risk of PCa and reduced progression-free survival [197–199]; however others report no significant association [200, 201]. The preclinical animal studies showed controversial results as well. Some studies suggest a diet high in SFA does not increase PCa growth or survival in mice [202]; however, others argue the opposite. Escobar and coworkers compared isocaloric diets of only 7% fat, and discovered that rats on a lard-derived diet displayed significantly increased prostate weight, testosterone, cell proliferation, and AR expression compared to rats on a linseed oil-derived diet [which is rich in α-linolenic acid (α–LNA)], suggesting that lard, which is rich in palmitic acid and oleic acid has cancer-promoting effects compared to an omega 3 PUFA-enriched diet [203]. SFA can be acquired through diet and produced de novo by FA synthesis. Despite several in vitro studies demonstrated that FASN inhibition-mediated PCa cell death could be rescued by the addition of palmitic acid, the primary product of FASN [34, 204], the rescue of FASN inhibition by dietary FAs has not been described in any preclinical models. Thus, it is possible that the cellular pools of dietary and de novo FAs supply different cellular processes, or perhaps the high rate of FA synthesis, which is required for PCa cell survival, cannot be compensated for by the diet.
Moreover, early epidemiological studies showed a lower incidence of certain cancers in mediterranean regions [205] consuming a diet rich in olive oil, whose main component is the oleic acid (delta-9 MUFA), however the effect of dietary MUFAs on PCa risk is still unclear, since there may be other components of the mediterranean diet that contribute to the reduced incidence [reviewed in 13]. Akin to the enigma concerning the role of dietary versus de novo SFAs in tumor maintance, dietary MUFAs and de novo MUFAs may also have distinct roles in PCa and no study so far has investigated the link between dietary MUFAs and SCD1. In contrast to SFAs and MUFAs, the two main types of PUFAs, omega-6 (n-6) and omega-3 (n-3), cannot be synthesized de novo in mammals and therefore need to be obtained through the diet. Linoleic acid (LA) is the most common dietary omega-6 PUFA and can be converted to arachidonic acid (AA), which is a precursor to many different eicosanoids [206]. LA is enriched in certain oils such as that of corn, sunflower, and safflower [207]. α-LNA is the major precursor to long-chain omega-3 PUFAs but its conversion is limited [208]. Therefore, omega-3 derived eicosanoids are primarily derived from dietary eicosapentaenoic acid (EPA) and docosanoids from docosahexaenoic acid (DHA). Although still largely unclear, multiple mechanisms by which PUFAs exert their effects in PCa have been proposed, including eicosanoid synthesis, angiogenesis, immune cell regulation, and membrane structure and function [reviewed in 13]. Moreover, despite a large body of literature supports that omega-6 PUFAs (except for omega-6 PUFA dihomolinolenic acid) increase PCa risk and disease progression while omega-3 PUFAs show a protective effect, the subject of good versus bad PUFAs is still debated [reviewed in 13, 209]. Recently, Wang and coworkers have shown that omega-3 PUFAs inhibit PTEN-null castration-resistant PCa in part by accelerating proteasome-degradation of the AR protein and suggested that omega-3 PUFAs supplementation in conjunction with androgen ablation may significantly delay the development of castration resistant PCa and have a beneficial effect on androgen ablation-induced osteoporosis [210]. Moreover, no study has refuted the benefits of a balanced PUFA diet and several animal studies have described an anticancer benefit from diet containing a low omega-6 to omega-3 ratio [211, 212], thus suggesting that the quality of dietary fat rather than quantity might impact PCa development.
Concluding Remarks
Thanks to the advent of lipidomic technologies and re-kindled interest in the field, significant insights on the role of lipid metabolism in cancer are accumulating. It’s becoming clear that both de novo and dietary lipids are important players in the development and progression of PCa. In particular, de novo lipogenesis, by modulating membrane lipid composition of cancer cells affects cancer cell biology far beyond the mere increase in membrane biogenesis, playing significant roles in processes such as including signal transduction, nutrient transport, ion channel activity, cell death signaling and energy metabolism. Furthermore, several evidence has underscored that the dysregulation of lipid metabolism in PCa is not confined to increased lipid biosynthesis but include aberrant lipid remodeling, FA catabolism, and synthesis of mevalonate-pathway intermediates, which are highly involved in the development and progression of cancer and particularly of hormonally-dependent cancers such as that of the prostate. A more detailed understanding of the implications of these changes is destined to place lipid metabolism at center stage of future cancer research, including PCa, and may open new therapeutic and diagnostic avenues.
Highlights.
Switch in lipid metabolism during PCa development and progression
Targeting of de novo lipogenesis as therapeutic strategy for PCa
Acetate and choline as successful tracers for PET imaging of PCa
Systemic metabolic disorders, including metabolic syndrome, increase the risk of PCa
The metabolism of dietary lipids versus de novo lipids synthesized intra-tumor
Acknowledgments
Prostate Cancer Foundation, the DF/HCC SPORE in Prostate Cancer (NIH/NCI P50 CA90381), and NIH grant RO1CA131945 to ML. GZ was supported by fellowships from the American Italian Cancer Foundation and from the Andrea e Libi Lorini Foundation.
Footnotes
Conflict of Interest
The authors declare no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Djulbegovic M, Neuberger MM, Dahm P. Prostate-cancer mortality after PSA screening. The New England journal of medicine. 2012;366:2228–2229. doi: 10.1056/NEJMc1204298. author reply 2230–2221. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. The lancet oncology. 2009;10:981–991. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bracarda S, Logothetis C, Sternberg CN, Oudard S. Current and emerging treatment modalities for metastatic castration-resistant prostate cancer. BJU international. 2011;107(Suppl 2):13–20. doi: 10.1111/j.1464-410X.2010.10036.x. [DOI] [PubMed] [Google Scholar]
- 5.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. The New England journal of medicine. 2004;351:1513–1520. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 6.Sartor AO. Progression of metastatic castrate-resistant prostate cancer: impact of therapeutic intervention in the post-docetaxel space. Journal of hematology & oncology. 2011;4:18. doi: 10.1186/1756-8722-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 8.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 9.Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6379–6383. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. International journal of cancer Journal international du cancer. 2002;98:19–22. doi: 10.1002/ijc.10127. [DOI] [PubMed] [Google Scholar]
- 11.Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, Bubley G, Balk S, Loda M. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Molecular cancer research: MCR. 2003;1:707–715. [PubMed] [Google Scholar]
- 12.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature reviews Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 13.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Current opinion in clinical nutrition and metabolic care. 2006;9:358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 15.Ramirez de Molina A, Gutierrez R, Ramos MA, Silva JM, Silva J, Bonilla F, Sanchez JJ, Lacal JC. Increased choline kinase activity in human breast carcinomas: clinical evidence for a potential novel antitumor strategy. Oncogene. 2002;21:4317–4322. doi: 10.1038/sj.onc.1205556. [DOI] [PubMed] [Google Scholar]
- 16.Ramirez de Molina A, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez J, Bonilla F, Rosell R, Lacal J. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochemical and biophysical research communications. 2002;296:580–583. doi: 10.1016/s0006-291x(02)00920-8. [DOI] [PubMed] [Google Scholar]
- 17.Ramirez de Molina A, Gallego-Ortega D, Sarmentero J, Banez-Coronel M, Martin-Cantalejo Y, Lacal JC. Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer research. 2005;65:5647–5653. doi: 10.1158/0008-5472.CAN-04-4416. [DOI] [PubMed] [Google Scholar]
- 18.Gallego-Ortega D, Ramirez de Molina A, Ramos MA, Valdes-Mora F, Barderas MG, Sarmentero-Estrada J, Lacal JC. Differential role of human choline kinase alpha and beta enzymes in lipid metabolism: implications in cancer onset and treatment. PloS one. 2009;4:e7819. doi: 10.1371/journal.pone.0007819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nature reviews Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 20.Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Science’s STKE: signal transduction knowledge environment. 2006;2006:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 21.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nature reviews Molecular cell biology. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 22.Nadolski MJ, Linder ME. Protein lipidation. The FEBS journal. 2007;274:5202–5210. doi: 10.1111/j.1742-4658.2007.06056.x. [DOI] [PubMed] [Google Scholar]
- 23.Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS letters. 2010;584:1923–1930. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 24.Bergum C, Zoratti G, Boerner J, List K. Strong expression association between matriptase and its substrate prostasin in breast cancer. Journal of cellular physiology. 2012;227:1604–1609. doi: 10.1002/jcp.22877. [DOI] [PubMed] [Google Scholar]
- 25.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–452. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 26.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, Takada S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Developmental cell. 2006;11:791–801. doi: 10.1016/j.devcel.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Fiorentino M, Zadra G, Palescandolo E, Fedele G, Bailey D, Fiore C, Nguyen PL, Migita T, Zamponi R, Di Vizio D, Priolo C, Sharma C, Xie W, Hemler ME, Mucci L, Giovannucci E, Finn S, Loda M. Overexpression of fatty acid synthase is associated with palmitoylation of Wnt1 and cytoplasmic stabilization of beta-catenin in prostate cancer. Lab Invest. 2008;88:1340–1348. doi: 10.1038/labinvest.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 30.Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annual review of immunology. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 31.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate cancer and prostatic diseases. 2006;9:230–234. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 33.Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, Van Poppel H, Baert L, Goossens K, Heyns W, Verhoeven G. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int J Cancer. 2000;88:176–179. doi: 10.1002/1097-0215(20001015)88:2<176::aid-ijc5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 34.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101:519–532. doi: 10.1093/jnci/djp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vance DE, Vance JE. Biochemistry of Lipids, Lipoproteins and Membranes. 4. Elsevier Science; 2002. [Google Scholar]
- 36.Enoch HG, Catala A, Strittmatter P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. The Journal of biological chemistry. 1976;251:5095–5103. [PubMed] [Google Scholar]
- 37.Li J, Ding SF, Habib NA, Fermor BF, Wood CB, Gilmour RS. Partial characterization of a cDNA for human stearoyl-CoA desaturase and changes in its mRNA expression in some normal and malignant tissues. International journal of cancer Journal international du cancer. 1994;57:348–352. doi: 10.1002/ijc.2910570310. [DOI] [PubMed] [Google Scholar]
- 38.Lu J, Pei H, Kaeck M, Thompson HJ. Gene expression changes associated with chemically induced rat mammary carcinogenesis. Molecular carcinogenesis. 1997;20:204–215. doi: 10.1002/(sici)1098-2744(199710)20:2<204::aid-mc7>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 39.Thai SF, Allen JW, DeAngelo AB, George MH, Fuscoe JC. Detection of early gene expression changes by differential display in the livers of mice exposed to dichloroacetic acid. Carcinogenesis. 2001;22:1317–1322. doi: 10.1093/carcin/22.8.1317. [DOI] [PubMed] [Google Scholar]
- 40.Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Nagai R, Ishibashi S, Kadowaki T, Makuuchi M, Ohnishi S, Osuga J, Yamada N. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. 2005;41:1316–1322. doi: 10.1016/j.ejca.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 41.Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avances C, Allory Y, de la Taille A, Culine S, Blancou H, Cristol JP, Michel F, Sardet C, Fajas L. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Molecular cancer therapeutics. 2010;9:1740–1754. doi: 10.1158/1535-7163.MCT-09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, Ashida S, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer research. 2009;69:8133–8140. doi: 10.1158/0008-5472.CAN-09-0775. [DOI] [PubMed] [Google Scholar]
- 43.Jayakumar A, Tai MH, Huang WY, al-Feel W, Hsu M, Abu-Elheiga L, Chirala SS, Wakil SJ. Human fatty acid synthase: properties and molecular cloning. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8695–8699. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer research. 1953;13:27–29. [PubMed] [Google Scholar]
- 45.Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition. 2000;16:202–208. doi: 10.1016/s0899-9007(99)00266-x. [DOI] [PubMed] [Google Scholar]
- 46.Santos CR, Schulze A. Lipid metabolism in cancer. The FEBS journal. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 47.Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA-519) as a predictor of progression of prostate cancer. Human pathology. 1996;27:917–921. doi: 10.1016/s0046-8177(96)90218-x. [DOI] [PubMed] [Google Scholar]
- 48.Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer research. 2007;67:1262–1269. doi: 10.1158/0008-5472.CAN-06-1794. [DOI] [PubMed] [Google Scholar]
- 49.Swinnen JV, Esquenet M, Goossens K, Heyns W, Verhoeven G. Androgens stimulate fatty acid synthase in the human prostate cancer cell line LNCaP. Cancer research. 1997;57:1086–1090. [PubMed] [Google Scholar]
- 50.Swinnen JV, Heemers H, Deboel L, Foufelle F, Heyns W, Verhoeven G. Stimulation of tumor-associated fatty acid synthase expression by growth factor activation of the sterol regulatory element-binding protein pathway. Oncogene. 2000;19:5173–5181. doi: 10.1038/sj.onc.1203889. [DOI] [PubMed] [Google Scholar]
- 51.Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Molecular cancer research: MCR. 2012;10:133–142. doi: 10.1158/1541-7786.MCR-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer research. 2003;63:3799–3804. [PubMed] [Google Scholar]
- 53.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer research. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 54.Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer research. 2005;65:6719–6725. doi: 10.1158/0008-5472.CAN-05-0571. [DOI] [PubMed] [Google Scholar]
- 55.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer research. 2007;67:8180–8187. doi: 10.1158/0008-5472.CAN-07-0389. [DOI] [PubMed] [Google Scholar]
- 57.Moon JS, Jin WJ, Kwak JH, Kim HJ, Yun MJ, Kim JW, Park SW, Kim KS. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. The Biochemical journal. 2011;433:225–233. doi: 10.1042/BJ20101104. [DOI] [PubMed] [Google Scholar]
- 58.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Fu YM, Meadows GG. Differential effects of specific amino acid restriction on glucose metabolism, reduction/oxidation status and mitochondrial damage in DU145 and PC3 prostate cancer cells. Oncology letters. 2011;2:349–355. doi: 10.3892/ol.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G. Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochemical and biophysical research communications. 2003;302:898–903. doi: 10.1016/s0006-291x(03)00265-1. [DOI] [PubMed] [Google Scholar]
- 63.Van de Sande T, Roskams T, Lerut E, Joniau S, Van Poppel H, Verhoeven G, Swinnen JV. High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. The Journal of pathology. 2005;206:214–219. doi: 10.1002/path.1760. [DOI] [PubMed] [Google Scholar]
- 64.Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer research. 2003;63:7330–7337. [PubMed] [Google Scholar]
- 65.Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. Journal of cellular physiology. 2011;226:299–308. doi: 10.1002/jcp.22400. [DOI] [PubMed] [Google Scholar]
- 66.Hochachka PW. Living Without Oxygen. Harvard University Press; Cambridge, USA: 1980. [Google Scholar]
- 67.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, Vanderhoydonc F, Smans K, Waelkens E, Verhoeven G, Swinnen JV. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer research. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 69.Staubach S, Hanisch FG. Lipid rafts: signaling and sorting platforms of cells and their roles in cancer. Expert review of proteomics. 2011;8:263–277. doi: 10.1586/epr.11.2. [DOI] [PubMed] [Google Scholar]
- 70.Willemarck N, Rysman E, Brusselmans K, Van Imschoot G, Vanderhoydonc F, Moerloose K, Lerut E, Verhoeven G, van Roy F, Vleminckx K, Swinnen JV. Aberrant activation of fatty acid synthesis suppresses primary cilium formation and distorts tissue development. Cancer research. 2010;70:9453–9462. doi: 10.1158/0008-5472.CAN-10-2324. [DOI] [PubMed] [Google Scholar]
- 71.Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer. Cancer research. 2006;66:6463–6467. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- 72.Plotnikova OV, Golemis EA, Pugacheva EN. Cell cycle-dependent ciliogenesis and cancer. Cancer research. 2008;68:2058–2061. doi: 10.1158/0008-5472.CAN-07-5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Igal RA. Stearoyl-CoA desaturase-1: a novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis. 2010;31:1509–1515. doi: 10.1093/carcin/bgq131. [DOI] [PubMed] [Google Scholar]
- 74.Schaffer JE. Lipotoxicity: when tissues overeat. Current opinion in lipidology. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 75.Leonard AE, Pereira SL, Sprecher H, Huang YS. Elongation of long-chain fatty acids. Progress in lipid research. 2004;43:36–54. doi: 10.1016/s0163-7827(03)00040-7. [DOI] [PubMed] [Google Scholar]
- 76.Kihara A. Very long-chain fatty acids: elongation, physiology and related disorders. Journal of biochemistry. 2012;152:387–395. doi: 10.1093/jb/mvs105. [DOI] [PubMed] [Google Scholar]
- 77.Suneja SK, Osei P, Cook L, Nagi MN, Cinti DL. Enzyme site-specific changes in hepatic microsomal fatty acid chain elongation in streptozotocin-induced diabetic rats. Biochimica et biophysica acta. 1990;1042:81–85. doi: 10.1016/0005-2760(90)90059-7. [DOI] [PubMed] [Google Scholar]
- 78.Farese RV, Jr, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Price DT, Coleman RE, Liao RP, Robertson CN, Polascik TJ, DeGrado TR. Comparison of [18 F]fluorocholine and [18 F]fluorodeoxyglucose for positron emission tomography of androgen dependent and androgen independent prostate cancer. The Journal of urology. 2002;168:273–280. [PubMed] [Google Scholar]
- 81.Effert PJ, Bares R, Handt S, Wolff JM, Bull U, Jakse G. Metabolic imaging of untreated prostate cancer by positron emission tomography with 18fluorine-labeled deoxyglucose. The Journal of urology. 1996;155:994–998. [PubMed] [Google Scholar]
- 82.Liu Y, Zuckier LS, Ghesani NV. Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach. Anticancer research. 2010;30:369–374. [PubMed] [Google Scholar]
- 83.Zha S, Ferdinandusse S, Hicks JL, Denis S, Dunn TA, Wanders RJ, Luo J, De Marzo AM, Isaacs WB. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. The Prostate. 2005;63:316–323. doi: 10.1002/pros.20177. [DOI] [PubMed] [Google Scholar]
- 84.Luo J, Zha S, Gage WR, Dunn TA, Hicks JL, Bennett CJ, Ewing CM, Platz EA, Ferdinandusse S, Wanders RJ, Trent JM, Isaacs WB, De Marzo AM. Alpha-methylacyl-CoA racemase: a new molecular marker for prostate cancer. Cancer research. 2002;62:2220–2226. [PubMed] [Google Scholar]
- 85.Kumar-Sinha C, Shah RB, Laxman B, Tomlins SA, Harwood J, Schmitz W, Conzelmann E, Sanda MG, Wei JT, Rubin MA, Chinnaiyan AM. Elevated alpha-methylacyl-CoA racemase enzymatic activity in prostate cancer. The American journal of pathology. 2004;164:787–793. doi: 10.1016/s0002-9440(10)63167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. The Journal of biological chemistry. 1997;272:28875–28881. doi: 10.1074/jbc.272.46.28875. [DOI] [PubMed] [Google Scholar]
- 87.Arver S. Zinc and zinc ligands in human seminal plasma. III. The principal low molecular weight zinc ligand in prostatic secretion and seminal plasma. Acta physiologica Scandinavica. 1982;116:67–73. doi: 10.1111/j.1748-1716.1982.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 88.Ford WC, Harrison A. The role of citrate in determining the activity of calcium ions in human semen. International journal of andrology. 1984;7:198–202. doi: 10.1111/j.1365-2605.1984.tb00777.x. [DOI] [PubMed] [Google Scholar]
- 89.Marengo S. Prostate Physiology and regulation. In: Resnick MI, Thompson IM, editors. Advanced Therapy of Prostate Disease. Hamilton, Ontario, Canada: Bs Decker Inc; 2000. pp. 92–100. [Google Scholar]
- 90.Delvecchio FC, Brizuela RM, Khan SR, Byer K, Li Z, Zhong P, Preminger GM. Citrate and vitamin E blunt the shock wave-induced free radical surge in an in vitro cell culture model. Urological research. 2005;33:448–452. doi: 10.1007/s00240-005-0506-2. [DOI] [PubMed] [Google Scholar]
- 91.Costello LC, Franklin RB. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: connecting the dots. Molecular cancer. 2006;5:17. doi: 10.1186/1476-4598-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Costello LC, Franklin RB. The intermediary metabolism of the prostate: a key to understanding the pathogenesis and progression of prostate malignancy. Oncology. 2000;59:269–282. doi: 10.1159/000012183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. The Journal of biological chemistry. 2006;281:37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 94.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochimica et biophysica acta. 2011;1807:726–734. doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 95.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. The Journal of clinical investigation. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pelton K, Freeman MR, Solomon KR. Cholesterol and prostate cancer. Current opinion in pharmacology. 2012;12:751–759. doi: 10.1016/j.coph.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. The Journal of clinical investigation. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Llaverias G, Danilo C, Wang Y, Witkiewicz AK, Daumer K, Lisanti MP, Frank PG. A Western-type diet accelerates tumor progression in an autochthonous mouse model of prostate cancer. The American journal of pathology. 2010;177:3180–3191. doi: 10.2353/ajpath.2010.100568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 100.Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86:839–848. doi: 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 101.Chen M Hughes-Fulford. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2, International journal of cancer. Journal international du cancer. 2001;91:41–45. doi: 10.1002/1097-0215(20010101)91:1<41::aid-ijc1009>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 102.Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, Nelson CC. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer research. 2004;64:2212–2221. doi: 10.1158/0008-5472.can-2148-2. [DOI] [PubMed] [Google Scholar]
- 103.Freeman MR, Yang W, Di Vizio D. Caveolin-1 and prostate cancer progression. Advances in experimental medicine and biology. 2012;729:95–110. doi: 10.1007/978-1-4614-1222-9_7. [DOI] [PubMed] [Google Scholar]
- 104.Williams TM, Hassan GS, Li J, Cohen AW, Medina F, Frank PG, Pestell RG, Di Vizio D, Loda M, Lisanti MP. Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. The Journal of biological chemistry. 2005;280:25134–25145. doi: 10.1074/jbc.M501186200. [DOI] [PubMed] [Google Scholar]
- 105.Freeman MR, Solomon KR. Cholesterol and prostate cancer. Journal of cellular biochemistry. 2004;91:54–69. doi: 10.1002/jcb.10724. [DOI] [PubMed] [Google Scholar]
- 106.Roy M, Kung HJ, Ghosh PM. Statins and prostate cancer: role of cholesterol inhibition vs. prevention of small GTP-binding proteins. American journal of cancer research. 2011;1:542–561. [PMC free article] [PubMed] [Google Scholar]
- 107.Mener DJ. Prostate specific antigen reduction following statin therapy: Mechanism of action and review of the literature. IUBMB life. 2010;62:584–590. doi: 10.1002/iub.355. [DOI] [PubMed] [Google Scholar]
- 108.Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31:4967–4978. doi: 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- 109.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, Knudsen B, Hess DL, Nelson CC, Matsumoto AM, Bremner WJ, Gleave ME, Nelson PS. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer research. 2007;67:5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 110.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer research. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 111.Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Molecular and cellular endocrinology. 2008;295:115–120. doi: 10.1016/j.mce.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mostaghel EA, Solomon KR, Pelton K, Freeman MR, Montgomery RB. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PloS one. 2012;7:e30062. doi: 10.1371/journal.pone.0030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer discovery. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang YA, Han WF, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Experimental cell research. 2002;279:80–90. doi: 10.1006/excr.2002.5600. [DOI] [PubMed] [Google Scholar]
- 116.Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. British journal of cancer. 2009;100:1369–1372. doi: 10.1038/sj.bjc.6605007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 119.Van de Sande T, De Schrijver E, Heyns W, Verhoeven G, Swinnen JV. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer research. 2002;62:642–646. [PubMed] [Google Scholar]
- 120.Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR. Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene. 2005;24:3574–3582. doi: 10.1038/sj.onc.1208463. [DOI] [PubMed] [Google Scholar]
- 121.Tomek K, Wagner R, Varga F, Singer CF, Karlic H, Grunt TW. Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Molecular cancer research : MCR. 2011;9:1767–1779. doi: 10.1158/1541-7786.MCR-10-0467. [DOI] [PubMed] [Google Scholar]
- 122.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of clinical investigation. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell metabolism. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. The Journal of biological chemistry. 2002;277:33895–33900. doi: 10.1074/jbc.M204681200. [DOI] [PubMed] [Google Scholar]
- 127.Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 128.Signoretti S, Montironi R, Manola J, Altimari A, Tam C, Bubley G, Balk S, Thomas G, Kaplan I, Hlatky L, Hahnfeldt P, Kantoff P, Loda M. Her-2-neu expression and progression toward androgen independence in human prostate cancer. Journal of the National Cancer Institute. 2000;92:1918–1925. doi: 10.1093/jnci/92.23.1918. [DOI] [PubMed] [Google Scholar]
- 129.Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, Chinnaiyan AM. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer research. 2003;63:132–139. [PubMed] [Google Scholar]
- 130.Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, Park BW, Kim KS. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. The Journal of biological chemistry. 2007;282:26122–26131. doi: 10.1074/jbc.M702854200. [DOI] [PubMed] [Google Scholar]
- 131.Alli PM, Pinn ML, Jaffee EM, McFadden JM, Kuhajda FP. Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene. 2005;24:39–46. doi: 10.1038/sj.onc.1208174. [DOI] [PubMed] [Google Scholar]
- 132.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes & cancer. 2010;1:617–628. doi: 10.1177/1947601910379132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D. Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. The Journal of biological chemistry. 2010;285:36267–36274. doi: 10.1074/jbc.M110.141606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morrish F, Neretti N, Sedivy JM, Hockenbery DM. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008;7:1054–1066. doi: 10.4161/cc.7.8.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804:581–591. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 137.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell metabolism. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grundmark B, Garmo H, Loda M, Busch C, Holmberg L, Zethelius B. The metabolic syndrome and the risk of prostate cancer under competing risks of death from other causes. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:2088–2096. doi: 10.1158/1055-9965.EPI-10-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morales R, Suarez C, Ropero J, Nunez I, Planas J, Placer J, María Valverde C, Maldonado X, Morote J, Carles J. The influence of metabolic syndrome on prostate cancer risk detection and its aggressiveness. J Clin Oncol. 2012;30 (suppl 5; abstr 226) [Google Scholar]
- 141.Luo Z, Saha AK, Xiang X, Ruderman NB. AMPK, the metabolic syndrome and cancer. Trends in pharmacological sciences. 2005;26:69–76. doi: 10.1016/j.tips.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 142.Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, Takahashi C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer cell. 2009;15:255–269. doi: 10.1016/j.ccr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 143.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen-Dale AL, Levine AJ, Bargonetti J, Prives C. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–258. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer research. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 145.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010;6:551–562. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. The Journal of pathology. 2011;223:283–294. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer research. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- 148.Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Molecular cancer therapeutics. 2012;11:1925–1935. doi: 10.1158/1535-7163.MCT-12-0095. [DOI] [PubMed] [Google Scholar]
- 149.Liu G, Lynch JK, Freeman J, Liu B, Xin Z, Zhao H, Serby MD, Kym PR, Suhar TS, Smith HT, Cao N, Yang R, Janis RS, Krauser JA, Cepa SP, Beno DW, Sham HL, Collins CA, Surowy TK, Camp HS. Discovery of potent, selective, orally bioavailable stearoyl-CoA desaturase 1 inhibitors. Journal of medicinal chemistry. 2007;50:3086–3100. doi: 10.1021/jm070219p. [DOI] [PubMed] [Google Scholar]
- 150.Koltun DO, Zilbershtein TM, Migulin VA, Vasilevich NI, Parkhill EQ, Glushkov AI, McGregor MJ, Brunn SA, Chu N, Hao J, Mollova N, Leung K, Chisholm JW, Zablocki J. Potent, orally bioavailable, liver-selective stearoyl-CoA desaturase (SCD) inhibitors. Bioorganic & medicinal chemistry letters. 2009;19:4070–4074. doi: 10.1016/j.bmcl.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 151.Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. The Journal of pharmacology and experimental therapeutics. 2011;336:496–505. doi: 10.1124/jpet.110.174870. [DOI] [PubMed] [Google Scholar]
- 152.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care. 2006;29:1990–1991. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- 154.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer causes & control : CCC. 2009;20:1617–1622. doi: 10.1007/s10552-009-9407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 2008;1:369–375. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 157.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 158.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF, Giorgetti-Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer research. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 159.Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. doi: 10.1053/j.gastro.2006.07.032. author reply 974–975. [DOI] [PubMed] [Google Scholar]
- 160.Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 161.Moretti RM, Montagnani Marelli M, Motta M, Limonta P. Oncostatic activity of a thiazolidinedione derivative on human androgen-dependent prostate cancer cells. International journal of cancer Journal international du cancer. 2001;92:733–737. doi: 10.1002/1097-0215(20010601)92:5<733::aid-ijc1254>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 162.Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochemical and biophysical research communications. 2004;321:161–167. doi: 10.1016/j.bbrc.2004.06.133. [DOI] [PubMed] [Google Scholar]
- 163.Guigas B, Sakamoto K, Taleux N, Reyna SM, Musi N, Viollet B, Hue L. Beyond AICA riboside: in search of new specific AMP-activated protein kinase activators. IUBMB life. 2009;61:18–26. doi: 10.1002/iub.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 165.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 166.Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. The Journal of biological chemistry. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. The Journal of biological chemistry. 2007;282:32539–32548. doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- 168.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiological reviews. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 169.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. The Biochemical journal. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 170.Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS letters. 2008;582:2650–2654. doi: 10.1016/j.febslet.2008.06.044. [DOI] [PubMed] [Google Scholar]
- 171.Garcia-Garcia C, Fumarola C, Navaratnam N, Carling D, Lopez-Rivas A. AMPK-independent down-regulation of cFLIP and sensitization to TRAIL-induced apoptosis by AMPK activators. Biochem Pharmacol. 79:853–863. doi: 10.1016/j.bcp.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 172.Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, Shao W, Su MB, Li JY, Nan FJ, Li J. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. The Journal of biological chemistry. 2008;283:16051–16060. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee KH, Hsu EC, Guh JH, Yang HC, Wang D, Kulp SK, Shapiro CL, Chen CS. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. The Journal of biological chemistry. 2011;286:39247–39258. doi: 10.1074/jbc.M111.264598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bui T, Thompson CB. Cancer’s sweet tooth. Cancer cell. 2006;9:419–420. doi: 10.1016/j.ccr.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 175.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nature reviews Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 176.Plathow C, Weber WA. Tumor cell metabolism imaging. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2008;49(Suppl 2):43S–63S. doi: 10.2967/jnumed.107.045930. [DOI] [PubMed] [Google Scholar]
- 177.Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, Cheson BD, O’Shaughnessy J, Guyton KZ, Mankoff DA, Shankar L, Larson SM, Sigman CC, Schilsky RL, Sullivan DC. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- 178.Czernin J, Benz MR, Allen-Auerbach MS. PET Imaging of Prostate Cancer Using C-Acetate. PET clinics. 2009;4:163–172. doi: 10.1016/j.cpet.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Emonds KM, Swinnen JV, van Weerden WM, Vanderhoydonc F, Nuyts J, Mortelmans L, Mottaghy FM. Do androgens control the uptake of 18F-FDG, 11C-choline and 11C-acetate in human prostate cancer cell lines? European journal of nuclear medicine and molecular imaging. 2011;38:1842–1853. doi: 10.1007/s00259-011-1861-6. [DOI] [PubMed] [Google Scholar]
- 180.Fox JJ, Schoder H, Larson SM. Molecular imaging of prostate cancer. Current opinion in urology. 2012;22:320–327. doi: 10.1097/MOU.0b013e32835483d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vavere AL, Kridel SJ, Wheeler FB, Lewis JS. 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J Nucl Med. 2008;49:327–334. doi: 10.2967/jnumed.107.046672. [DOI] [PubMed] [Google Scholar]
- 182.Nobes JP, Langley SE, Laing RW. Metabolic syndrome and prostate cancer: a review. Clin Oncol (R Coll Radiol) 2009;21:183–191. doi: 10.1016/j.clon.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 183.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nature reviews Cancer. 2011;11:886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 184.Freedland SJ. Obesity and prostate cancer: a growing problem. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:6763–6766. doi: 10.1158/1078-0432.CCR-05-1305. [DOI] [PubMed] [Google Scholar]
- 185.Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med. 2008;14:741–751. doi: 10.2119/2008-00058.Rabe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grossmann ME, Mizuno NK, Bonorden MJ, Ray A, Sokolchik I, Narasimhan ML, Cleary MP. Role of the adiponectin leptin ratio in prostate cancer. Oncology research. 2009;18:269–277. doi: 10.3727/096504009x12596189659367. [DOI] [PubMed] [Google Scholar]
- 187.Michalakis K, Williams CJ, Mitsiades N, Blakeman J, Balafouta-Tselenis S, Giannopoulos A, Mantzoros CS. Serum adiponectin concentrations and tissue expression of adiponectin receptors are reduced in patients with prostate cancer: a case control study. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007;16:308–313. doi: 10.1158/1055-9965.EPI-06-0621. [DOI] [PubMed] [Google Scholar]
- 188.Li H, Stampfer MJ, Mucci L, Rifai N, Qiu W, Kurth T, Ma J. A 25-year prospective study of plasma adiponectin and leptin concentrations and prostate cancer risk and survival. Clinical chemistry. 2010;56:34–43. doi: 10.1373/clinchem.2009.133272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bub JD, Miyazaki T, Iwamoto Y. Adiponectin as a growth inhibitor in prostate cancer cells. Biochemical and biophysical research communications. 2006;340:1158–1166. doi: 10.1016/j.bbrc.2005.12.103. [DOI] [PubMed] [Google Scholar]
- 190.Zadra G, Priolo C, Patnaik A, Loda M. New strategies in prostate cancer: targeting lipogenic pathways and the energy sensor AMPK. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:3322–3328. doi: 10.1158/1078-0432.CCR-09-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Morote, Ropero J, Planas J, Bastaros JM, Delgado G, Placer J, Celma A, de Torres IM, Carles J, Reventos J, Doll A. Metabolic syndrome increases the risk of aggressive prostate cancer detection. BJU international. 2012 doi: 10.1111/j.1464-410X.2012.11406.x. [DOI] [PubMed] [Google Scholar]
- 192.Horikoshi M, Hara K, Ohashi J, Miyake K, Tokunaga K, Ito C, Kasuga M, Nagai R, Kadowaki T. A polymorphism in the AMPKalpha2 subunit gene is associated with insulin resistance and type 2 diabetes in the Japanese population. Diabetes. 2006;55:919–923. doi: 10.2337/diabetes.55.04.06.db05-0727. [DOI] [PubMed] [Google Scholar]
- 193.Matsui H, Suzuki K, Ohtake N, Nakata S, Takeuchi T, Yamanaka H, Inoue I. Genomewide linkage analysis of familial prostate cancer in the Japanese population. Journal of human genetics. 2004;49:9–15. doi: 10.1007/s10038-003-0099-y. [DOI] [PubMed] [Google Scholar]
- 194.Nguyen PL, Ma J, Chavarro JE, Freedman ML, Lis R, Fedele G, Fiore C, Qiu W, Fiorentino M, Finn S, Penney KL, Eisenstein A, Schumacher FR, Mucci LA, Stampfer MJ, Giovannucci E, Loda M. Fatty acid synthase polymorphisms, tumor expression, body mass index, prostate cancer risk, and survival. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28:3958–3964. doi: 10.1200/JCO.2009.27.0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, Bray F. International variation in prostate cancer incidence and mortality rates. European urology. 2012;61:1079–1092. doi: 10.1016/j.eururo.2012.02.054. [DOI] [PubMed] [Google Scholar]
- 196.Dennis LK, Snetselaar LG, Smith BJ, Stewart RE, Robbins ME. Problems with the assessment of dietary fat in prostate cancer studies. American journal of epidemiology. 2004;160:436–444. doi: 10.1093/aje/kwh243. [DOI] [PubMed] [Google Scholar]
- 197.Whittemore AS, Kolonel LN, Wu AH, John EM, Gallagher RP, Howe GR, Burch JD, Hankin J, Dreon DM, West DW, et al. Prostate cancer in relation to diet, physical activity, and body size in blacks, whites, and Asians in the United States and Canada. Journal of the National Cancer Institute. 1995;87:652–661. doi: 10.1093/jnci/87.9.652. [DOI] [PubMed] [Google Scholar]
- 198.Strom SS, Yamamura Y, Forman MR, Pettaway CA, Barrera SL, DiGiovanni J. Saturated fat intake predicts biochemical failure after prostatectomy. International journal of cancer Journal international du cancer. 2008;122:2581–2585. doi: 10.1002/ijc.23414. [DOI] [PubMed] [Google Scholar]
- 199.Park SY, Wilkens LR, Henning SM, Le Marchand L, Gao K, Goodman MT, Murphy SP, Henderson BE, Kolonel LN. Circulating fatty acids and prostate cancer risk in a nested case-control study: the Multiethnic Cohort. Cancer causes & control : CCC. 2009;20:211–223. doi: 10.1007/s10552-008-9236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Crowe FL, Allen NE, Appleby PN, Overvad K, Aardestrup IV, Johnsen NF, Tjonneland A, Linseisen J, Kaaks R, Boeing H, Kroger J, Trichopoulou A, Zavitsanou A, Trichopoulos D, Sacerdote C, Palli D, Tumino R, Agnoli C, Kiemeney LA, Bueno-de-Mesquita HB, Chirlaque MD, Ardanaz E, Larranaga N, Quiros JR, Sanchez MJ, Gonzalez CA, Stattin P, Hallmans G, Bingham S, Khaw KT, Rinaldi S, Slimani N, Jenab M, Riboli E, Key TJ. Fatty acid composition of plasma phospholipids and risk of prostate cancer in a case-control analysis nested within the European Prospective Investigation into Cancer and Nutrition. The American journal of clinical nutrition. 2008;88:1353–1363. doi: 10.3945/ajcn.2008.26369. [DOI] [PubMed] [Google Scholar]
- 201.Jackson MD, Walker SP, Simpson-Smith CM, Lindsay CM, Smith G, McFarlane-Anderson N, Bennett FI, Coard KC, Aiken WD, Tulloch T, Paul TJ, Wan RL. Associations of whole-blood fatty acids and dietary intakes with prostate cancer in Jamaica. Cancer causes & control : CCC. 2012;23:23–33. doi: 10.1007/s10552-011-9850-4. [DOI] [PubMed] [Google Scholar]
- 202.Lloyd JC, Antonelli JA, Phillips TE, Masko EM, Thomas JA, Poulton SH, Pollak M, Freedland SJ. Effect of isocaloric low fat diet on prostate cancer xenograft progression in a hormone deprivation model. The Journal of urology. 2010;183:1619–1624. doi: 10.1016/j.juro.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Escobar EL, Gomes-Marcondes MC, Carvalho HF. Dietary fatty acid quality affects AR and PPARgamma levels and prostate growth. The Prostate. 2009;69:548–558. doi: 10.1002/pros.20905. [DOI] [PubMed] [Google Scholar]
- 204.Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Molecular cancer research: MCR. 2012;10:133–142. doi: 10.1158/1541-7786.MCR-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Trichopoulou A, Lagiou P, Kuper H, Trichopoulos D. Cancer and Mediterranean dietary traditions. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2000;9:869–873. [PubMed] [Google Scholar]
- 206.Berquin IM, Edwards IJ, Kridel SJ, Chen YQ. Polyunsaturated fatty acid metabolism in prostate cancer. Cancer metastasis reviews. 2011;30:295–309. doi: 10.1007/s10555-011-9299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Beare-Rogers JL, Gray L, Nera EA, Levin OL. Nutritional properties of poppyseed oil relative to some other oils. Nutrition and metabolism. 1979;23:335–346. doi: 10.1159/000176272. [DOI] [PubMed] [Google Scholar]
- 208.Burdge GC, Calder PC. Conversion of alpha-linolenic acid to longer-chain polyunsaturated fatty acids in human adults. Reproduction, nutrition, development. 2005;45:581–597. doi: 10.1051/rnd:2005047. [DOI] [PubMed] [Google Scholar]
- 209.Chavarro JE, Stampfer MJ, Li H, Campos H, Kurth T, Ma J. A prospective study of polyunsaturated fatty acid levels in blood and prostate cancer risk. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2007;16:1364–1370. doi: 10.1158/1055-9965.EPI-06-1033. [DOI] [PubMed] [Google Scholar]
- 210.Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, Cramer SD, Thomas MJ, Perry DL, Edwards IJ, Mucci LA, Sinnott JA, Loda MF, Sui G, Berquin IM, Chen YQ. Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer. Carcinogenesis. 2012;33:404–412. doi: 10.1093/carcin/bgr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, Thomas MJ, Thornburg T, Kulik G, Smith A, Edwards IJ, D’Agostino R, Zhang H, Wu H, Kang JX, Chen YQ. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. The Journal of clinical investigation. 2007;117:1866–1875. doi: 10.1172/JCI31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Akinsete JA, Ion G, Witte TR, Hardman WE. Consumption of high omega-3 fatty acid diet suppressed prostate tumorigenesis in C3(1) Tag mice. Carcinogenesis. 2012;33:140–148. doi: 10.1093/carcin/bgr238. [DOI] [PMC free article] [PubMed] [Google Scholar]