

Abstract
Drug addiction involves potentially life-long behavioral abnormalities that are caused in vulnerable individuals by repeated exposure to a drug of abuse. The persistence of these behavioral changes suggests that long-lasting changes in gene expression, within particular regions of the brain, may contribute importantly to the addiction phenotype. Work over the past decade has demonstrated a crucial role for epigenetic mechanisms in driving lasting changes in gene expression in diverse tissues, including brain. This has prompted recent research aimed at characterizing the influence of epigenetic regulatory events in mediating the lasting effects of drugs of abuse on the brain in animal models of drug addiction. This review provides a progress report of this still early work in the field. As will be seen, there is robust evidence that repeated exposure to drugs of abuse induces changes within the brain’s reward regions in three major modes of epigenetic regulation—histone modifications such as acetylation and methylation, DNA methylation, and non-coding RNAs. In several instances, it has been possible to demonstrate directly the contribution of such epigenetic changes to addiction-related behavioral abnormalities. Studies of epigenetic mechanisms of addiction are also providing an unprecedented view of the range of genes and non-genic regions that are affected by repeated drug exposure and the precise molecular basis of that regulation. Work is now needed to validate key aspects of this work in human addiction and evaluate the possibility of mining this information to develop new diagnostic tests and more effective treatments for addiction syndromes.
Keywords: Cocaine, opiates, histone acetylation, histone methylation, DNA methylation, microRNA
Introduction
Drug addiction can be viewed as maladaptive neural plasticity that occurs in vulnerable individuals in response to repeated exposure to a drug of abuse. That vulnerability is determined roughly half by genetic factors (although few specific causative genes have as yet been identified) and half by non-genetic factors which include environmental exposures as well as stochastic events during development. Once formed, in turn, addiction can drive life-long behavioral abnormalities.
These features of addiction suggest an important role for epigenetic mechanisms. The term epigenetics has several definitions; this review utilizes a broad one, which defines epigenetics as a series of biochemical processes through which changes in gene expression are achieved throughout the lifecycle of an organism without a change in DNA sequence (Jaenisch and Bird, 2003). Epigenetics can thus be viewed as the vehicle through which environment interacts with an individual’s genome to determine all aspects of function, in health and disease. A subset of epigenetic changes are very stable, which makes them ideal mediators both of addiction vulnerability and of drug-induced brain maladaptations that underlie an addiction syndrome.
Within this context, there are three general roles that epigenetic mechanisms likely play in addiction (Tsankova et al., 2007; Robison and Nestler, 2011). First, repeated exposure to a drug of abuse in adolescence or adulthood causes addiction in vulnerable individuals by inducing stable changes in gene expression through epigenetic regulation of those specific genes. Such epigenetic regulation involves alterations in the steady state expression levels of a set of genes as well as changes in other genes’ inducibility—both sensitization (priming) and desensitization—without a change in steady state expression. Regulation of gene inducibility can be seen as “latent” in that it would not be apparent by analysis of mRNA or protein levels. Epigenetic regulation of genes also alters the expression of splice isoforms of a gene, which is usually not apparent from traditional microarray analyses of expressed mRNAs. Second, epigenetic regulation mediates changes in steady state gene expression or inducibility of genes that occur throughout an individual’s lifetime in response to a host of environmental exposures, which help determine that individual’s vulnerability to drug exposure and addiction later in life (Hiroi and Agatsuma, 2005). Third, there is the possibility that drugs or other environmental exposures induce epigenetic changes in sperm or ova, which are then passed on to offspring and alter their vulnerability to addiction. Such trans-generational epigenetic inheritance of addiction vulnerability remains controversial.
Finally, because the large majority of investigations of epigenetic mechanisms have been carried out on cultured cells or peripheral tissues, studies of epigenetic regulation in addiction models will teach the field fundamental principles about epigenetics in the developing and adult nervous system. In this way, such work provides the first ever look at mechanisms of transcriptional regulation in brain, and will likely have enormous impact on the field.
Overview of Mechanisms of Epigenetic Regulation
The 3 billion nucleotides of DNA in a mammalian genome would be ~2 meters long if stretched out linearly, yet fits within a microscopic cell nucleus due to its extraordinary degree of organization and compaction in chromatin—nuclear material composed of DNA, histones, and non-histone proteins (Jaenisch and Bird, 2003). The fundamental unit of chromatin is the nucleosome, which consists of ~147 base pairs of DNA wrapped around a core histone octamer (~1.65 turns). Each octamer contains two copies each of the histones H2A, H2B, H3, and H4 (Fig. 1A). Epigenetic mechanisms control the spacing of nucleosomes and the degree to which they are condensed, which thereby determines gene activity. In simplified terms, chromatin exists in an inactivated, condensed state (heterochromatin), which does not allow transcription of genes, and in an activated, open state (euchromatin), which allows individual genes to be transcribed (Fig. 2). In reality, chromatin exists in many states in between these two extremes. Regulation of the state of chromatin around specific genes, as well as in non-genic regions, is controlled by complex biochemical processes, involving diverse types of post-translational modifications of histones, methylation of DNA itself, large families of chromatin remodeling proteins, and non-coding RNAs, which are described briefly here.
Figure 1. Scheme of post-translational modifications of histones.
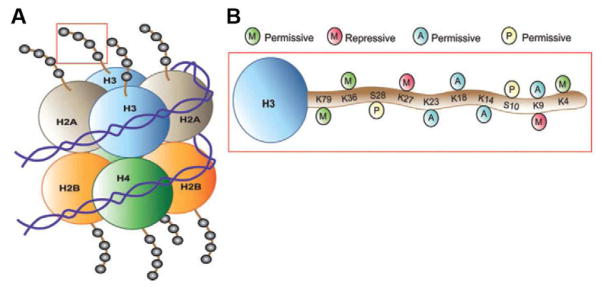
(A) The nucleosome is the functional unit of chromatin, composed of 147 bp of DNA wrapped around a core octamer of histone proteins (two copies each of H2A, H2B, H3, and H4). The N-terminal tails of these histones face outward from the nucleosome. (B) Combinations of acetylation, phosphorylation, methylation, etc., on histone tails (here, H3 is depicted) alter chromatin compaction and regulate gene expression. Histone modifications that weaken the interaction between histones and DNA or that promote the recruitment of transcriptional activating complexes (e.g., H3 acetylation at K23, K18, K14, and K9, as well as methylation at K79, K36, and K4 or phosphorylation at S28 and S10) correlate with permissive gene expression. Histone deacetylation, which strengthens histone:DNA contacts, or histone methylation on K27 or K9, which recruits repressive complexes to chromatin, promote a state of transcriptional repression. Adapted from Tsankova et al. (2007) and Maze and Nestler (2011) with permission.
Figure 2. Epigenetic regulation by drugs of abuse.
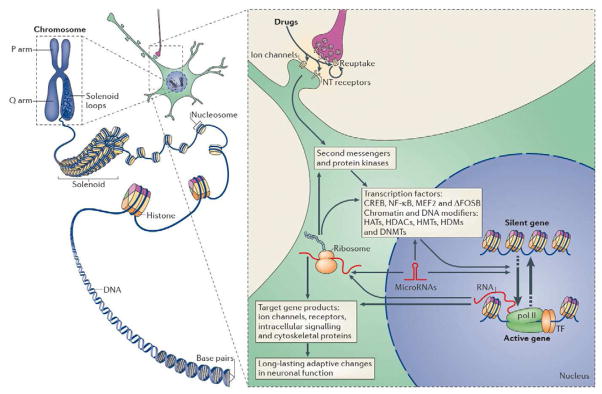
In eukaryotic cells, DNA wraps around histone octomers to form nucleosomes, which are then further organized and condensed to form chromosomes (right). Unraveling compacted chromatin makes the DNA of a specific gene accessible to the transcriptional machinery. Drugs of abuse (left) act through synaptic targets to alter intracellular signaling cascades, which leads to the activation or inhibition of transcription factors and of many other nuclear proteins; the detailed mechanisms involved in the latter remain poorly understood. This leads to the induction or repression of particular genes, including those for noncoding RNAs; altered expression of some of these genes can in turn further regulate gene transcription. It is hypothesized that some of these drug-induced changes at the chromatin level are extremely stable and thereby underlie the long-lasting behaviors that define addiction. CREB, cAMP response element binding protein; DNMTs, DNA methyltransferases; HATs, histone acetyltransferases; HDACs, histone deacetylases; HDMs, histone demethylases; HMTs, histone methyltransferases; MEF2, myocyte enhancing factor-2; NFκB, nuclear factor κB; pol II, RNA polymerase II. From Robison and Nestler (2011) with permission.
Histones
The best characterized chromatin remodeling mechanism in brain is the post-translational, covalent modification of histones at distinct amino acid residues on their N-terminal tails (Jeuwein and Allis, 2001). Such modifications include acetylation, ubiquitination, or SUMOylation at lysine (K) residues, methylation at lysine or arginine (R) residues, phosphorylation at serine (S) or threonine (T) residues, and ADP-ribosylation at glutamate (E) residues (e.g., Fig. 1B). Acetylation generally promotes decondensation of chromatin and increases gene activity by negating the positive charge of K residues in histone tails and increases spacing between nucleosomes. In contrast, histone methylation can either promote or repress gene activity, depending on the residue undergoing methylation. Phosphorylation of histones is also associated with chromatin inhibition or activation. The roles of histone ubiquitylation, SUMOylation, and ADP ribosylation are less well understood. The diversity of histone modifications supports the “histone code hypothesis,” which posits that the sum of modifications at a particular gene defines a specific epigenetic state of gene activation or silencing (Jenuwein and Allis, 2001). However, as will be seen, such codes are likely to be highly complex and have yet to be identified.
The enzymes that mediate these various covalent modifications of histones can be understood as “writers” and “erasers,” respectively. For example, histone acetyltransferases (HATs) catalyze acetylation and histone deacetylases (HDACs) catalyze deacetylation, while histone methyltransferases (HMTs) catalyze methylation and histone demethylases (HDMs) catalyze demethylation. The specificity of numerous HATs and HDACs for specific K residues remains incompletely understood. In contrast, distinct HMTs and HDMs control the methylation of specific K and R residues and even the valence of methylation, i.e., mono-, di-, or tri-methylated states. The functional consequences of histone modifications are mediated partly through “readers”—proteins that bind to specific modified residues and effect transcriptional change (Jenuwein and Allis, 2001; Jaenisch and Bird, 2003). For example, different families of chromatin remodeling proteins, which use ATP-derived energy to alter nucleosome spacing and condensation, recognize specific forms of modified histones and enhance or repress the activity of nearby genes. Ultimately, hundreds of proteins are thought to be recruited to a gene in concert with its activation or repression, again emphasizing the extraordinary complexity of epigenetic mechanisms.
DNA methylation
DNA methylation occurs with the addition of a methyl group to the C5 position of cytosine (5-mC) predominantly at CpG sites (Klose and Bird, 2006). It plays a pivotal role in cell differentiation, imprinting, and X chromosome inactivation. DNA methylation generally exerts a repressive effect on gene transcription. It can either prevent the association of DNA-binding factors with their target sequence or bind to methyl-CpG-binding proteins to recruit transcriptional co-repressors to modify the surrounding chromatin into a silenced state. Compared with histone tail modifications, most of which are considered readily reversible, DNA methylation is viewed as a more stable epigenetic change.
DNA methylation is catalyzed by DNA methyltransferases (DNMTs); DNMT3a is the only DNMT expressed in postnatal brain (Moore et al., 2013). Despite evidence for the dynamic control of DNA methylation in adult brain, including its reversibility, the enzymatic basis of demethylation remains incompletely understood. Putative demethylases are enzymes best studied for their role in DNA repair, such as the growth arrest and DNA damage (GADD45) family of proteins. Similarly, members of ten-eleven translocation (TET) proteins oxidize 5-mC into 5-hydroxymethylcytosine (5-hmC), and subsequently into 5-formylcytosine and 5-carboxylcytosine (Kriaucionis et al., 2009; Tahiliani et al., 2009; Wu and Zhang, 2011; Moore et al., 2013). Through deamination, glycosylation, or base excision repair, these newly discovered forms of cytosine modification can then be converted back into an unmethylated state. Interestingly, 5-mC oxidation derivatives are expressed at highest levels in neurons. In contrast to the repressive effect of 5-mC on gene expression, 5-hmC is more correlated with transactivation (Szulwach et al., 2011). Most studies of DNA methylation to date have not distinguished between 5-mC and 5-hmC, which is clearly a major need in the field.
Noncoding RNA
The complete sequencing of the mammalian genome and its transcriptional products has revealed a surprisingly large number of expressed RNAs that are not translated into proteins. Such non-coding RNAs have been shown to play crucial regulatory roles in cell function (Dunham et al., 2012; Rinn and Chang, 2012). Most studied are microRNAs (miRNAs), typically 20–25 nt, which by binding to targeted mRNAs either inhibit their translation or induce their breakdown. More recently, long non-coding RNAs (lncRNAs), defined as having a length >200 nt, are emerging as key regulators of gene transcription. They modulate chromatin-modifying complexes through direct interactions with transcription factors and other nuclear proteins.
Role of Histone Modifications in Addiction
Increasing evidence is defining the mechanisms by which chronic exposure to a drug of abuse alters the steady state levels of mRNAs through chromatin regulatory mechanisms or primes or desensitizes additional genes for altered expression after some period of withdrawal (Fig. 3).
Figure 3. Gene priming and desensitization.

In addition to regulating the steady-state expression levels of certain genes, cocaine induces latent effects at many other genes, which alter their inducibility in response to a subsequent stimulus. A. Analysis of mRNA expression after acute or repeated cocaine. Heat maps marked with an asterisk (*) show all genes that are upregulated in the NAc 1 hr after a cocaine challenge in naïve animals (acute), in animals treated repeatedly with cocaine (repeated + acute), or in animals after 1 wk of withdrawal from repeated cocaine (repeated wd + acute). Associated heat maps show how the same genes are affected under the other two conditions. Desensitized transcriptional responses after repeated cocaine are indicated (***). B. Early evidence suggests that epigenetic mechanisms are important in mediating such gene priming and desensitization and that many such changes are latent, meaning that they are not reflected by stable changes in steady-state mRNA levels. Rather, such changes alter chromatin structure such that later drug challenge induces a given gene to a greater (primed) or lesser (desensitized) extent based on the epigenetic modifications induced by previous chronic drug exposure. A major goal of current research is to identify the chromatin signatures that underlie such regulation. From Robison and Nestler (2011) with permission.
Histone acetylation
Histone acetylation is by far the most studied chromatin modification in drug abuse models, with the majority of work to date focused on stimulants (Robison and Nestler, 2011; Rogge and Wood, 2013). Acute or repeated exposure to cocaine or other stimulants increases global (i.e., total cellular) levels of acetylated histone H3 and H4—as detected by Western blotting or immunocytochemistry—in the nucleus accumbens (NAc), a key brain reward region (Kumar et al., 2005; Schroeder et al., 2008; Shen et al., 2008). There has not yet been a detailed accounting of which of several K residues in these histones display this regulation. Likewise, little is known about the intracellular signaling pathways through which stimulants, presumably through activation of dopamine receptors, leads to such global changes in histone acetylation, including which of numerous subtypes of HATs and HDACs are responsible for modifying the various K residues that display regulation. The HAT, CBP (CREB-binding protein), and Class II HDAC, HDAC5, have been implicated (Levine et al., 2005; Renthal et al., 2007; Malvaez et al., 2011; Taniguchi et al., 2012).
Experimental manipulation of histone acetylation in NAc has variable effects on stimulant-elicited behavioral responses (e.g., Kumar et al., 2005; Levine et al., 2005, 2011; Kalda et al., 2007; Renthal et al., 2007; Kim et al., 2008; Romieu et al., 2008; Schroeder et al., 2008; Sun et al., 2008; Malvaez et al., 2010; Wang et al., 2010). Short-term increases in histone acetylation generally promote behavioral responses to the drugs, while sustained increases oppose cocaine’s effects, based on the actions of systemic or intra-NAc administration of HDAC inhibitors. Recent work has provided insight into this time-dependent regulation, and indicates an important role for repressive histone methylation (see next section) (Kennedy et al., 2013). There has not yet been a systematic evaluation of numerous HAT and HDAC subtypes in regulating these behavioral responses. Heterozygous CBP knockout mice show attenuated locomotor and rewarding responses to repeated cocaine exposure (Levine et al., 2005; Malvaez et al., 2011), whereas homozygous HDAC5 knockout mice show hypersensitivity (Renthal et al., 2007). Conversely, overexpression of HDAC4 or 5, but not other Class II HDACs, in NAc attenuates cocaine responses (Kumar et al., 2005; Renthal et al., 2007; Wang et al., 2010). Mice with targeted NAc-selective deletions of HDAC1, but not of two other Class I HDACs (HDAC2 or 3), show attenuated behavioral responses to cocaine (Kennedy et al., 2013). In contrast, NAc-specific deletion of HDAC3 facilitates extinction of cocaine place conditioning (Malvaez et al., 2013). These observations highlight the need to study the effect of individual HDACs in NAc in a broad range of behavioral models, in particular, drug self-administration and relapse procedures.
Altered histone acetylation has been demonstrated at several candidate genes in the NAc in response to stimulants, and these changes correlate with their altered expression. For example, H4 acetylation is increased at the c-Fos promoter acutely, with no changes seen chronically, consistent with desensitization of c-Fos expression after chronic drug exposure (Kumar et al., 2005; Renthal et al., 2008) (Fig. 4). In contrast, the BDNF (brain-derived neurotrophic factor) and Cdk5 (cyclin-dependent kinase-5) promoters show H3 acetylation only after chronic cocaine, consistent with induction of these genes by chronic drug exposure (Kumar et al., 2005). Induction of CaMKIIα (Ca2+/calmodulin-dependent protein kinase IIα) in NAc by cocaine is also associated with H3 acetylation at the CaMKIIα gene (Wang et al., 2010; Robison et al., 2013). A genome-wide study utilizing ChIP-chip—chromatin immunoprecipitation (ChIP) with antibodies against pan-acetylated H3 or H4 followed by promoter microarrays—has provided a more complete map of genes in NAc that display altered histone acetylation after chronic cocaine (Renthal et al., 2009). Numerous gene promoters were found to be hyper- or hypoacetylated, with minimal overlap between genes that display alterations in H3 versus H4. While many of the genes that showed altered histone acetylation in response to cocaine exhibit commensurate changes in mRNA expression—with hyperacetylation associated with increased expression and hypoacetylation decreased expression—most genes did not follow this pattern. These observations indicate that any histone code for gene regulation is likely to be very complex, with histone acetylation contributing just a fraction of all epigenetic information that determines a gene’s activity. It will be important to repeat these genome-wide determinations for each of the many individual sites of histone acetylation to better understand the role of each in gene regulation. It will also be important to study the role of histone acetylation in several other brain reward regions (e.g., prefrontal cortex [PFC]), where initial reports of cocaine-induced changes in acetylation at specific genes (e.g., BDNF) have been reported (Sadri-Vakili et al., 2010; Schmidt et al., 2012).
Figure 4. Epigenetic basis of drug regulation of gene expression.

The figure is based on the mechanisms by which chronic cocaine, through ΔFosB, activates the Cdk5 gene (top) and represses the c-Fos gene (bottom). Top: ΔFosB binds to the Cdk5 gene and recruits several co-activators, including CBP (CREB binding protein) — a type of histone acetyltransferase (HAT) leading to increased histone acetylation, BRG1 (brahma-related gene 1) — a type of chromatin remodeling factor — and SUG1 (proteasome 26S ATPase subunit 5), another type of chromatin regulatory protein. ΔFosB also represses G9a expression, leading to reduced repressive histone methylation at the Cdk5 gene. The net result is gene activation and increased CDK5 expression. Bottom: In contrast, ΔFosB binds to the c-Fos gene and recruits several co-repressors, including HDAC1 (histone deacetylase 1) and SIRT1 (sirtuin 1). The gene also shows increased G9a binding and repressive histone methylation (despite global decreases in these marks). The net result is c-Fos gene repression. As transcriptional regulatory complexes contain dozens or hundreds of proteins, much further work is needed to further define the activational and repressive complexes that cocaine recruits to particular genes to mediate their transcriptional regulation and to explore the range of distinct activational and repressive complexes involved in cocaine action. From Robison and Nestler (2011) with permission.
Chronic cocaine also induces NAc expression of two sirtuins, SIRT1 and SIRT2 (Renthal et al., 2009), which are Class III HDACs that deacetylate numerous non-histone proteins as well. SIRT regulation by cocaine was discovered on ChIP-chip arrays, where cocaine was found to increase levels of histone acetylation at the SIRT1 and SIRT2 genes. Such SIRT induction promotes behavioral responses to cocaine, including cocaine self-administration, based on experiments that overexpress or knockout the SIRTs locally within the NAc or pharmacologically regulate SIRT activity (Renthal et al., 2009; Ferguson et al., 2012). Current efforts are focused on identifying the protein targets of SIRTs that mediate these behavioral effects.
Many fewer studies have examined histone acetylation in the context of other drugs of abuse. Delivery of HDAC inhibitors into the NAc enhances rewarding responses to opiates (Sheng et al., 2011), whereas such drugs, intra-PFC, promote extinction of conditioned place aversion during opiate withdrawal (Wang et al., 2012). Opiate regulation of several target genes, including those encoding several K+ channel subunits and the mu opioid receptor, is associated with altered histone acetylation in several brain regions (Hwang et al., 2009; Mazei-Robison et al., 2011). There are numerous reports linking chronic ethanol exposure and altered histone acetylation (Starkman et al., 2012; Ron and Messing, 2013). For example, ethanol increases global levels of H3 and H4 acetylation in PFC (Pascual et al., 2012), and reduces HDAC activity in whole brain (Botia et al., 2012), effects associated with increased rewarding effects of the drug. Ethanol also increases CBP levels and global H3 and H4 acetylation in amygdala, with HDAC inhibitors reducing anxiety-like symptoms observed during ethanol withdrawal (Sakharkar et al., 2012). These effects are observed in concert with increased histone acetylation at the NPY and pro-nociceptin genes in this brain region (D’Addario et al., 2013; Sakharkar et al., 2012). Work is needed to more fully characterize such regulation of histone acetylation by these other drugs of abuse and understand their contribution to addiction.
Histone K methylation
More recent research has implicated repressive histone K methylation in the actions of several drugs of abuse. G9a and GLP (G9a-like protein), two HMTs that catalyze the di-methylated state of K9 of H3 (H3K9me2), are downregulated in NAc by chronic cocaine (Maze et al., 2010) and opiate (Sun et al., 2012a) administration, along with decreases in global levels of this mark. A similar downregulation is seen in NAc of animals that self-administer these drugs. In contrast to G9a and GLP, numerous other types of HMTs and HDMs are not affected by drug exposure. Ethanol also downregulates G9a expression in cultured cortical neurons (Qiang et al., 2011). Genetic or pharmacological blockade of G9a in the NAc potentiates behavioral responses to cocaine and opiates, whereas increasing G9a function exerts the opposite effect (Maze et al., 2010; Sun et al., 2012a). Such drug-induced downregulation of G9a and H3K9me2 also sensitizes animals to the deleterious effects of subsequent chronic stress (Covington et al., 2011). Downregulation of G9a increases the dendritic arborization of NAc neurons, and is associated with increased expression of numerous proteins implicated in synaptic function, which directly connects altered G9a/H3K9me2 in the synaptic plasticity associated with addiction (Maze et al., 2010).
G9a appears to be a critical control point for epigenetic regulation in NAc, as we know it functions in two negative feedback loops. It opposes the induction of ΔFosB, a long-lasting transcription factor important for drug addiction (Robison and Nestler, 2011), while ΔFosB in turn suppresses G9a expression (Maze et al., 2010; Sun et al., 2012a). Interestingly, a prior history of cocaine exposure, followed by one month of withdrawal, leads to enhanced inducibility of the FosB gene in response to a subsequent cocaine challenge (Damez-Werno et al., 2012a). This priming is not associated with changes in the upstream signaling pathways that control FosB expression, thus pointing to a chromatin mechanism. Indeed, the priming is associated with reduced H3K9me2 binding at the FosB gene as well as with enrichment of a particular phosphorylated form of RNA polymerase II which has been associated with gene priming in cell culture systems (Damez-Werno et al., 2012a). FosB gene priming thus represents an important example of latent regulation that is mediated via chromatin mechanisms, although much further work is needed to understand the underlying mechanisms involved.
Also, G9a is induced in NAc upon prolonged HDAC inhibition, which explains the paradoxical attenuation of cocaine’s behavioral effects seen under these conditions, as noted above (Kennedy et al., 2013). GABAA receptor subunit genes are among those that are controlled by this feedback loop. Thus, chronic cocaine, or prolonged HDAC inhibition, induces several GABAA receptor subunits in NAc, which is associated with increased frequency of inhibitory postsynaptic currents (IPSCs). In striking contrast, combined exposure to cocaine and HDAC inhibition, which triggers the induction of G9a and increased global levels of H3K9me2, leads to blockade of GABAA receptor and IPSC regulation. Our hypothesis is that the combination of cocaine and HDAC inhibition results in excessive hyperacetylation, which causes—as a classic negative feedback mechanism—induction of G9a to counteract the transcriptional-activating effects of increased acetylation (Fig. 5).
Figure 5. Schema depicting regulation of GABAA receptor subunit gene expression in NAc through cross-talk between histone acetylation and repressive methylation.
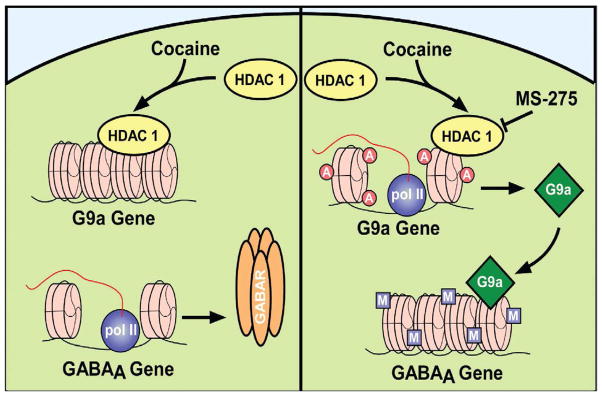
Repeated cocaine targets HDAC1 to the G9a/GLP (G9a like protein) promoters, leading to decreased G9a/GLP gene expression and decreased binding of these histone methyltransferases (HMTs) at the promoters of certain GABAA receptor subunit genes. The resulting decreased repressive histone methylation (reduced H3K9me2) allows for increased transcription of the GABAA receptor subunits and increased inhibitory tone in the NAc. Chronic cocaine plus chronic intra-NAc infusion of MS-275, by inhibiting HDAC1, promotes excessive histone acetylation and leads to the induction of G9a/GLP gene expression. These HMTs then catalyze increased H3K9me2 at GABAA receptor subunit gene promoters to block cocaine-induced transcriptional activation of the GABAA subunits and increased inhibitory tone. From Kennedy et al. (2013) with permission.
Genome-wide maps, utilizing ChIP-chip or the more powerful ChIP-seq (ChIP followed by deep sequencing), have been obtained for altered H3K9me2 binding in NAc after chronic cocaine or opiates (Renthal et al., 2009; Sun et al., 2012a). As with histone acetylation, regulation of H3K9me2 is associated with altered gene expression, but is not in itself deterministic. Current research is focused on examining the numerous genes whose expression is controlled in NAc in the drug-treated state by alterations in H3K9me2 enrichment.
Chronic cocaine administration also downregulates global NAc levels of H3K9me3, a heterochromatic mark associated with gene silencing (Maze et al., 2011). Coincident with such regulation is an increase in the size of NAc neuronal nuclei after cocaine exposure, which emphasizes the profound effect that cocaine exerts on the genome of this cell type. ChIP-seq reveals, as expected, that virtually all H3K9me3 binding in NAc is at non-genic regions, with cocaine reducing this binding at numerous loci (Maze et al., 2011). Among such regulated sites are repetitive elements, such as long-interspersed nuclear element-1 (LINE-1), which show commensurate increases in expression after cocaine. While global levels of H3K9me3 are not altered in NAc by chronic opiate administration, we did observe thousands of loci in non-genic regions with altered levels of H3K9me2 (Sun et al., 2012a), again suggesting a unappreciated role of these repetitive regions in drug action, a possibility that requires further study.
It would be interesting moving forward to perform ChIP-seq for numerous other sites of K methylation in drug abuse models, as just some examples, H3K4me1 (associated in simpler systems with gene enhancer function), H3K4me3 (associated with promoter activation), H3K27me3 (associated with gene repression), H3K36me3 (associated with transcriptional elongation), and H4K20me3 (associated with gene repression). Such work will likely help identify genes regulated by drugs of abuse and the underlying chromatin mechanisms involved.
Other histone mechanisms
Another important area of future investigation is to characterize the effect of chronic drug exposure on many other forms of histone modifications. There are preliminary reports that chronic cocaine exposure, for example, alters levels of histone phosphorylation (Brami-Cherrier et al., 2005, 2009; Bertran-Gonzalez et al., 2008; Stipanovich et al., 2008), R methylation (Damez-Werno et al., 2012b), and poly-ADP ribosylation (Scobie et al., 2012) and the enzymes that catalyze these changes, as well as levels of several specific chromatin modifying proteins such as brahma related gene-1 (BRG1), ATP-utilizing chromatin assembly and remodeling factor-1 (ACF1), and Williams syndrome transcription factor (WSTF) (Hwang et al., 2009; Sun et al., 2012b). These early findings suggest the broad range of histone modifications that likely contribute to addiction-related phenomena.
Role of DNA Methylation in Addiction
Given the hypothesized importance of DNA methylation in mediating sustained transcriptional change, it is unfortunate that there have not been more studies of this epigenetic mechanism in drug abuse models. Expression levels of DNMT3a in NAc are differentially altered by acute versus chronic cocaine exposure, and during extended withdrawal (Anier et al., 2010; LaPlant et al., 2010). Local knockout of DNMT3a from the NAc, or local infusion of the DNMT inhibitors RG108 or zebularine, increases behavioral responses to cocaine, whereas DNMT3a overexpression in NAc has the opposite effect. DNMT3a overexpression also increases dendritic arborizations of NAc neurons (LaPlant et al., 2010). In addition, NAc knockout of MeCP2 (methyl CpG binding protein-2) enhances amphetamine reward (Deng et al., 2010). In contrast, chronic cocaine increases MeCP2 expression in dorsal striatum, where local knockdown of the protein attenuates cocaine self-administration (Im et al., 2010). These findings underscore the complexity of drug regulation of DNMT3a and MeCP2 and the importance of reconciling effects in NAc versus dorsal striatum and in contingent versus non-contingent drug administration paradigms.
Moreover, such actions of DNMT3a or MeCP2 do not necessarily indicate a role for altered DNA methylation, since these proteins could serve different functions, and information on the regulation of DNA methylation in addiction models remains limited. A small number of studies have investigated DNA methylation changes at particular genes of interest (Nielsen et al., 2009, 2012; Anier et al., 2010; Ponomarev et al., 2012), but there has not yet been a genome-wide mapping of such regulation. Genome-wide measures of DNA methylation are challenging technically: most existing methods focus on gene promoter regions only and do not distinguish between several forms of DNA methylation, as noted earlier. True genome-wide maps of DNA methylation, with single base resolution based on deep sequencing, are still too expensive to be practical. Nevertheless, such maps are critically needed to understand the dynamic regulation of DNA methylation at particular genomic sites over a broad time course of drug exposure, and to what extent such changes are reversible during drug abstinence.
In preliminary work (J. Feng and E. Nestler, unpublished observations), we have found that chronic cocaine administration decreases TET1 expression in NAc, an effect seen in human cocaine addicts as well. Such downregulation of TET1 potentiates behavioral responses to cocaine. We have also obtained preliminary genome-wide maps of 5-hmc in NAc after cocaine exposure and found that changes in 5-hmc generally correlate with increased gene expression. These findings further underscore the importance of obtaining genome-wide maps of several forms of DNA methylation in drug abuse models.
Role of Noncoding RNAs in Addiction
Multiple miRNAs are reported to be up- or downregulated by drugs of abuse. For instance, cocaine increases levels of miR-181a and decreases miR-124 and let-7d in rat striatum (Chandrasekar et al., 2009, 2011; Schaefer et al., 2010; Hwang et al., 2012; Sartor et al., 2012), and mimicking the direction of each of these changes enhances cocaine reward. Since miRNAs function via base-pairing with complementary sequences within mRNA molecules, it is possible to infer target mRNAs of drug-altered miRNAs through computational predictions, although such methods can yield false positive and negative results. Recent work is encouraging. miR-212 is induced in rat dorsal striatum after cocaine self-administration, and serves to inhibit cocaine intake (Hollander et al., 2010). This action was attributed to the ability of miR-212 to indirectly lead to the activation of CREB, a transcription factor that antagonizes cocaine reward (Robison and Nestler, 2011). Several additional genes implicated in addiction models, such as ΔFosB, dopamine transporter, and glutamate receptor subunits, have also been related to drug-triggered alterations in specific miRNAs (Chandrasekar et al., 2011; Saba et al., 2012). Next generation sequencing recently identified tens of miRNAs that are altered in NAc whole extracts and purified striatal post-synaptic densities after chronic cocaine (Eipper-Mains et al., 2011). Pietrzykowski et al. (2008) reported that chronic ethanol suppresses the expression of BK channels via induction of miR9 in striatum and preoptic area of hypothalamus. Future work is needed to identify the definitive mRNA targets for each regulated miRNA as well as to explore still un-annotated, novel miRNAs that are altered by drugs of abuse using available next generation sequencing datasets. Another area ripe for investigation is drug regulation of lncRNAs, as a recent microarray study revealed several to be altered in the NAc of human heroin addicts examined postmortem (Michelhaugh et al., 2011).
Future Directions
Although still in relatively early stages, work to date has demonstrated that many forms of epigenetic regulation are altered in brain reward regions by drugs of abuse and in turn serve to regulate drug action. Already these initial studies have raised several key questions that will need to be addressed moving forward.
One surprising finding is that drug exposure alters global levels of several histone modifications, such as increased histone acetylation or decreased H3K9me2 and H3K9me3 in NAc. Genome-wide studies have confirmed that a greater number of genomic sites show increased acetylation (Renthal et al., 2009) or reduced H3K9 methylation (Maze et al., 2011; Sun et al., 2012a), however, hundreds of genes show opposite changes in these marks, and most genes show no alterations after drug exposure. This raises the question of what determines whether, and in what direction, a specific gene is modified in the face of a global change in a histone-modifying enzyme and its mark. Based on early ChIP-seq work, one possibility is that global changes may be more reflected in non-genic regions of the genome (e.g., Maze et al., 2011; Sun et al., 2012a), where they might control more general features of chromatin structure, expression of repetitive elements, etc.
Another important question is how epigenetic regulation is translated into transcriptional change, not only steady state alterations in expression but also inducibility in response to a subsequent challenge as well as changes in alternative splicing, which are thought to be under the control of epigenetic regulation (Luco et al., 2011). As noted earlier, no single modification examined to date is deterministic for a change in gene activity. In fact, modifications that are most clearly associated with a functional change, for example, H3K4me3 in promoting transcription, are associated with no change or even opposite changes in transcription at many genes. Such findings are consistent with the required involvement of numerous modifications that work in concert. Indeed, there are increasing reports from simpler systems, and more recently brain, that certain combinations of epigenetic changes do converge at specific genes to mediate their activation or repression. Deciphering such a code, or chromatin signatures, will be a very difficult, yet also highly important, goal for future research. A technical challenge in this effort is the heterogeneous cell population of even brain micronuclei, which makes it impossible to derive data as clear-cut as for cell culture systems. Methodologies are underway to isolate specific cell types from brain (Jiang et al., 2008) and to perform genome-wide ChIP-seq, RNA-seq, and DNA methylation assays on much less starting material (e.g., Adli and Bernstein, 2011). In the meantime, nearly all bioinformatic tools for genome-wide analysis have been developed based on simpler cell culture data, which are not optimal to detect the more subtle signals from terminally differentiated neurons, particularly with the high background noise unavoidable with in vivo studies. Improved analytical tools will require creative collaborations between biologists and bioinformaticians.
Another surprising finding, based on reports that epigenetic modifications can be stable, is that most chromatin changes observed to date are highly labile, reverting to control levels within hours or days of drug withdrawal. At the same time, certain changes are long-lived. To date, this has only been investigated for a small number of candidate genes, and needs to be examined with genome-wide assays. A goal of such work is to determine the time course of multiple types of epigenetic changes in brain reward regions during a course of drug self-administration and subsequent withdrawal and in response to subsequent drug challenges. Important questions to answer include: What determines whether a chromatin modification at a particular gene or non-genic region is stable? And is the stability or instability of any given change predictable based on coincident chromatin signatures at that locus? Initial ChIP-seq studies suggest that global changes in histone marks are not the determining factor of stability, since many histone modifications persist long after such global changes revert to normal.
We also need to better understand the intracellular signaling pathways through which synaptic transmission and neural activity are translated into epigenetic modifications. We still know very little about these steps, with only a small number of examples reported to date (see Fig. 2). For example, ERK (extracellular signal regulated kinase) and its downstream activation of MSK1 (mitogen and stress-activated kinase) have been implicated in the phosphorylation of Ser10 of histone H3 in NAc, as has DARPP32 regulation of nuclear protein phosphatase 1 (Stipanovich et al., 2008). These effects appear selective for the subtype of NAc medium spiny neuron that expresses predominantly D1 dopamine receptors. As another example, protein kinase A, CDK5, and CaMKII have been implicated in cocaine regulation of HDAC5 in NAc (Renthal et al., 2007; Taniguchi et al., 2012). Work is also needed to understand the mechanisms through which epigenetic regulation is targeted to individual genomic loci, as stated earlier.
A major limitation in the field is the difficulty in relating chromatin modifications at a given gene to a functional outcome. Studies to date have relied by necessity on overexpressing or knocking out, or pharmacologically inhibiting, a chromatin modifying enzyme (e.g., an HDAC, HMT, or DNMT) within a given brain region like the NAc and studying the behavioral consequences. However, such manipulations regulate the targeted chromatin modification at hundreds or thousands of genes. One approach to overcome this limitation is to use synthetic zinc finger proteins (ZFPs) or sequence-specific transcription activator-like effectors (TALEs), coupled to an enzymatic moiety, to target a particular chromatin modification to a given gene of interest within a region of adult brain. While still very early in development, such approaches would represent a huge advance in the field, to test, for example, whether H3K9me2 at a particular gene truly regulates that gene and resulting behaviors. Preliminary results are encouraging, in that we can target increased H3K9me2 to the FosB gene in NAc in vivo and consequently blunt cocaine’s behavioral effects (Heller et al., 2012), but more work is needed to validate and extend this approach.
Finally, we need to do more to study the role of epigenetic regulation in mediating lifelong effects of drug exposure or other environmental stimuli (e.g., stress) on subsequent vulnerability to addiction. There are early reports of long-lasting histone modifications being induced at a small number of candidate genes in brain reward regions as a consequence of prenatal exposure to cannabinoids, and such changes correlate with increased susceptibility for heroin self-administration later in life (DiNieri et al., 2011; Tomasiewicz et al., 2012). Likewise, fetal exposure to alcohol is reported to alter the methylation status of the POMC (proopiomelanocortin) gene (Govorko et al., 2012). We also need to ascertain whether any drug-induced epigenetic modifications are transferred to offspring to influence their susceptibility to drug abuse or other conditions. Such trans-generational transmission would require drug-induced epigenetic changes in sperm or ova to persist in the fertilized embryo and to influence adult brain function. There is an early report that this may be the case (Vassoler et al., 2013), however, much further work is needed to demonstrate definitively an epigenetic basis of such transmission and to understand the underlying mechanisms involved.
Conclusions
The ultimate goal of epigenetic studies of addiction is to understand how repeated exposure to a drug of abuse changes the brain in sustained ways to cause the lasting syndrome of addiction. Such studies are also important to understand how an individual’s life experiences establish stable changes at genomic loci, which then help determine that individual’s vulnerability to the addiction-causing effects of subsequent drug exposure. It is our expectation that these studies will reveal a host of genes whose products play important roles in the pathogenesis of addiction and could serve as a template of drug targets to evaluate in future drug discovery efforts. It would also be interesting to determine whether drug effects on epigenetic endpoints in peripheral tissues (e.g., blood) might serve as useful biomarkers for clinical features of addiction, even if those changes in blood are different from those in brain. In these ways, epigenetic approaches promise unprecedented advances in our understanding, diagnosis, and treatment of drug addiction.
Highlights.
Epigenetic regulation mediates adaptations to the environment such as abused drugs.
Epigenetic regulation includes post-translational modifications of histones.
Epigenetic regulation also includes DNA methylation and non-coding RNAs.
Each of these epigenetic mechanisms are influenced by drugs of abuse.
In turn, these epigenetic mechanisms control behavioral responses to the drugs.
Acknowledgments
Preparation of this review was supported by grants from the National Institute on Drug Abuse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 2010;35:2450–2461. doi: 10.1038/npp.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botia B, Legastelois R, Alaux-Cantin S, Naassila M. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS One. 2012;7:e47527. doi: 10.1371/journal.pone.0047527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Hervé D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 2011;6:1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Roze E, Girault JA, Betuing S, Caboche J. Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J Neurochem. 2009;108:1323–1335. doi: 10.1111/j.1471-4159.2009.05879.x. [DOI] [PubMed] [Google Scholar]
- Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci. 2009;42:350–362. doi: 10.1016/j.mcn.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology. 2011;36:1149–1164. doi: 10.1038/npp.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, III, Maze I, Sun HS, Wu EY, Dietz D, Lobo MK, Ghose S, Neve R, Tamminga CA, Nestler EJ. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Addario C, Caputi FF, Ekström TJ, Di Benedetto M, Maccarrone M, Romualdi P, Candeletti S. Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J Mol Neurosci. 2013;49:312–319. doi: 10.1007/s12031-012-9829-y. [DOI] [PubMed] [Google Scholar]
- Damez-Werno D, LaPlant Q, Dietz DM, Sun SH, Scobie KN, Walker I, Koo JW, Mouzon E, Russo SJ, Nestler EJ. Drug experience epigenetically primes fosB gene inducibility in rat nucleus accumbens and caudate putamen. J Neurosci. 2012a;32:10267–10272. doi: 10.1523/JNEUROSCI.1290-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damez-Werno D, Scobie KN, Sun H, Dietz DM, Dias CM, Casadio F, Neve RL, Nestler EJ. Histone arginine methylation in the nucleus accumbens in response to chronic cocaine and stress. Soc Neurosci Abs 69.09 2012b [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNieri JA, Wang X, Szutorisz H, Spano SM, Kaur J, Casaccia P, Dow-Edwards D, Hurd YL. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol Psychiatry. 2011;70:763–769. doi: 10.1016/j.biopsych.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eipper-Mains JE, Kiraly DD, Palakodeti D, Mains RE, Eipper BA, Graveley BR. microRNA-Seq reveals cocaine-regulated expression of striatal microRNAs. RNA. 2011;17:1529–1543. doi: 10.1261/rna.2775511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson D, Shao NY, Koo JW, Feng J, Vialou V, Dietz DM, Maze I, Liu XC, Kennedy P, Renthal W, Neve R, Shen L, Sartorelli V, Nestler EJ. Next generation sequencing using ChIP-Seq highlights essential role for SIRT1 in emotional plasticity. Soc Neurosci Abs. 2012:778.09. [Google Scholar]
- Govorko D, Bekdash RA, Zhang C, Sarkar DK. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry. 2012;72:378–388. doi: 10.1016/j.biopsych.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller E, Sun H, Cates H, Knight S, Zhang F, Zhang S, Nestler EJ. Bidirectional regulation of the fosB gene using synthetic zinc-finger transcription factors for the study of addiction and depression. Soc Neurosci Abs 69.07 2012 [Google Scholar]
- Hiroi N, Agatsuma S. Genetic susceptibility to substance dependence. Mol Psychiatry. 2005;10:336–344.3. doi: 10.1038/sj.mp.4001622. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature. 2010;466:197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, Law PY, Wei LN, Loh HH. Epigenetic programming of mu-opioid receptor gene in mouse brain is regulated by MeCP2 and Brg1 chromatin remodelling factor. J Cell Mol Med. 2009;13(9B):3591–3615. doi: 10.1111/j.1582-4934.2008.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CK, Wagley Y, Law PY, Wei LN, Loh HH. MicroRNAs in opioid pharmacology. J Neuroimmune Pharmacol. 2012;7:808–819. doi: 10.1007/s11481-011-9323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Heidmets LT, Shen HY, Zharkovsky A, Chen JF. Histone deacetylase inhibitors modulates the induction and expression of amphetamine induced behavioral sensitization partially through an associated learning of the environment in mice. Behav Brain Res. 2007;181:76–84. doi: 10.1016/j.bbr.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PJ, Robison AJ, Maze I, Feng J, Badimon A, Bassel-Duby R, Olson EN, Nestler EJ. HDAC1 inhibition blocks cocaine-induced plasticity through targeted changes in histone methylation. Nat Neurosci. 2012 doi: 10.1038/nn.3354. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Kim S, Kim JH. Chronic microinjection of valproic acid into the nucleus accumbens attenuates amphetamine-induced locomotor activity. Neurosci Lett. 2008;432:54–57. doi: 10.1016/j.neulet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, Russo SJ, LaPlant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren B, Maze I, Dietz DM, Watts EL, Iñiquez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren YH, Eisch AJ, Bolaños CA, Kabbaj M, Xiao GH, Neve RL, Hurd YL, Oosting RS, Fan GP, Morrison JH, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci USA. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine A, Huang Y, Drisaldi B, Griffin EA, Jr, Pollak DD, Xu S, Yin D, Schaffran C, Kandel DB, Kandel ER. Molecular mechanism for a gateway drug: epigenetic changes initiated by nicotine prime gene expression by cocaine. Science Transl Med. 2011;3:107ra109. doi: 10.1126/scitranslmed.3003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, Sanchis-Segura C, Vo D, Lattal KM, Wood MA. Modulation of chromatin modification facilitates extinction of cocaine-induced conditioned place preference. Biol Psychiatry. 2010;67:36–43. doi: 10.1016/j.biopsych.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, Mhillaj E, Matheos DP, Palmery M, Wood MA. CBP in the nucleus accumbens regulates cocaine-induced histone acetylation and is critical for cocaine-associated behaviors. J Neurosci. 2011;31:16941–16948. doi: 10.1523/JNEUROSCI.2747-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, McQuown SC, Rogge GA, Astarabadi M, Jacques V, Carreiro S, Rusche JR, Wood MA. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc Natl Acad Sci USA. 2013 doi: 10.1073/pnas.1213364110. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Nestler EJ. The epigenetic landscape of addiction. Ann NY Acad Sci. 2011;1216:99–113. doi: 10.1111/j.1749-6632.2010.05893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Covington HE, III, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren YH, Sampath SC, Hurd YL, Greengard P, Tarakovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Feng J, Wilkinson MB, Sun HS, Shen L, Nestler EJ. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci USA. 2011;108:3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazei-Robison MS, Koo JW, Friedman A, Lansink CS, Robison AJ, Vinish M, Krishnan V, Kim S, Siuta MA, Galli MA, Niswender KD, Appasani R, Horvath MC, Neve RL, Worley PF, Snyder SH, Hurd YL, Cheer JF, Han MH, Russo SJ, Nestler EJ. Role for mTOR signaling and neuronal activity in morphine-induced adaptations in ventral tegmental area dopamine neurons. Neuron. 2011;72:977–990. doi: 10.1016/j.neuron.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelhaugh SK, Lipovich L, Blythe J, Jia H, Kapatos G, Bannon MJ. Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem. 2011;116:459–466. doi: 10.1111/j.1471-4159.2010.07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, Kreek MJ. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873. doi: 10.1038/npp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Huang W, Hamon SC, Maili L, Witkin BM, Fox RG, Cunningham KA, Moeller FG. Forced abstinence from cocaine self-administration is associated with DNA methylation changes in myelin genes in the corpus callosum: a preliminary study. Front Psychiatry. 2012;3:60. doi: 10.3389/fpsyt.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Do Couto BR, Alfonso-Loeches S, Aguilar MA, Rodriguez-Arias M, Guerri C. Changes in histone acetylation in the prefrontal cortex of ethanol-exposed adolescent rats are associated with ethanol-induced place conditioning. Neuropharmacology. 2012;62:2309–2319. doi: 10.1016/j.neuropharm.2012.01.011. [DOI] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, Siegelmann HT, Treistman SN. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Denny A, Lieu M, Carreon S, Li J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics. 2011;6:1095–1104. doi: 10.4161/epi.6.9.16924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, Xiao GH, Kumar A, Russo SJ, Graham A, Tsankova N, Kerstetter KA, Kippin TE, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, III, Truong HT, Alibhai I, Kumar A, Olson EN, Nestler EJ. ΔFosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Kumar A, Xiao GH, Wilkinson M, Convington HE, III, Maze I, Sikder D, Robison AJ, LaPlant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ. Genome wide analysis of chromatin regulation by cocaine reveals a novel role for sirtuins. Neuron. 2009;62:335–348. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, Wee SM, Koob G, Turecki G, Neve R, Thomas M, Nestler EJ. Behavioral and structural responses to chronic cocaine require a feed-forward loop involving ΔFosB and CaMKII in the nucleus accumbens shell. J Neurosci. 2013 doi: 10.1523/JNEUROSCI.5192-12.2013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge GA, Wood MA. The role of histone acetylation in cocaine-induced neural plasticity and behavior. Neuropsychopharmacology. 2013;38:94–110. doi: 10.1038/npp.2012.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu P, Host L, Gobaille S, Sandner G, Aunis D, Zwiller J. Histone deacetylase inhibitors decrease cocaine but not sucrose self-administration in rats. J Neurosci. 2008;28:9342–9348. doi: 10.1523/JNEUROSCI.0379-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Messing RO. Signaling pathways mediating alcohol effects. Curr Top Behav Neurosci. 2013;13:87–126. doi: 10.1007/7854_2011_161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Storchel PH, Aksoy-Aksel A, Kepura F, Lippi G, Plant TD, Schratt GM. Dopamine-regulated microRNA MiR-181a controls GluA2 surface expression in hippocampal neurons. Mol Cell Biol. 2012;32:619–632. doi: 10.1128/MCB.05896-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Kumaresan V, Schmidt HG, Famous KR, Chawla P, Vassoler FM, Overland RP, Xia E, Bass CE, Terwilliger TF, Pierce RC, Cha JHJ. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res. 2012;36:61–71. doi: 10.1111/j.1530-0277.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor GC, St Laurent G, 3rd, Wahlestedt C. The emerging role of non-coding RNAs in drug addiction. Front Genet. 2012;3:106. doi: 10.3389/fgene.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Im HI, Veno MT, Fowler CD, Min A, Intrator A, Kjems J, Kenny PJ, O’Carroll D, Greengard P. Argonaute 2 in dopamine 2 receptor-expressing neurons regulates cocaine addiction. J Exp Med. 2010;207:1843–1851. doi: 10.1084/jem.20100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Sangrey GR, Darnell SB, Schassburger RL, Cha JH, Pierce RC, Sadri-Vakili G. Increased brain-derived neurotrophic factor (BDNF) expression in the ventral tegmental area during cocaine abstinence is associated with increased histone acetylation at BDNF exon I-containing promoters. J Neurochem. 2012;120:202–209. doi: 10.1111/j.1471-4159.2011.07571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FA, Penta KL, Matevossian A, Jones SR, Konradi C, Tapper AR, Akbarian S. Drug-induced activation of dopamine D(1) receptor signaling and inhibition of class I/II histone deacetylase induce chromatin remodeling in reward circuitry and modulate cocaine-related behaviors. Neuropsychopharmacology. 2008;33:2981–2992. doi: 10.1038/npp.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scobie KN, Damez-Werno D, Sun H, Neve RL, Caiafa P, Nestler EJ. Role of poly(ADP-ribosyl)ation in drug-seeking behavior. Soc Neurosci Abs 69.12 2012 [Google Scholar]
- Shen HY, Kalda A, Yu L, Ferrara J, Zhu J, Chen JF. Additive effects of histone deacetylase inhibitors and amphetamine on histone H4 acetylation, cAMP responsive element binding protein phosphorylation and DeltaFosB expression in the striatum and locomotor sensitization in mice. Neuroscience. 2008;157:644–655. doi: 10.1016/j.neuroscience.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Sheng J, Lv Z, Wang L, Zhou Y, Hui B. Histone H3 phosphoacetylation is critical for heroin-induced place preference. Neuroreport. 2011;22:575–580. doi: 10.1097/WNR.0b013e328348e6aa. [DOI] [PubMed] [Google Scholar]
- Starkman BG, Sakharkar AJ, Pandey SC. Epigenetics-beyond the genome in alcoholism. Alcohol Res. 2012;34:293–305. doi: 10.35946/arcr.v34.3.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanovich A, Valjent E, Matamales M, Nishi A, Ahn JH, Maroteaux M, Bertran-Gonzalez J, Brami-Cherrier K, Enslen H, Corbillé AG, Filhol O, Nairn AC, Greengard P, Hervé D, Girault JA. A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 2008;453:879–884. doi: 10.1038/nature06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HS, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D, Neve RL, Zachariou V, Shen L, Nestler EJ. Morphine epigenetically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci. 2012a;32:17454–17464. doi: 10.1523/JNEUROSCI.1357-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HS, Damez-Werno D, Scobie K, Dias C, Koo JW, Kennedy P, Dietz DM, Neve R, Varga-Weisz P, Nestler EJ. Role of chromatin remodelers in the mouse nucleus accumbens in preclinical models of depression and cocaine addiction. Soc Neurosci Abs 69.10 2012b [Google Scholar]
- Sun J, Wang L, Jiang B, Hui B, Lv Z, Ma L. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self administration in rats. Neurosci Lett. 2008;441:72–76. doi: 10.1016/j.neulet.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, Vasanthakumar A, Godley LA, Chang Q, Cheng X, He C, Jin P. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW. Histone Deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron. 2012;73:108–120. doi: 10.1016/j.neuron.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiewicz HC, Jacobs MM, Wilkinson MB, Wilson SP, Nestler EJ, Hurd YL. Proenkephalin mediates the enduring effects of adolescent cannabis exposure associated with adult opiate vulnerability. Biol Psychiatry. 2012;72:803–810. doi: 10.1016/j.biopsych.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nature Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Vassoler FM, White SL, Schmidt HD, Sadri-Vakili G, Pierce RC. Epigenetic inheritance of a cocaine-resistance phenotype. Nat Neurosci. 2013;16:42–47. doi: 10.1038/nn.3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WS, Kang S, Liu WT, Li M, Liu Y, Yu C, Chen J, Chi ZQ, He L, Liu JG. Extinction of aversive memories associated with morphine withdrawal requires ERK-mediated epigenetic regulation of brain-derived neurotrophic factor transcription in the rat ventromedial prefrontal cortex. J Neurosci. 2012;32:13763–13775. doi: 10.1523/JNEUROSCI.1991-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]