

Abstract
Objective
The purpose of this study was to determine whether cyclooxygenase inhibition improves vascular dysfunction of adipose microvessels from obese humans.
Design and Methods
In 20 obese subjects (age 37±12 yrs, BMI 47±8 kg/m2) we collected subcutaneous and visceral fat during bariatric surgery and characterized adipose depot-specific gene expression, endothelial cell phenotype, and microvascular function. Vasomotor function was assessed in response to endothelium-dependent agonists using videomicroscopy of small arterioles from fat.
Results
Arterioles from visceral fat exhibited impaired endothelium-dependent, acetylcholine-mediated vasodilation, compared to the subcutaneous depot (p<0.001). Expression of mRNA transcripts relevant to the cyclooxygenase pathway were upregulated in visceral compared to subcutaneous fat. Pharmacological inhibition of cyclooxygenase with indomethacin improved endothelium-dependent vasodilator function of arterioles from visceral fat by 2-fold (p=0.01), whereas indomethacin had no effect in the subcutaneous depot. Indomethacin increased activation via serine-1177 phosphorylation of endothelial nitric oxide synthase in response to acetylcholine in endothelial cells from visceral fat. Inhibition of endothelial nitric oxide synthase with Nω-nitro-L-arginine methyl ester abrogated the effects of cyclooxygenase-inhibition suggesting that vascular actions of indomethacin were related to increased nitric oxide bioavailability.
Conclusions
Our findings suggest that cyclooxygenase-mediated vasoconstrictor prostanoids partly contribute to endothelial dysfunction of visceral adipose arterioles in human obesity.
Keywords: adiposity, endothelium, vasodilation, arteries, inflammation
Introduction
Obesity is closely linked to premature vascular disease deaths from ischemic heart disease and stroke (1, 2). Central obesity and accumulation of intra-abdominal visceral fat, in particular, have been closely linked to cardiometabolic risk, possibly owing to increased elaboration of proatherogenic mediators from the visceral compartment (3–5). We recently demonstrated that the visceral adipose microenvironment is intrinsically toxic to the vasculature with upregulation of pro-inflammatory, oxidative-stress related, and hypoxia-induced adipocytokines that severely impair arteriolar endothelial vasodilator function in visceral compared to subcutaneous depots (6).
Experimental studies suggest that therapeutic modulation with anti-inflammatory and anti-oxidant strategies improves adipose tissue arteriolar responses (7, 8). Our group and others have demonstrated that cyclooxygenase (COX)-driven production of vasoconstrictor prostanoids and reactive oxygen species are important in the pathogenesis of endothelial dysfunction observed in obesity-related conditions such as diabetes, hypertension and atherosclerosis (9–16). However, the role of the eicosanoid/cyclooxygenase pathway in regulating vasomotor dysfunction in arterioles from human fat has not been previously examined. In this study, we sought to investigate fat depot-specific expression patterns of COX-related gene products and determine whether pharmacological COX inhibition modulates endothelium-dependent microvascular dilator responses in subcutaneous and visceral adipose depots in severely obese humans.
Methods and Procedures
Study subjects
We enrolled obese men and women (BMI≥30 kg/m2, age ≥18 years) scheduled to undergo bariatric surgery at Boston Medical Center. Subjects with unstable medical conditions such as active coronary syndromes, congestive heart failure, systemic infection, acute illness, malignancy or pregnancy were excluded. The study was approved by Boston Medical Center Institutional Review Board and all subjects gave written informed consent. For each subject we recorded blood pressure, weight, BMI, and waist circumference. Biochemical analyses including lipids, glucose, insulin, homeostasis model assessment of insulin resistance (HOMA), glycosylated hemoglobin (HbA1c), high-sensitivity C-reactive protein (hs-CRP) were quantified from blood samples collected in a fasting state.
Adipose tissue collection and vessel preparation
Subcutaneous and visceral adipose tissue biopsies were collected intra-operatively during planned bariatric surgery. Subcutaneous adipose tissue was harvested from the lower abdominal wall and visceral tissue secured from the greater omentum, respectively. Specimens were prepared immediately after surgical collection as previously described (6). Briefly, using a tissue dissection microscope, adipose arterioles (75–250 μm internal diameters) were carefully removed of surrounding fat and suspended immediately in an organ chamber containing Krebs solution. Arterioles were cannulated with glass micropipettes, pressure equilibrated and continuously perfused with Krebs buffer aerated with a gas mixture of 5% O2, 21% CO2 and 74% N2. The organ chamber was then mounted onto a stage of an inverted microscope (magnification x200) and video camera monitor (model VIA-100; Boeckler Instruments, Inc, Tucson, AZ) for vascular diameter measurements using videomicroscopy.
Assessment of adipose arteriolar function
The internal arterial diameter of each vessel was initially measured at a steady state followed by administration of endothelin-1 (ET-1, 2×10−6 M, Sigma-Aldrich, St. Louis, MO) to preconstrict vessels to 50–70% of their internal diameter. Endothelial nitric oxide synthase (eNOS)-dependent vasodilation was assessed by measuring arteriolar change in diameter to increasing doses of receptor-mediated NO-agonist acetylcholine (Ach, 10−10 to 10−5 M, Sigma-Aldrich) as previously described (6). To determine the role of COX-mediated prostanoids, indomethacin (Indo, Sigma-Aldrich) was administrated to the vessel chamber after initial baseline measurement of Ach-induced vasodilation of adipose arterioles. A dose of 10−5 M for Indo was chosen based on data from previous ex vivo studies (13, 17). Ach-mediated vascular responses were repeated following 30-minute incubation with indomethacin. The effect of indomethacin on Ach-induced vasodilation was also examined in the presence Nω-nitro-L-arginine methyl ester (L-NAME, 10−4 M, Sigma-Aldrich) an inhibitor of NOS isoforms. Endothelium-independent relaxation was determined using papaverine (Pap, 2×10−4 M, Sigma-Aldrich). All pharmacological agents were added to the external bathing solution of the organ chamber, and the indicated concentration represents the final chamber molar concentration.
Endothelial cell isolation from whole adipose tissue
Subcutaneous and visceral adipose tissue biopsies were collected during bariatric surgery and placed immediately into cold DMEM (Gibco life technology, Grand Island, NY) supplemented with sodium pyruvate, penicillin, and streptomycin (Gibco life technology). Tissue was cut into small pieces, minced and digested in collagenase I (2.5ug/ml, Sigma-Aldrich) for 1-hour in 37°C water bath in a 90 rpm rotation and passed through 100-uM filter to remove any remaining undigested tissue. Cells were then centrifuged at 400 rpm at 4°C for 10 minutes to separates adipocytes (top layer), lysed for red blood cells using 1 X RBC lysis buffer (R&D Systems, Minneapolis, MN), and remaining cells were passed through 40-uM filter in DMEM. Collected cells were labeled with CD31 microbeads (Miltenyi Biotech, Auburn, CA) before being loaded into the autoMACS Pro Separator. Isolated CD31+ endothelial cells were plated on a fibronectin coated (Fisher Scientific, Pittsburg, PA) coated 4-well chamber slides (BD Bioscience). Cells were allowed to settle for 1-hour and pretreated with 10−5 M Indomethacin for 30-minutes and with and without 10−4 M acetylcholine for 30-minutes. Cells were then fixed immediately in 4% paraformaldehyde.
Endothelial cell protein expression by quantitative immunofluorescence
Stimulatory activation via phosphorylation of endothelial nitric oxide synthase (p-eNOS) at serine 1177 in response to acetylcholine was assessed as previously described (18). Briefly, fixed samples were rehydrated with 50 mmol/L glycine (Sigma-Aldrich), permeabilized with 0.1% Triton-X and blocked with 0.5% bovine serum albumin (BSA). Slides were incubated for an hour at 37°C with primary antibodies against p-eNOS at serine 1177 (1:200 dilution; Millipore, Billerica, MA) and von Willebrand factor (vWF, 1:300 dilution, Dako Carpinteria, CA) to select endothelial cells and used analogous Alexa Fluor-488 and Alexa Fluor-594 antibodies (1:200 dilution, Invitrogen, Carlsbad, CA) for the secondary antibodies. Cells were mounted under glass coverslips with Vectasheild (Vector Laboratories, Burlingame, CA) containing DAPI to identify nuclei. Slides were imaged using a fluorescent microscope (x20 magnification, Nikon Eclipse TE2000-E) and digital images were captured using a Photometric CoolSnap HQ2 Camera (Photometrics, Tucson, AZ). Exposure time was kept constant and fluorescent intensity (corrected for background fluorescence) was quantified by NIS Elements AR Software (Nikon Instruments Inc, Melville, NY). Fluorescent intensity was quantified in 20 cells from each depot/subject and averaged. To control for batch-to-batch staining variability, fluorescence intensity for each sample was normalized to the intensity of human aortic endothelial cells (HAEC) staining performed simultaneously. Data are expressed in arbitrary units (a.u.) calculated by dividing the average fluorescent intensity of the subject sample by the intensity of the HAEC sample multiplied by 100.
Adipose tissue and arteriole gene expression
Adipose tissue was collected, placed immediately in RNALater (Qiagen, Germantown, MD) and stored at −80°C until further processing. Blood vessels were isolated from visceral and subcutaneous adipose tissue as described above and snap frozen in liquid nitrogen immediately. Adipose tissue total RNA was extracted using Qiagen RNA lipid easy kit (Germantown, MD) and total RNA from blood vessels was extracted using Qiagen RNeasy micro kit. Synthesis of cDNA for both adipose tissue and vessel RNA was completed by using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and quantitative real-time PCR reactions were performed using TaqMan gene expression assays (Applied Biosystems). Data normalized to GAPDH were compared using the ΔΔct method and expressed as fold-induction in gene expression in visceral adipose tissue compared to subcutaneous fat, or fold-induction in gene expression in visceral compared to subcutaneous blood vessels.
Adipose tissue culture and prostanoid production
Freshly harvested human adipose tissue was minced and digested with collagenase (300 U/ml, Worthington Biochemical Corporation, NJ) for 30-minutes and then placed in sterile EGM-2-MV media (Lonza, Allendale, NJ) for 48-hours. Supernatants were collected and stored at −80°C until further processing. Prostaglandin E2 (PGE2), 6-keto prostaglandin F1α (the degradation product of prostacyclin), and thromboxane B2 (TBX2, a metabolite of thromboxane metabolism) were assayed using EIA kits (Cayman Chemical, Ann Arbor, Michigan).
Statistical Analysis
Repeated measures analysis of variance (ANOVA) was used to compare vasodilation to increasing doses of Ach between adipose depots and under different conditions. Group differences in clinical characteristics, blood vessel gene expression, endothelial cell Ach-induced change in pENOS, and prostanoid production were examined by independent t-test or Mann–Whitney–Wilcoxon based on normality and Chi square tests, for continuous and categorical variables, respectively. Whole adipose tissue gene expression differences between depots were examined by paired t-tests. Associations between vascular dilator responses (defined by dose-response area under the curve), adipose tissue gene expression, and clinical data were examined using Spearman’s rank correlation analyses. Statistical significance was defined as p<0.05. All data are expressed as mean ± SD, unless otherwise indicated. All data were analyzed using SPSS for Windows, version 13.1.
Results
We harvested a total of 20 adipose tissue arterioles from subcutaneous (n=8) and visceral (n=12) fat depots in 20 severely obese subjects. The clinical characteristics of subjects that provided adipose samples during bariatric surgery are summarized in Table 1. No significant differences in clinical parameters were observed between subjects that provided vessels from subcutaneous or visceral depots. The average internal resting diameter of arterioles isolated from subcutaneous fat was 134±64 Mm and 165±29 Mm in visceral adipose vessels (p=0.20). Endothelium-dependent vasodilation was then assessed with dose-response relationship to increasing Ach concentrations (10−10–10−5 M). In agreement with our previous findings (6), dose-response to Ach-mediated, endothelium-dependent vasodilation was severely impaired in visceral arterioles compared to subcutaneous microvessels (p<0.001 by ANOVA) while responses to papaverine were similar indicating impairment at the level of the vascular endothelium (figure 1). To specifically evaluate depot-specific activation of eNOS, we quantified stimulatory eNOS phosphorylation at serine 1177. Corroborating our findings in intact adipose arterioles, we observed a significant impairment in Ach-mediated activation of p-eNOS in vascular endothelial cells isolated from visceral compared to subcutaneous fat (p<0.05, Supplemental figure I).
Table 1.
Study population characteristics
| Clinical parameter | n=20 |
|---|---|
| Age (yrs) | 37±12 |
| Female (%) | 76% |
| BMI (kg/m2) | 47±8 |
| Waist circumference (cm) | 135±15 |
| Weight (kg) | 130±29 |
| Systolic BP (mmHg) | 126±12 |
| Diastolic BP (mmHg) | 73±6 |
| Insulin (mU/ml) | 17±13 |
| Glucose (mg/dl) | 97±23 |
| HOMA | 4±5 |
| Triglycerides (mg/dl) | 99±31 |
| Total cholesterol (mg/dl) | 170±27 |
| HDL-C (mg/dl) | 43±7 |
| LDL-C (mg/dl) | 108±26 |
| hs-CRP (mg/dl) | 7.8 ±7.4 |
| HbA1c (%) | 6.0 ±1.3 |
| Diabetes (%) | 37% |
| Hypertension (%) | 48% |
| Hypercholesterolemia (%) | 23% |
Figure 1. Effect of Indomethacin treatment on adipose tissues arteriolar responses.
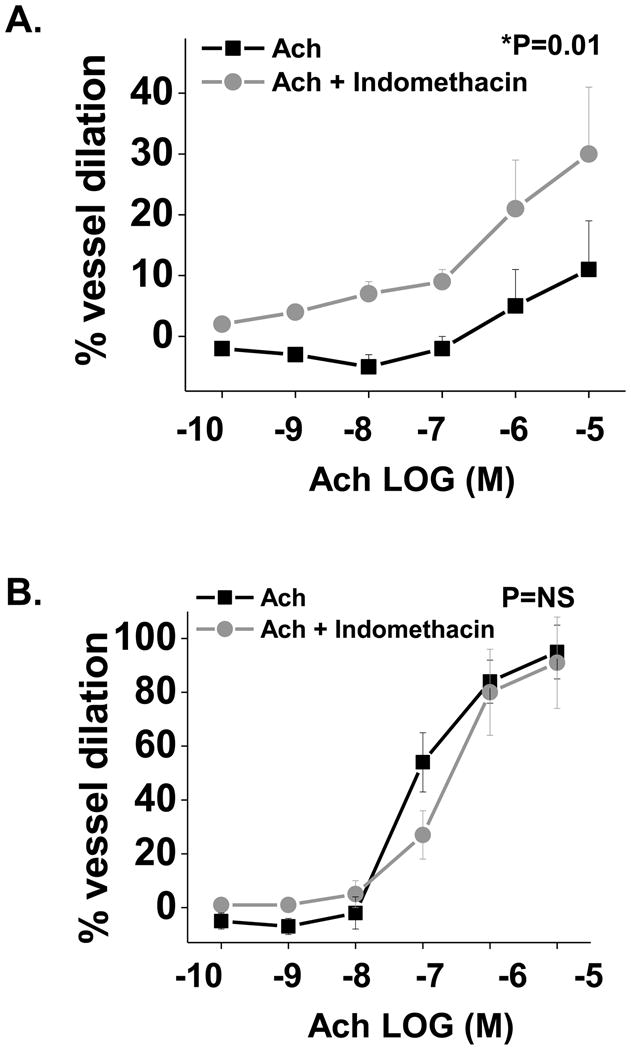
A) Indomethacin (10−5 M) treatment of microvessels from visceral adipose tissue improves Ach-induced vasodilation by 2-fold (n=12, p=0.01 by ANOVA). B) In contrast, indomethacin had no effect on subcutaneous adipose tissue arteriolar responses (n=8, p=NS). Data presented as mean ± SEM.
Vascular responses to indomethacin
To determine the role of COX-mediated prostanoids in the regulation of vascular function in adipose microvessels, Ach-mediated, endothelium-dependent vasodilation was assessed before and after treatment with indomethacin (Indo, 10−5 M), a COX-specific inhibitor (19). As shown in figure 1A, treatment with Indo significantly improved Ach-mediated vasodilation in the visceral adipose arterioles by 2-fold (p=0.01) suggesting that COX-derived vasoconstrictor prostanoids play a role in vascular impairment within the visceral depot. In contrast, Indo had no significant effect on Ach-mediated dilation in subcutaneous arterioles (figure 1B) which exhibited markedly better responses compared to the visceral depot. There was no differential vascular effect of COX inhibition in subjects with (53%) and without (47%) metabolic syndrome or median HOMA value cut-points.
To determine whether improved endothelial function in response to Indo related to endothelial nitric oxide synthase (eNOS) bioaction in the visceral compartment, we examined vasodilation to Ach ± Indo (10−5 M) in the presence of L-NAME (10−4 M) in a subset of vessels. As shown in figure 2, treatment of blood vessels with L-NAME fully abolished indomethacin-induced improvement in Ach-mediated dilation (n=10, p<0.01 by ANOVA). To specifically examine the effect of Indo on eNOS activation, we quantified p-eNOS in visceral endothelial cells at basal levels and after Ach stimulation ± Indo (10−5 M). Treatment with Indo did not alter the basal levels of p-eNOS. In contrast, Indo significantly improved Ach-induced p-eNOS in visceral endothelial cells (p<0.05, figures 3A and 3B), supporting the biological mechanism observed in our whole vessel physiological studies. Collectively, these data suggest that the endothelial dysfunction of visceral microvessels is partly due to the negative effects of COX-derived products, and impaired of eNOS activation and nitric oxide bioaction.
Figure 2. Effect of L-NAME on adipose vasomotor responses after treatment with indomethacin.

Treatment of visceral arterioles with L-NAME (light gray, triangle symbol) completely abolished indomethacin-induced improvement in Ach-mediated dilation (dark grey, circle symbol; n=10, p<0.01). Data presented as mean ± SEM.
Figure 3. Effect of indomethacin on acetylcholine-induced eNOS phosphorylation in endothelial cells isolated from visceral adipose tissue.

A) Representative quantitative immunofluorescence of isolated adipose endothelial cells demonstrating increased stimulatory eNOS phosphorylation at serine 1177 following indomethacin exposure compared to control. For each panel, left upper quadrant demonstrates merged image of an endothelial cell, right upper DAPI nuclear staining (blue), left lower von Willebrand factor staining (green), and right lower p-eNOS at serine 1177 (red). B) Quantification display demonstrating increased Ach-mediated change in eNOS phosphorylation at serine 1177 compared to control condition (*p<0.05).
Adipose tissue characterization
We examined depot-specific mRNA transcripts relevant to the prostanoid pathway. As shown in figure 4, we found significant upregulation of COX-1 and COX-2, rate limiting enzymes involved in prostaglandin synthesis, in the visceral compared to subcutaneous fat. Both COX- isoforms produce biologically active prostaglandin H2 (PGH2) that serves as a substrate for downstream generation of mediators including prostacyclin (prostaglandin I2, PGI2) or thromboxane (TXA2), which exert their actions via specific prostaglandin I receptor (IP) and TXA2-prostanoid receptors (TP) (20). Additionally, we demonstrated increased expression of downstream synthases including prostaglandin D synthase (PGDS) and prostaglandin E synthase (PGES) that catalyze production of prostaglandin D2 (PGD2) and prostaglandin E2 (PGE2), respectively that are known to promote inflammation and vascular dysfunction (20). In agreement with transcriptomic data, higher levels of 6-keto prostaglandin F1α (metabolite of PGI2) and PGE2 were released from visceral adipose tissue compared to subcutaneous fat after 48-hours of culture (P<0.05). A directionally similar trend was observed for TBX2 (Supplemental figure II). In addition, mRNA expression analysis specifically performed in isolated vessels for COX enzymes and select synthases yielded similar results, identifying COX-1 as the predominantly expressed isoform in visceral vessels compared to subcutaneous (Supplemental figure III). Ach-mediated vasodilation correlated negatively with adipose expression of COX-1 (r=−0.54, p=0.02). We previously demonstrated increased mRNA transcripts for proinflammatory tumor necrosis factor (TNF)-α and pro-oxidant NADPH-oxidase-2 (NOX-2) in the visceral compared to subcutaneous fat (6). In our present cohort, TNF-α expression correlated positively with COX-1 (r=0.4, p= 0.03), COX-2 (r= 0.37, p=0.049), PGES (r= 0.43, p=0.02), and thromboxane synthase (TBXAS, r= 0.44, p=0.02), while NOX-2 correlated with TBXAS (r= 0.4, p=0.03).
Figure 4. Adipose tissue gene expression.
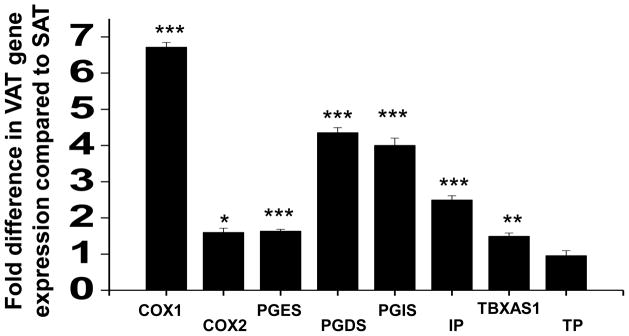
Expression of cyclooxygenases and synthases important in prostanoid production were upregulated in visceral adipose tissue (VAT) compared to subcutaneous adipose tissue (SAT, n=17). COX1= Cyclooxygenase 1, COX2 = Cyclooxygenase 2, PGES = Prostaglandin E synthase, PGDS = Prostaglandin D synthase, PGIS = Prostaglandin I synthase, IP = Prostaglandin I receptor, TBXAS = Thromboxane synthase, TP = Thromboxane prostanoid receptor. ***p<0.001, **p<0.01 and *p<0.05
Discussion
In the present study, we demonstrate that vascular endothelial function is severely impaired in visceral compared to subcutaneous fat in severely obese humans. We provide novel data by demonstrating upregulated expression of transcripts involving the cyclooxygenase pathway in visceral compared to subcutaneous fat that are involved in the production of vasoconstrictive prostanoids and reactive oxygen species known to impair vascular tone. Pharmacological inhibition of COX with indomethacin significantly improved endothelium-dependent vasodilation of visceral adipose vessels via increased local NO bioavailability, but had no significant effect on subcutaneous arterioles. We corroborated our whole vessel physiological findings by demonstrating that indomethacin improved acetylcholine-mediated eNOS phosphorylation in vascular endothelial cells harvested from visceral depots. Collectively, the findings suggest that endothelial dysfunction in visceral fat is driven in part by increased activity of the eicosanoid/cyclooxygenase pathway in human obesity.
While prostaglandins are clinically recognized as pain mediators in the human body, their dysregulation is also implicated in pathological conditions such as hypertension, cancer, inflammation, and cardiovascular disease. The role of cyclooxygenase products in the regulation of adipose arteriolar function in the context of human obesity has not been previously examined, although clinical studies of obesity-related diseases such as hypertension and diabetes show a clear role for vasoconstrictor prostanoids in the impairment of vascular function (9, 12, 15, 21, 22). Experimental animal models demonstrate impaired endothelium-dependent relaxation or contraction in metabolic disease states that can be ameliorated with cyclooxygenase inhibition (14, 23–25). In human disease, indomethacin restores Ach-induced NO-mediated vasodilation in the forearm vasculature of patients with essential hypertension, suggesting that COX products impair NO bioaction (12). Similarly, diclofenac improves vasodilator responses to methacoline in patients with chronic renal failure (26), and selective COX-2 inhibition reverses brachial artery endothelial dysfunction in hypertensive subjects (15). These vascular effects are likely specific to both the disease state and vascular bed as manipulation of COX activity paradoxically adversely modulates coronary physiology in CAD patients (27–29).
In the present study, we demonstrate differential upregulation of cyclooxygenase-driven mediators that along with pro-inflammatory and pro-oxidant adipocytokines act in concert to impair vasomotor function in the visceral adipose microenvironment (6). Prior studies show that ex vivo TNF-α antagonism or antioxidant treatment reverses vasoconstriction in visceral arterioles (7). In our study, we observed a significant beneficial effect of indomethacin on acetylcholine-mediated responses, suggesting that COX-driven products are physiologically active and regulate vascular phenotype in human adipose tissue. Interestingly, indomethacin had no effect on subcutaneous microvessels where vasodilator function was relatively preserved and mRNA profiles less proatherogenic compared to the visceral depot. We thus postulate that an imbalance between endothelium-derived constrictive and relaxing factors exists in the visceral depot, driven in part by cyclooxygenase-mediated generation of vasoconstrictor mediators that impair vascular tone. While expression of both COX enzymes was altered in the visceral depot, the greatest difference was observed for COX1 in both whole fat and arteriolar tissue. Both isoforms are implicated in mechanisms of endothelial dysfunction through generation of vasoconstrictor prostanoids in animal models although isoform specificity relates to disease phenotype (11, 30–32).
Cyclooxygenase products resulting from arachidonic acid metabolism are highly heterogeneous, and may have differential actions depending on the vascular bed and disease state. As such, it is likely that vascular dysfunction is caused by a mixture of products rather than a single mediator (24). In our study, we observed increased expression of vasoconstrictive thromboxane synthase (TBXAS), which was paralleled by increased transcription of PGI2 synthase in visceral fat. Generally, PGI2 synthase promotes vasodilation via prostacyclin generation. While this may be compensatory in this disease state of obesity, a pathogenic role may also be implicated owing to paradoxical inhibition of its activity by tyrosine nitration by peroxynitrite, a potent oxidant formed by the interaction of superoxide anion and NO (33). Clinical studies support this concept as PGI2 synthase nitration and inactivation favors a vasoconstrictive milieu and is associated with carotid atherosclerosis (34). Additionally, PGI2 and its precursor PGH2 can elicit vasoconstriction via the thromboxane receptor (TP) at high concentrations in diseased vessels (25, 35). This raises the possibility that pharmacological inhibition of the thromboxane receptor may serve as a target for treatment in vascular disease (36–38). We further demonstrated overexpression of PGES and increased production of PGE2 in visceral fat known to promote an inflammatory phenotype. As such, we found a significant correlation between adipose TNF-α expression and several prostanoid transcripts including PGES in our adipose samples, suggesting a pathophysiological connection (39).
The improvement in visceral vasodilation with indomethacin in our experimental model was blunted by co-administration of L-NAME. This implies that inhibition of eNOS activity abrogates the favorable effects of COX-inhibition. Possible mechanisms include direct effect of indomethacin on substrate bioavailability, eNOS enzymatic activity, or scavenging of oxygen-derived free radicals that impair nitric oxide action. The latter mechanism is supported by recent studies demonstrating that cyclooxygenase inhibition reverses NADPH oxidase-driven reactive oxygen species production and improves vasorelaxation in hypertensive animal models (40). Additionally, our data in isolated endothelial cells from visceral fat suggests that COX inhibition augments Ach-induced activation of eNOS by increasing phosphorylation at serine 1177, mechanistically supporting the vasodilator findings in our whole vessel experiments.
Our current study has several potential limitations. First, our study population was severely obese with long-standing class III–IV obesity referred for bariatric surgery. Thus our findings may not be applicable to a population with milder degrees of excess weight. This limitation is counterbalanced by the clinical feasibility of accessing both subcutaneous and visceral adipose depots simultaneously in obese humans. Secondly, our experimental model involved ex vivo pharmacological manipulation and may not fully recapitulate the in vivo adipose microenvironment. However, vascular studies and endothelial cell experiments were performed immediately following surgical harvest and under physiological conditions. Lastly, while we attributed endothelial dysfunction in visceral adipose arterioles to increased production of vasoconstrictor prostanoids, we cannot exclude that other mechanisms are operative.
In conclusion, we demonstrate that adipose tissue vascular endothelial function is severely impaired in visceral compared to subcutaneous compartments in severely obese humans. We provide evidence that COX-derived vasoconstrictor prostanoids play a role in modulating the pathogenesis of vasomotor dysfunction in visceral arterioles. Manipulation of the eicosanoid-cyclooxygenase pathway may be considered as a therapeutic target in obesity related vascular disease.
Supplementary Material
What is already known about this subject
Accumulation of visceral adiposity is closely linked to cardiovascular disease risk.
We have previously published that the visceral adipose tissue microenvironment is toxic to the vasculature with upregulation of pro-inflammatory and oxidative-stress related mediators that severely impair arteriolar endothelial vasodilator function.
Cyclooxygenase (COX)-driven vasoconstrictor prostanoids modulate the pathogenesis of vascular endothelial dysfunction and cardiovascular disease, but their depot-specific role in mediating adipose vascular dysfunction is completely unknown.
What this study adds
We demonstrated upregulation of the cyclooxygenase pathway in visceral compared to subcutaneous human fat which are involved in the production of vasoconstrictive prostanoids that differentially impair vasomotor function in visceral fat.
Pharmacological inhibition of COX with indomethacin significantly improved endothelium-dependent vasodilation of visceral adipose vessels via increased stimulatory eNOS phosphorylation at serine 1177 and nitric oxide bioavailability.
These findings suggest that endothelial dysfunction in visceral fat is driven in part by increased activity of the eicosanoid/cyclooxygenase pathway in human obesity that may serve as a therapeutic target.
Acknowledgments
MGF conceived the study, performed all the vascular experiments, collected adipose samples during surgery, analyzed data, and prepared the manuscript. ST recruited and consented study subjects, and collected adipose samples during surgery. SK assisted with recruitment and in intellectual design. DTMN quantified prostanoids secretion from adipose tissue and assisted in data analysis. BC and DTH were involved in patient recruitment and provided clinical surgical specimens. MAZ and KW performed gene expression analyses and assisted with data analysis and interpretation. JLF assisted with analysis of data. NMH and JAV assisted with data interpretation, scientific direction, and manuscript preparation. CMA was involved in patient recruitment and assisted with intellectual design. NG conceived the overall experimental strategy and design, provided scientific direction, edited the manuscript, and oversaw all aspects of the experimental studies.
Sources of Funding:
Dr. Gokce is supported by National Institutes of Health (NIH) grants HL1145675, and HL084213. Dr. Farb is supported by an American Heart Association Postdoctoral Fellowship grant 12POST11780028. Dr. Walsh is supported by NIH grants HL068758 and AG034972. Dr. Vita is supported by NIH grants HL083801, HL083269, HL75795, and K12 HL083781. Dr. Hamburg is supported by NIH grants HL109790 and HL102299. Dr. Fetterman is supported by NIH grant T32 HL07224. Drs. Walsh, Gokce, and Vita are jointly supported by NIH grant P01 HL081587.
Footnotes
Conflict of Interest Statement:
The authors have no conflicts of interest with the current manuscript.
Reference List
- 1.de Berrington GA, Hartge P, Cerhan JR, Flint AJ, Hannan L, MacInnis RJ, et al. Body-mass index and mortality among 1.46 million white adults. N Engl J Med. 2010;363(23):2211–9. doi: 10.1056/NEJMoa1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J, et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373(9669):1083–96. doi: 10.1016/S0140-6736(09)60318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116(1):39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- 4.Parikh NI, Keyes MJ, Larson MG, Pou KM, Hamburg NM, Vita JA, et al. Visceral and subcutaneous adiposity and brachial artery vasodilator function. Obesity (Silver Spring) 2009;17(11):2054–9. doi: 10.1038/oby.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, et al. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation. 2007;116(11):1234–41. doi: 10.1161/CIRCULATIONAHA.107.710509. [DOI] [PubMed] [Google Scholar]
- 6.Farb MG, Ganley-Leal L, Mott M, Liang Y, Ercan B, Widlansky ME, et al. Arteriolar function in visceral adipose tissue is impaired in human obesity. Arterioscler Thromb Vasc Biol. 2012;32(2):467–73. doi: 10.1161/ATVBAHA.111.235846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Virdis A, Santini F, Colucci R, Duranti E, Salvetti G, Rugani I, et al. Vascular generation of tumor necrosis factor-alpha reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol. 2011;58(3):238–47. doi: 10.1016/j.jacc.2011.01.050. [DOI] [PubMed] [Google Scholar]
- 8.Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119(12):1661–70. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 9.Campia U, Choucair WK, Bryant MB, Quyyumi AA, Cardillo C, Panza JA. Role of cyclooxygenase products in the regulation of vascular tone and in the endothelial vasodilator function of normal, hypertensive, and hypercholesterolemic humans. Am J Cardiol. 2002;89(3):286–90. doi: 10.1016/s0002-9149(01)02229-9. [DOI] [PubMed] [Google Scholar]
- 10.Chenevard R, Hurlimann D, Bechir M, Enseleit F, Spieker L, Hermann M, et al. Selective COX-2 inhibition improves endothelial function in coronary artery disease. Circulation. 2003;107(3):405–9. doi: 10.1161/01.cir.0000051361.69808.3a. [DOI] [PubMed] [Google Scholar]
- 11.Pratico D, Tillmann C, Zhang ZB, Li H, FitzGerald GA. Acceleration of atherogenesis by COX-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98(6):3358–63. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taddei S, Virdis A, Ghiadoni L, Magagna A, Salvetti A. Cyclooxygenase inhibition restores nitric oxide activity in essential hypertension. Hypertension. 1997;29(1 Pt 2):274–9. doi: 10.1161/01.hyp.29.1.274. [DOI] [PubMed] [Google Scholar]
- 13.Tesfamariam B, Brown ML, Deykin D, Cohen RA. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J Clin Invest. 1990;85(3):929–32. doi: 10.1172/JCI114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traupe T, Lang M, Goettsch W, Munter K, Morawietz H, Vetter W, et al. Obesity increases prostanoid-mediated vasoconstriction and vascular thromboxane receptor gene expression. J Hypertens. 2002;20(11):2239–45. doi: 10.1097/00004872-200211000-00024. [DOI] [PubMed] [Google Scholar]
- 15.Widlansky ME, Price DT, Gokce N, Eberhardt RT, Duffy SJ, Holbrook M, et al. Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension. 2003;42(3):310–5. doi: 10.1161/01.HYP.0000084603.93510.28. [DOI] [PubMed] [Google Scholar]
- 16.Xiang L, Dearman J, Abram SR, Carter C, Hester RL. Insulin resistance and impaired functional vasodilation in obese Zucker rats. Am J Physiol Heart Circ Physiol. 2008;294(4):H1658–H1666. doi: 10.1152/ajpheart.01206.2007. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Y, Varadharaj S, Zhao X, Parinandi N, Flavahan NA, Zweier JL. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am J Physiol Heart Circ Physiol. 2005;289(3):H1027–H1032. doi: 10.1152/ajpheart.00226.2005. [DOI] [PubMed] [Google Scholar]
- 18.Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, et al. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127(1):86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50 (Suppl):S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helmersson J, Vessby B, Larsson A, Basu S. Association of type 2 diabetes with cyclooxygenase-mediated inflammation and oxidative stress in an elderly population. Circulation. 2004;109(14):1729–34. doi: 10.1161/01.CIR.0000124718.99562.91. [DOI] [PubMed] [Google Scholar]
- 22.Taddei S, Virdis A, Mattei P, Ghiadoni L, Fasolo CB, Sudano I, et al. Hypertension causes premature aging of endothelial function in humans. Hypertension. 1997;29(3):736–43. doi: 10.1161/01.hyp.29.3.736. [DOI] [PubMed] [Google Scholar]
- 23.Kukreja RC, Kontos HA, Hess ML, Ellis EF. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res. 1986;59(6):612–9. doi: 10.1161/01.res.59.6.612. [DOI] [PubMed] [Google Scholar]
- 24.Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122(2):140–9. doi: 10.1016/j.pharmthera.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Tesfamariam B, Cohen RA. Role of superoxide anion and endothelium in vasoconstrictor action of prostaglandin endoperoxide. Am J Physiol. 1992;262(6 Pt 2):H1915–H1919. doi: 10.1152/ajpheart.1992.262.6.H1915. [DOI] [PubMed] [Google Scholar]
- 26.Annuk M, Fellstrom B, Lind L. Cyclooxygenase inhibition improves endothelium-dependent vasodilatation in patients with chronic renal failure. Nephrol Dial Transplant. 2002;17(12):2159–63. doi: 10.1093/ndt/17.12.2159. [DOI] [PubMed] [Google Scholar]
- 27.Edlund A, Berglund B, van DD, Kaijser L, Nowak J, Patrono C, et al. Coronary flow regulation in patients with ischemic heart disease: release of purines and prostacyclin and the effect of inhibitors of prostaglandin formation. Circulation. 1985;71(6):1113–20. doi: 10.1161/01.cir.71.6.1113. [DOI] [PubMed] [Google Scholar]
- 28.Friedman PL, Brown EJ, Jr, Gunther S, Alexander RW, Barry WH, Mudge GH, Jr, et al. Coronary vasoconstrictor effect of indomethacin in patients with coronary-artery disease. N Engl J Med. 1981;305(20):1171–5. doi: 10.1056/NEJM198111123052002. [DOI] [PubMed] [Google Scholar]
- 29.Pacold I, Hwang MH, Lawless CE, Diamond P, Scanlon PJ, Loeb HS. Effects of indomethacin on coronary hemodynamics, myocardial metabolism and anginal threshold in coronary artery disease. Am J Cardiol. 1986;57(11):912–5. doi: 10.1016/0002-9149(86)90729-0. [DOI] [PubMed] [Google Scholar]
- 30.Bagi Z, Erdei N, Toth A, Li W, Hintze TH, Koller A, et al. Type 2 diabetic mice have increased arteriolar tone and blood pressure: enhanced release of COX-2-derived constrictor prostaglandins. Arterioscler Thromb Vasc Biol. 2005;25(8):1610–6. doi: 10.1161/01.ATV.0000172688.26838.9f. [DOI] [PubMed] [Google Scholar]
- 31.Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D, et al. Involvement of cyclo-oxygenase-1-mediated prostacyclin synthesis in the vasoconstrictor activity evoked by ACh in mouse arteries. Exp Physiol. 2012;97(2):277–89. doi: 10.1113/expphysiol.2011.062034. [DOI] [PubMed] [Google Scholar]
- 32.Tang EH, Ku DD, Tipoe GL, Feletou M, Man RY, Vanhoutte PM. Endothelium-dependent contractions occur in the aorta of wild-type and COX2−/− knockout but not COX1−/− knockout mice. J Cardiovasc Pharmacol. 2005;46(6):761–5. doi: 10.1097/01.fjc.0000187174.67661.67. [DOI] [PubMed] [Google Scholar]
- 33.Zou MH. Peroxynitrite and protein tyrosine nitration of prostacyclin synthase. Prostaglandins Other Lipid Mediat. 2007;82(1–4):119–27. doi: 10.1016/j.prostaglandins.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 34.He C, Choi HC, Xie Z. Enhanced tyrosine nitration of prostacyclin synthase is associated with increased inflammation in atherosclerotic carotid arteries from type 2 diabetic patients. Am J Pathol. 2010;176(5):2542–9. doi: 10.2353/ajpath.2010.090783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams SP, Dorn GW, Rapoport RM. Prostaglandin I2 mediates contraction and relaxation of vascular smooth muscle. Am J Physiol. 1994;267(2 Pt 2):H796–H803. doi: 10.1152/ajpheart.1994.267.2.H796. [DOI] [PubMed] [Google Scholar]
- 36.Belhassen L, Pelle G, Dubois-Rande JL, Adnot S. Improved endothelial function by the thromboxane A2 receptor antagonist S 18886 in patients with coronary artery disease treated with aspirin. J Am Coll Cardiol. 2003;41(7):1198–204. doi: 10.1016/s0735-1097(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 37.Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, et al. Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation. 2005;111(3):334–42. doi: 10.1161/01.CIR.0000153386.95356.78. [DOI] [PubMed] [Google Scholar]
- 38.Zuccollo A, Shi C, Mastroianni R, Maitland-Toolan KA, Weisbrod RM, Zang M, et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation. 2005;112(19):3001–8. doi: 10.1161/CIRCULATIONAHA.105.581892. [DOI] [PubMed] [Google Scholar]
- 39.Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci U S A. 2012;109(12):E715–E724. doi: 10.1073/pnas.1111600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Revelles S, Avendano MS, Garcia-Redondo AB, Alvarez Y, Aguado A, Perez-Giron JV, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18(1):51–65. doi: 10.1089/ars.2011.4335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.