

Abstract
Background
Disulfide bond formation is a key posttranslational modification, with implications for structure, function and stability of numerous proteins. While disulfide bond formation is a necessary and essential process for many proteins, it is deleterious and disruptive for others. Cells go to great lengths to regulate thiol-disulfide bond homeostasis, typically with several, apparently redundant, systems working in parallel. Dissecting the extent of oxidation and reduction of disulfides is an ongoing challenge due, in part, to the facility of thiol/disulfide exchange reactions.
Scope of the review
In the present account, we briefly survey the toolbox available to the experimentalist for the chemical determination of thiols and disulfides. We have chosen to focus on the key chemical aspects of current methodology, together with identifying potential difficulties inherent in their experimental implementation.
Major conclusions
While many reagents have been described for the measurement and manipulation of the redox status of thiols and disulfides, a number of these methods remain underutilized. The ability to effectively quantify changes in redox conditions in living cells presents a continuing challenge.
General Significance
Many unresolved questions in the metabolic interconversion of thiols and disulfides remain. For example, while pool sizes of redox pairs and their intracellular distribution are being uncovered, very little is known about the flux in thiol-disulfide exchange pathways. New tools are needed to address this important aspect of cellular metabolism.
Keywords: Detection, Modification, Redox, Exchange, Nucleophile
1. Introduction
1.1 Thiol-disulfide exchange
Thiol-disulfide exchange reactions play critical roles in many aspects of cellular function. In these reactions, a nucleophilic thiolate attacks one of the two sulfur atoms of the target disulfide bond (Fig. 1). Since the reactivity of the sulfhydryl group is dominated by that of its deprotonated form, we first briefly address aspects of the acidity (pKa) and nucleophilicity of thiolates. The protonated forms of simple alkyl thiols are practically unreactive as nucleophiles under normal conditions; reacting some 1010–fold slower than their corresponding thiolates [1,2]. As the concentration of the thiolate is derived from the Henderson-Hasselbalch the equation, for the pH dependency of the reaction rate for thiol-disulfide exchanges is governed by the following equation for reaction kinetic relationship [3]:
Fig. 1.

Thiol-disulfide exchange. The attack of a thiolate (the nucleophile, S-n) on a disulfide bond takes place through a linear transition state where the central sulfur atom (Sc) will participate in a new disulfide bond and resolution of a new leaving group thiolate (S-lg). Which of the two sulfur atoms participating in the disulfide bond will eventually act as leaving group is dependent on steric, electrostatic and intrinsic acidity of the thiolate species involved.
Here kobs is the observed rate constant at a given pH, and k is the corresponding limiting rate constant for the thiolate at high pH values. Thus, kobs is one half of the limiting rate constant at the pKa, but falls to 1/104 of the maximal reactivity at 4 pH units below the pKa.
Biological thiols show a very wide range of pKa values (from about 3 to 11, thus corresponding to an 8-order of magnitude shift in the deprotonation equilibrium [4]). The factors contributing to this profound modulation of thiol pKa's are under, which so profoundly modulate thiol pKa's, are under active investigation, and include solvation, electrostatic effects with neighboring charges and dipoles, as well as H-bonding interactions [5-7]. It is important to note that the pKa of thiols has two distinct effects on reactivity. Obviously, as noted above, a lower thiol pKa increases the fraction of thiol in its reactive thiolate form, however, the intrinsic reactivity of fully-formed thiolates (at the high pH limit) typically declines with decreasing thiol pKa for a series of structurally-related thiols [8-10]. The ability of a thiol sulfur atom to retain a proton is to some extent a reflection of its intrinsic nucleophilicity, thus illustrating the correlation between nucleophilicity and pKa.
Although it might seem unnecessary in terms of populating the thiolate, some enzymes have evolved to have pKa's far below the predominant pH of a typical cellular environment. Such low pKa values might, however, suppress oxidative side reactions that would otherwise compromise catalysis. Another reason is that marked differences in acidity allow the equilibrium constant for thiol disulfide exchange to be tuned by thiol pKa values. Thus, in the thiol-disulfide exchange reaction:
-- lowering the pKa of R2-SH with respect to R1-SH will improve the leaving-group properties of R2-SH and bias the equilibrium to the right [5,11].
Two consecutive thiol/disulfide exchange reactions accompany the overall redox reaction shown below:
Knowing the stability of one disulfide, together with the magnitude of Kox, allows the stability of the other disulfide to be directly calculated. Again the magnitude of Kox will be dependent on a combination of effects including steric, electrostatic and pKa values of the thiol species involved [12].
Finally, the rates of thiol-disulfide exchange reactions are influenced by the requirement for a linear arrangement of the three sulfur atoms in the transition state [13,14]. In proteins, the two sulfur atoms of the disulfide bonds often differ markedly in their accessibility to an attacking thiolate nucleophile generating a single mixed disulfide intermediate. In the event that both disulfide sulfur atoms are exposed, the outcome of disulfide exchange may largely reflect discrimination based on pKa values (see above).
1.2 Overall principles for thiol-disulfide detection and quantifications
Thiols are typically detected directly by virtue of their relatively high reactivity compared to most other common species in biological systems. Disulfides, on the other hand, have no strong chemical signature, and are hence most commonly detected after reduction to their corresponding thiols. Thus, the most common methodologies for thiol and disulfide quantification involve determination of free thiol concentration, followed by alkylation, reduction of disulfide bonds, and subsequent quantification of the additional exposed thiols. The processes of reduction and alkylation are thus pivotal for thiol quantification. In the determination of disulfides, the complete removal of the reducing species prior to detection is crucial so that no cross-reaction takes place between the reductant and the reagent used for thiol detection.
2. Quenching of thiol oxidation and exchange
2.1 Thiol alkylation
Alkylation of cysteine thiols with iodo, bromo or chloro substituted acetic acid or acetamide is a classic approach that has been exploited since the 1930′s. The relative reaction rates between glutathione and these halogenated acetates are 100:60:1 for iodo-, bromo- and chloro-acetates respectively [15]. Although iodoacetic acid or iodoacetamide are by far the most widely used haloalkanes for thiol alkylation, they can show significant reactivity towards other nucleophilic side chains. While such side-reactions may be fairly innocuous for most analytical applications, they become major problems in proteomic approaches involving the identification of reacted modified species by mass spectrometry. Thus iodoacetate treatment was shown to significantly modify lysine residues as demonstrated by mass spectroscopy [16]. The substitution of chloroacetic acid alleviated this problem but this solution cannot be adopted when thiol residues must be quenched rapidly.
Maleimides are very widely used reagents for the alkylation of thiols. The reaction represents a Michael addition of the thiolate on the electrophilic double bond of the maleimide (Fig. 2, reaction A). The enone functionality of N-ethylmaleimide (NEM) shows an extinction coefficient of 620 M−1cm−1 at 302 nm allowing reactions with nucleophiles to be conveniently followed spectrophotometrically [17]. A notable additional advantage of maleimides is that they react rapidly with thiols at neutral or slightly acidic pH values with rate constants that are some 3 to 4 orders of magnitude faster than iodoacetamide under comparable conditions [1,18,19]. Despite their utility, several reactions may complicate the use of maleimides, particularly at pH values above 7. Firstly, while maleimides are frequently characterized in the literature as irreversible thiol-modifying reagents, the adducts are subject to base-catalyzed reverse Michael reactions (Fig. 2, reaction B and C) leading to the possible migration of the maleimide between thiol targets [20]. Further maleimide adducts, particularly those where N-R represents an aniline functionality, are prone to ring-open by hydrolysis, yielding the isomeric products shown in Fig. 2, reaction D [21]. Such ring-opening reactions have been used to identify maleimide-labeled peptides, [22,23]. In aggregate, these secondary reactions may play an important role in modulating the stability of maleimide conjugates in vivo [20,21].
Fig. 2.

Addition of thiols to maleimides together with selected exchange and ring opening reactions. The reaction of thiol (R1-SH) with maleimides (in this case NEM) is reversible, albeit shifted strongly toward the adduct formation (Reaction A). In the presence of excess thiols (R2-SH), however, reversal of the initial adduct leads to the accumulation of the alternative adduct (Reactions B and C). The adduct is susceptible to ring-opening by hydrolysis of the imide (Reaction D).
Mammalian cultured cells are permeable to NEM and this has encouraged its use for quenching thiols in intact cells. However, the inclusion of a denaturant, such as SDS, may be necessary to ensure rapid labeling of all free cysteine residues because about 20% of total cellular protein thiols are not susceptible to modification by NEM under native conditions [24].
Vinyl pyridine, like NEM, reacts with thiols at the double bond and was previously widely used. Since vinyl pyridine reacts more than 500-fold slower than NEM, both high concentration and long reaction times are required for complete reaction [25,26].
Cyanylation using 1-cyano-4-dimethylamino-pyridinium salts (CDAP) represents an efficient means of thiol blocking [27-30] (Fig. 3A). The reaction is rapid at pH 4-5 leading to quantitative derivatization of thiols using low mM concentrations of CDAP [30,31]. These properties are useful because they allow efficient alkylation at low pH where thiol exchange is minimal. An additional feature of CDAP is that cyanylated peptide-thiol adducts are susceptible to specific cleavage in the presence of ammonia (Fig. 3B). Here, a cyclization involving the cyanylated side-chain results in cleavage of the peptide chain N-terminal to the target cysteine residue. In combination with mass spectroscopy this procedure allows for mapping of disulfide bond patterns in proteins [32]. On the other hand, the cyanylated proteins are intrinsically unstable above pH 7. CDAP itself is stable in polar aprotic solvents, such as acetonitrile, but is prone to hydrolysis in aqueous solutions above pH 5 [30].
Fig. 3.

Cyanylation using CDAP. Reaction (A) of CDAP with cysteinyl peptide results in formation of a cyanylated species. This species can react further in 1.5 M NH4OH to cleave the adjacent N-proximal peptide bond (B).
Although rapid and indiscriminate alkylation of thiols is often the desired outcome of labeling protocols, less reactive reagents have been recently used very effectively in proteomic approaches for the identification of proteins containing hyper-reactive cysteine residues [33,34].
3. Reduction of disulfide bonds
In the reduction of thiols for further analysis there are three major concerns: a) that the disulfide reduction is quantitative and rapid, b) that the reducing agent is specific, and does not show significant side reactions, and c) that the reductant selected does not complicate down-stream reactions and processes. Disulfide reduction is accomplished primarily by thiol exchange type reagents (like dithiothreitol, DTT, or 2-mercaptoethanol, ME) or by various substituted phosphines such as tris(2-carboxyethyl)phosphine, TCEP [35].
3.1 Phosphine- and Thiol-based reductants
Unlike thiol reagents, for all practical purposes phosphines are irreversible reductants of disulfide bonds in aqueous solutions (Fig. 4). Here the phosphine performs a nucleophilic attack on one of the two sulfur atoms forming a phosphonium ion sulfur adduct which is subsequently hydrolyzed yielding the corresponding phosphine oxide. This irreversibility contrasts with thiol-based reductants, which are typically needed in large excess and consequently must be removed, or quenched, prior to sample workup.
Fig. 4.
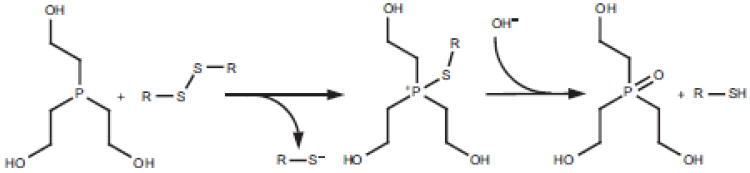
Reaction mechanism for the reduction of disulfide bonds by phosphines. Here, the reaction of tris-(2-hydroxyethyl) phosphine (THP) is shown. Several other water soluble phosphines are commercially available, or have been described in the literature.
TCEP is now a widely used substitute for DTT in a broad range of biochemical applications. Mono-, di- and tri-methyl esters of TCEP show progressively lower pKa values at phosphorus [36]. Thus, while the pKa of the nucleophilic center in TCEP is 7.5, that of trimethyl-TCEP is 4.7 [36]. While lowering the pKa promotes the expected decreases in intrinsic nucleophilicity, these water-soluble phosphine methyl esters are more facile reductants than TCEP at lower pH values. For example, at pH 5, their effectiveness in the reduction of a disulfide-bearing model peptide was tri- > di- > mono-methyl-TCEP > TCEP ≫ DTT [36]. Nevertheless, none of these phosphine-based reductants are likely to be useful at the low pH values needed to suppress H/D exchange in peptides liberated from peptic digests during the assessment of the surface accessibility and dynamics of proteins. (At pH values lower than 3, electrochemical or zinc metal reductions may provide viable substitutes; see below). Finally, neutral water-soluble phosphines may prove useful alternatives to DTT in the modulation of cellular redox poise. For example, the tmTCEP analog penetrates model lipid bilayers much more rapidly than DTT [36]: a standard reductant used to apply reductive stress in cell culture studies. This permeability, combined with the irreversibility noted earlier, may be advantageous for in vivo purposes when short pulses of reduction are desired.
Although TCEP is the most widely used of the water soluble phosphine reductants, the commercially-available tris(2-hydroxyethyl) phosphine (THP) is perhaps more useful. It is frequently more reactive as a disulfide reductant than TCEP [36,37]. Furthermore, THP can be added to solutions in mM amounts without the necessity for pH adjustment, and THP is less polar allowing better access to hydrophobic environments than TCEP. Stock solutions of THP, as well as other phosphine reagents, can be standardized by any one of the methods described later in this review.
In terms of the thiol-based reductants, while ME is very convenient to use, and may be adequate for e.g. reducing SDS-PAGE applications, ME is not more reducing than generic protein thiols. In large excess (for SDS-PAGE typically >0.5 M) it will, however, shift the equilibrium towards mixed ME-protein disulfides and possibly generate the reduced protein species. As the pKa of the ME thiol is 9.5 [38] it is essential that pH values ≥ 7 are used to obtain efficient reaction.
DTT, on the other hand, has a low redox potential due to formation of a six-membered ring upon disulfide exchange. Indeed the equilibrium constant for the reaction between DTT and GSSG is around 200 [39]. Consequently, commercial preparations of GSH (the best of which often contain up to 1% GSSG) cannot typically achieve any significant reduction of oxidized DTT; a property that must be kept in mind when establishing equilibria with very reducing disulfide bonds in proteins. While DTT is overwhelmingly the mostly widely used dithiol reductant for protein disulfides, a series of newer alternatives, exhibiting a range of ring sizes, polarities and redox potentials, have been synthesized and characterized by Whitesides [40,41] and Raines [42] and their coworkers.
3.2 Other methods for disulfide reduction
A number of other reducing agents and methods have been described. These include reduction with sodium borohydride, metallic zinc, and methods for electrochemical reduction. The advantage of the first two reductants is that excess reagent can be easily removed. Borohydride reductions take place at high pH, however, acidification of the reaction mixture completely discharges the reactive hydride, with the evolution of gaseous H2 [43,44]. A potential disadvantage of this procedure is that the elevated pH values required for disulfide reduction can generate undesirable side-reactions such as peptide bond cleavage and glutamine/asparagine deamidation.
Metallic zinc was widely used as a reductant for disulfides in the older literature and deserves renewed consideration. Reductions of peptides and certain proteins (using zinc dust in 1% TFA in aqueous acetonitrile mixtures; pH ∼ 1) are complete in minutes [45]. Moreover, millimolar levels of GSSG are rapidly reduced by a modest excess of metallic zinc in citrate buffer, pH 2.5 (CT, unpublished observations). Excess zinc metal can be easily removed by centrifugation, or the reductant might be incorporated in a stationary phase for in-line processing. Since the reduction of disulfides by zinc is facilitated by low pH values, the procedure might prove useful for high-throughput mass spectrometry applications. It should be noted, however, that zinc ions may be released into solution, both during reduction of disulfide bonds, and as an incidental consequence of exposure of the metal to acidic pH values.
Finally, studies on the reduction of small molecule and protein disulfides at a dropping mercury electrode, or via stirred mercury surfaces, were initiated more than 50 years ago. In some instances small proteins could be reduced directly, in other cases chaotrophes were used with or without the presence of small mediating thiols [46-48]. Frequently, a large electrochemical over-potential was necessary to ensure efficient kinetic reduction, although more modest potentials may achieve controlled reduction of subsets of protein disulfides [46]. More recent work has explored a range of derivatized surfaces in the electrochemical reduction of disulfides [49-51].
4. Thiol detection
Thiols may be detected by a variety of reagents and separation techniques. Thiol reaction may result in quantitative formation of a chromophore or fluorophore, but covalent thiol modification may also provide analyte discrimination during liquid chromatograpy and gel electrophoresis or mass spectrometry.
4.1 Colorimetric thiol detection
The classical chromogenic reagent for thiol detection is 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB; also known as Ellman's reagent [52]. This compound has a highly oxidizing disulfide bond, which is stoichiometrically reduced by free thiols in an exchange reaction, forming a mixed disulfide and releasing one molecule of 5-thio-2-nitrobenzoic acid (TNB; Fig. 5). TNB is an excellent leaving group with a thiol pKa of 4.5 [53]. If a second thiol R2-SH initially evades reaction with DTNB it may resolve the mixed analyte disulfide in Fig. 5, thereby releasing the second TNB and generating a new mixed disulfide (R1-S-S-R2). In either case, one TNB is released for every thiol oxidized upon DTNB treatment.
Fig. 5.

Detection of thiols using Ellman's reagent (DTNB). Due to the low pKa of the TNB it forms an extremely efficient leaving group. The net effect of this is that the analyte efficiently forms disulfide bonds if steric and concentration issues allow. Note that for each analyte thiol reacted, a TNB is formed.
While DTNB has weak absorption at 412 nm, the extinction coefficient for TNB is 14,100 M−1cm−1 at pH 7.3 [53], but drops steeply at pH below reflecting protonation of the orange thiolate species.
The extinction coefficient of the thiolate is slightly dependent on ionic strength. This is relevant when thiols are determined in 6 M guanidinium chloride where the extinction coefficient at 412 nm drops to 13,700 M−1cm−1.
One should also be aware that DTNB is fairly sensitive to hydrolysis at elevated temperatures and, in particular, at pH values > 7. Decomposition is initiated via hydrolytic scission of the activated disulfide of DTNB to yield the sulfenate species shown in Fig. 6. A more hydrolytically stable derivative of DTNB has been developed to address this instability [54].
Fig. 6.

Hydrolysis of DTNB. Hydroxyl ions will attack the disulfide bond of DTNB forming a sulfenic acid and a thiolate. This reaction is particularly relevant for activated disulfides like that found in DTNB.
An emerging alternative to DTNB is 4,4′-dithiodipyridine (4-DPS). While reduction of DTNB generates the TNB thiolate, 4-DPS reduction leads to formation of the strongly absorbing resonance-stabilized 4-thiopyridone tautomer (Fig. 7). The pH independent absorption of the pyridone (extinction coefficient 21,000 M−1cm−1 at 324 nm) over pH 3 – 7 then allows quantification of thiols at relatively low pH values. Under these conditions a number of unwanted side reactions are suppressed and hydrolytic scission of the disulfide bond is insignificant. While this is a conspicuous advantage of 4-DPS, the longer wavelength maximum for TNB, over the pyridone product of 4-DPS, makes DTNB more suitable for the quantification of thiols in solutions that strongly absorb in the near UV. The much lower solubility of the electroneutral 4-DPS (∼ 3 mM in water) compared to the dianion form of DTNB at pH 7 is another factor to consider. Consequently DTNB is considered membrane impermeant at neutral pH values [36], whereas 4-DPS can access thiols in hydrophobic environments and pass through biological membranes.
Fig. 7.

Structure of 4-DPS showing subsequent reaction with thiols. For each thiol oxidized a molecule of thiopyridone is made. The latter is a favored tautomer of the thiol, which absorbs strongly at 324 nm. Note that as the thiopyridone is uncharged even at low pH the absorbtion remains unaffected.
While these activated chromogenic reagents have proven extremely useful in the quantification of thiols, they must be used with an appreciation of their limitations. We mention one example for illustration: the potential interference in the determination of thiol concentrations that is associated with the presence of sulfite in biological, technical or environmental samples. Sulfite attacks disulfide bonds effecting their net scission with the release of -SH and -S-SO32− functionalities. While generic disulfides are not particularly sensitive to sulfitolysis, the highly reactive disulfides of DTNB and DPS leads to a stoichiometric reaction with sulfite, and to a consequent overestimation of thiol concentration in the presence of sulfite [55].
4.2 Fluorescent adducts
Many reagents for the fluorimetric detection of thiols have been developed and are the subject of extensive reviews e.g. [56,57]. Here we identify a few highlights of selected reagent classes. Monobromobimane (mBBr) and benzofurazans gain fluorescence as they react with thiols. Thus mBBr shows a blue fluorescence only after thioether formation (exciting at 380 nm and emitting at 480 nm [58]. The reagent has been used extensively both for detection of low molecular weight thiols as well as proteins modified for in-gel detection.
There are two commonly used varieties of benzofurazans: 7-fluorobenzo-2-oxa-1,3-diazole-4-sulfonate (SBD-F) and 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole (ABD-F) Fig. 8 [59,60]. Both reagents are non-fluorescent, but attain strong fluorescence at around 500 nm upon conjugation with thiols. Of particular interest is the observation that these reagents do not significantly cross-react with phosphines, allowing reduction of disulfides and labeling of total thiols to be conducted in a single reaction mixture [61]. While SBD-F requires rather harsh conditions (60 °C at pH 8.5 for 1 h), modification with ABD-F is complete at ambient temperatures over 10 min at pH 8. For this reason the use of ABD-F is probably preferable, although for some applications a certain lack of specificity may pose a problem [62]. ABD-F is furthermore fairly unstable in the presence of excess thiols [63]. A particularly useful application of benzofurazans is that several small-molecular weight thiol-adducts can be detected simultaneously following HPLC separation with fluorescence detection [64]. Recently a new class of benzofurazans has been described which are more reactive and which have potential application in fluorescence microscopy [65].
Fig. 8.

Reaction of benzofurazanes with thiols. The reaction products are highly fluorescent.
4.3 Formation of detectable thiol adducts using gel-shift assays
Numerous maleimide derivatives are available commercially, in which the ethyl group of NEM has been replaced by other substituents carrying fluorophores, affinity tags, solubility enhancing tags, or tags that introduce a large increase in molecular mass. We will use the abbreviation HMD to denote heavy maleimide derivatives when referring to this latter application. Here the bulky substituent, following thiol modification, results in a shift in mobility on SDS-PAGE detectable by staining or by immunoblotting. Nevertheless, the effect of modification by HMDs may be unpredictable and methods must be optimized for each cysteine and protein [66]. The most popular HMD reagents include 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; [67]) and polyethylene glycol maleimides (PEG-mal; [68]). The shift in mobility for AMS is typically equivalent to 0.5-1 kD. PEG-mal is commercially available in mono-disperse preparations with molecular weights of up to 40 kD, however, 2 and 5 kD reagents are often the most relevant for gel shift assays since modification with PEG-mal results in shifts that exceed the actual mass of the reagent. This is apparently due to the interaction between SDS and PEG and hence excess PEG-mal reagent must also be removed prior to SDS-PAGE [69].
4.4 Other reagents for thiol blocking
In addition to the thiol alkylating reagents mentioned earlier, S-methyl methanethiosulfonate (MMTS) has been widely employed to trap thiol groups. MMTS generates a methylthio-mixed disulfide with target thiols while simultaneously liberating the good leaving group methylsulfinic acid (Fig. 9). While MMTS has the advantage of introducing a very small thiol substituent, this reagent class needs to be used with appropriate caution if the intent is to quench thiol/disulfide exchange reactions. Thus under insufficiently rigorous reaction conditions, MMTS can promote intramolecular disulfides rather than quenching all thiols as their methyl disulfides [70]. Recently, a new thiourea-activated disulfide fluorescent reagent (FCAD) has been synthesized that reacts efficiently with thiols at pH 3-4 [71]. FCAD thus reacts under conditions that minimize thiol/disulfide exchange and may find uses in chromatography and gel-electrophoresis.
Fig. 9.

Reaction of MMTS with thiols. The formation of a methyldisulfide (Reaction A) is prone to shuffling with adjacent reactive thiols (Reaction B). This renders MMTS unreliable as a thiol quencher.
5. Practical considerations for determination of the thiol/disulfide redox state of proteins
Gel shift assays are frequently used to determine the thiol/disulfide redox status of a protein. In some cases the formation of a disulfide bond, particularly one from widely-spaced thiols, generates a gel-shift without the need for further derivatization using HMDs. However, it is always essential to include an alkylation step to block any free thiols to prevent any potential thiol/disulfide shuffling during exposure to SDS. Indeed, shuffling can be quantitative when samples are heated for two minutes at 95 °C in preparation for SDS-PAGE analyses (JRW, unpublished data).
Quite often the formation of short-range disulfide bonds in proteins does not result in significant mobility shifts on SDS-PAGE. Here, free thiols can be quenched with a small reagent, such as NEM, and, after removal of excess maleimide, the disulfide bonds are reduced so that the liberated thiols can be labeled with an HMD. If the redox status of only one pair of cysteines is investigated it may be tempting to label the thiol fraction with the HMD directly and leave the disulfides unmodified. It should be noted that maleimides are not rigorously specific, and can undergo side reactions with amines particularly at higher pH values [72-74]. Obviously alkylation of reactive amines with HMDs will introduce significant error into the estimation of thiol/disulfide status. If however, these amines are first reacted with NEM, which does not significantly decrease the electrophoretic mobility of the protein, only thiols liberated after subsequent reduction of the disulfide bonds will be modified by the HMD [75].
In practical terms, the initial alkylation with NEM leaves the sample containing a significant concentration of free NEM. This alkylating agent must be removed prior to the subsequent reduction step to avoid an underestimation of the disulfide-content of the protein. Although the alkylating potential of NEM is discharged with either DTT or phosphine treatment, this reaction may be slower than the de novo generation of thiols in the sample by these reductants. One way to remove NEM is to pre-treat the sample with a small excess of ME relative to the alkylating reagent prior to reduction [76]. ME is not a strong enough reductant to significantly shift the redox balance under denaturing conditions, but will still readily react with the alkylating reagent. NEM is relatively stable in 100% (w/v) aqueous solution of TCA, and can therefore conveniently be added to the TCA solution used for quenching (R.E. Hansen and JRW, unpublished data). Excess NEM can also be removed prior to reduction following precipitation with TCA and washing the pelleted protein with acetone.
The quantification of GSH and GSSG has recently been reviewed extensively [77]. In brief, the classical assay developed by Tietze [78] remains very popular and can detect low levels of both GSH and GSSG in both conventional cuvettes and using a 96-well microtiter plate format. The assay is based on the specific reduction of glutathione disulfide by glutathione reductase at the expense of the oxidation of NADPH. For the determination of total glutathione, the inclusion of DTNB oxidizes the GSH component, and allows the rate of enzymatic reduction of the disulfide to be followed via release of the TNB anion. A key feature of this assay is that at low glutathione concentration (much lower than the Km for glutathione reductase) the rate of TNB generation will be proportional to the glutathione concentration. This can be tested empirically by construction of a suitable standard curve leading to the determination of total glutathione. For the specific measurement of GSSG levels, the GSH component in the sample must be initially alkylated so that it is not able to cycle in the assay. 2-vinylpridine has been used extensively for this purpose as it is capable of trapping GSH while it is ineffective at intercepting the thiol-based chemistry of the active site of glutathione reductase [79]. More recently an alternative vinylpyridine derivative has been suggested which more efficiently quenches of GSH without inhibiting glutathione reductase [80].
5.1 Electrochemical detection
In-line HPLC detection of thiols and disulfides by amperometric or coulometric methods represent interesting alternatives to conventional chemical conversion followed by chromogenic or fluorogenic detection. Here the redox active state of the thiols and disulfides species are directly detected either as a change in current or potential over suitable electrodes [81]. Thus, there is no need for chemical modification of the sample. The external potential can be changed so that it optimally matches the species of interest or array detectors can be employed [51,82,83].
5.2 Thiol-disulfide sensing using GFP-based probes
GFP-based sensors for thiol-disulfide reactions were developed about 10 years ago independently in two laboratories [84,85]. The expanding toolbox of available sensors has recently been the subject of an excellent review by Meyer and Dick [86]).
All these sensors feature a pair of engineered cysteines at the surface of the protein in close proximity to the fluorophore. The formation of a disulfide results in a slight movement of adjacent β-strands, with an associated modulation of the fluorescence of the probe. For any sensor the useful dynamic range must cover relevant practical conditions, in this case redox potential. The other prerequisite is the ability to equilibrate with the species to be determined. Somewhat serendipitously, the initial set of probes all proved to be highly reducing and somewhat oxidized under steady-state conditions in the eukaryotic cytosol. On should note that any thiol-disulfide redox sensor is initially synthezised on the ribosome in its reduced state. Thus, any equilibration with the surrounding medium necessitates an oxidation to establish equilibrium. A crucial, and perhaps somewhat surprising discovery, was that equilibration with the cytosolic glutathione buffer is catalyzed by glutaredoxin, whereas the steric properties of the sensing thiols render them inaccessible to reaction with thioredoxin [87]. It was demonstrated that cellular depletion of glutaredoxin activity slowed down oxidation of the sensor and thus prolonged the time required to for newly synthesized redox sensor reach equilibrium in vivo. This glutathione-sensing ability can be further enhanced by covalent linkage of glutaredoxin [88,89] allowing for GFP-based sensors to be used under steady-state conditions. Since these sensors employ a disulfide-sensing mechanism they cannot be used to determine absolute concentrations of GSH and GSSG, however, they respond to the redox potential of the glutathione buffer (that is to the [GSH]2/[GSSG] ratio) according to the relationship:
The kinetic separation from the thioredoxin pathway ensures that the sensing is specific for the glutathione pool [90].
Interestingly, the redox potentials determined in this way [85,87] are dramatically lower than those determined from classical chemical approaches. Likely, the challenges inherent in quenching thiol/disulfide exchange reactions, and the possible contamination of samples with the contents of cellular compartments with widely differing redox potentials, contribute to this discrepancy [91].
6. Challenges and pitfalls
6.1 Quench by acidification
A key issue in the study of thiol/disulfide equilibria remains the adequacy of the methods used for quenching. Because the thiolate represents the reactive species in these exchange reactions, it is common practice to quench redox reaction by lowering the pH. However, some cysteine residues have very low pKa values (e.g. 3.2 for DsbA [3,92]) and their parent proteins may themselves be fairly stable at low pH [93]. Thus, acidification may perturb thiol/disulfide equilibria between thiols of widely different pKa values (as shown for DsbA [94]) before the reaction is effectively quenched by denaturation of the protein. It may thus be necessary to combine quenching with the inclusion of chaotrophes (e.g. the guanidinium ion at low pH, or 10-15% trichloroacetic acid). Flash freezing before quenching with acid may prove advantageous.
6.2 Quench by alkylation
In place of acidification, NEM is often used for in vivo quenching in situations where a certain degree of protein integrity is required (e.g. during co-immunoprecipitation). However, to our knowledge, the effectiveness of NEM penetration into cells and organelles has never been quantitatively evaluated. Indeed the high intracellular concentrations of reduced glutathione must surely delay the effectiveness of NEM as a trapping agent in vivo and this may even bias the equilibria that NEM is intended to quench.
6.3 Oxygen and metal ions
A further challenge to the estimation of thiol/disulfide equilibria is the artefactual generation of disulfide by oxidation by ambient molecular oxygen. This reaction is stimulated by traces of metals (Cu2+ > Fe3+ > Ni2+ ≫ Co2+ [95]) and is particularly prevalent with copper and iron [96] which are common contaminants of reagents and surfaces. Chelators, such as EDTA, attenuate, but do not entirely suppress, thiol oxidation. While lowering the pH significantly slows unwanted thiol oxidation, such conditions are not always compatible with alkylation reactions. It is therefore prudent to purge solutions with nitrogen or argon and, wherever possible, to manipulate solutions under a blanket of argon or within an anaerobic glove box.
7. Concluding remarks and perspectives
The low kinetic barriers associated with many thiol/disulfide exchange reactions pose potential pitfalls for scientists who wish to determine the concentrations of these critical partners in redox homeostasis. In addressing these challenges, it is important to appreciate the chemical fundamentals of these deceptively simple exchange reactions. In particular, it is critical to ensure that the methods selected to quantify the redox status of thiols and disulfides do not themselves perturb the very system under investigation. Thiol/disulfide interconversions, which are in rapid equilibrium, present particular difficulties in this regard; not least when relevant enzymes are around. Here, the outcome may be biased by the kinetic reactivity profiles of the trapping agent. It is thus advisable to verify that quenching procedures do not alter the result of the analysis by spiking samples with relevant thiols or disulfides or in other ways introducing stringent controls.
The methods summarized in this review have focused heavily on the determination of thiol/disulfide concentrations without regard to how these levels are maintained (e.g. whether these concentrations correspond to a system in rapid equilibrium, or to one reflecting the kinetics of the steady state). While merely determining cellular thiol/disulfide levels will continue to pose significant technical difficulties, these measurements only address one aspect of redox homeostasis. Thus, almost nothing is known about the flux in thiol/disulfide redox pathways. For example, how fast is the GSH pool turned over intracellularly by redox cycling? What percentage of the maximal capacity of glutathione reductase is employed in the typical exponentially growing yeast cell? Or what flux disulfides accompanies oxidative protein folding in the endoplasmic reticulum of a typical cell? The exploration of the fluxes accompanying thiol/disulfide homeostasis is an emerging frontier that will require new experimental approaches and a new toolbox of reagents and probes.
Highlights.
Efficient quenching is essential for accurate determination of thiols and disulfides
It is necessary to carefully consider rates of alkylation and reduction in the context of pH
Some alkylation reactions are subject to reversibility
Many alkylation reagents have side-reactions which are incompatible with down-stream procedures.
Acknowledgments
Work from the authors' laboratories was supported in part by the Danish Council for Independent Research (JRW) and by NIH GM26643 (CT).
Abbreviations
- ABD-F
4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- CDAP
1-cyano-4-dimethylamino-pyridinium
- 4-DPS
4,4′-dithiodipyridine
- DTNB
5,5′-dithiobis-(2-nitrobenzoic) acid
- EDTA
ethylenediamine tetraacetic acid
- ER
endoplasmic reticulum
- GSH
glutathione
- GSSG
glutathione disulfide
- HMD
heavy maleimide derivative
- MBBr
Monobromobimane
- ME
2-mercato ethanol
- MMTS
S-methyl methanethiosulfonate
- PAGE
polyacrylaminde gel electrophoresis
- PEG
polyethyleneglycol
- SDS
sodium dodecylsulfate
- TCEP
tris(2-carboxyethyl) phosphine
- SBD-F
7-fluorobenzo-2-oxa-1,3-diazole-4-sulfonate
- THP
tris(2-hydroxyethyl) phosphine
- TNB
5-thio-2-nitrobenzoic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bednar RA. Reactivity and pH-dependence of thiol conjugation to N-ethylmaleimide -Detection of a conformational change in chalcone isomerase. Biochemistry. 1990;29:3684–3690. doi: 10.1021/bi00467a014. [DOI] [PubMed] [Google Scholar]
- 2.Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson JW, Creighton TE. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry. 1994;33:5974–5983. doi: 10.1021/bi00185a039. [DOI] [PubMed] [Google Scholar]
- 4.Pinitglang S, Watts AB, Patel M, Reid JD, Noble MA, Gul S, Bokth A, Naeem A, Patel H, Thomas EW, Sreedharan SK, Verma C, Brocklehurst K. A classical enzyme active center motif lacks catalytic competence until modulated electrostatically. Biochemistry. 1997;36:9968–9982. doi: 10.1021/bi9705974. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 6.Hansen RE, Ostergaard H, Winther JR. Increasing the reactivity of an artificial dithiol-disulfide pair through modification of the electrostatic milieu. Biochemistry. 2005;44:5899–5906. doi: 10.1021/bi0500372. [DOI] [PubMed] [Google Scholar]
- 7.Roos G, Foloppe N, Messens J. Understanding the pKa of redox cysteines: The key role of hydrogen bonding. Antioxid Redox Signal. 2013;18:94–127. doi: 10.1089/ars.2012.4521. [DOI] [PubMed] [Google Scholar]
- 8.Bulaj G, Kortemme T, Goldenberg DP. Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry. 1998;37:8965–8972. doi: 10.1021/bi973101r. [DOI] [PubMed] [Google Scholar]
- 9.March J. Advanced Organic Chemistry. John Wiley; New York: 1985. [Google Scholar]
- 10.Shaked Z, Szajewski RP, Whitesides GM. Rates of thiol-disulfide interchange reactions involving proteins and kinetic measurements of thiol pKa values. Biochemistry. 1980;19:4156–4166. doi: 10.1021/bi00559a004. [DOI] [PubMed] [Google Scholar]
- 11.Iversen R, Andersen PA, Jensen KS, Winther JR, Sigurskjold BW. Thiol-disulfide exchange between glutaredoxin and glutathione. Biochemistry. 2010;49:810–820. doi: 10.1021/bi9015956. [DOI] [PubMed] [Google Scholar]
- 12.Houk J, Whitesides GM. Structure reactivity relations for thiol disulfide interchange. J Am Chem Soc. 1987;109:6825–6836. [Google Scholar]
- 13.Rosenfield RE, Parthasarathy R, Dunitz JD. Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J Am Chem Soc. 1977;99:4860–4862. [Google Scholar]
- 14.Bach RD, Dmitrenko O, Thorpe C. Mechanism of thiolate-disulfide interchange reactions in biochemistry. J Org Chem. 2008;73:12–21. doi: 10.1021/jo702051f. [DOI] [PubMed] [Google Scholar]
- 15.Dickens F. Interaction of halogenacetates and SH compounds. The reaction of halogenacetic acids with glutathione and cysteine. The mechanism of iodoacetate poisoning of glyoxalase. Biochem J. 1933;27:1141–1151. doi: 10.1042/bj0271141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen ML, Vermeulen M, Bonaldi T, Cox J, Moroder L, Mann M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat Methods. 2008;5:459–460. doi: 10.1038/nmeth0608-459. [DOI] [PubMed] [Google Scholar]
- 17.Riordan JF, Vallee BL. Reactions with N-ethylmaleimide and p-mercuribenzoate. Methods Enzymol. 1972;25:449–456. doi: 10.1016/S0076-6879(72)25040-6. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert HF. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 19.MacQuarrie R, Bernhard SA. Mechanism of alkylation of rabbit muscle glyceraldehyde 3-phosphate dehydrogenase. Biochemistry. 1971;10:2456–2466. doi: 10.1021/bi00789a005. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin AD, Kiick KL. Tunable degradation of maleimide-thiol adducts in reducing environments. Bioconjugate Chem. 2011;22:1946–1953. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin D, Saleh S, Liebler DC. Reversibility of covalent electrophile - Protein adducts and chemical toxicity. Chem Res Toxicol. 2008;21:2361–2369. doi: 10.1021/tx800248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borges CR, Watson JT. Recognition of cysteine-containing peptides through prompt fragmentation of the 4-dimethylaminophenylazophenyl-4′-maleimide derivative during analysis by MALDI-MS. Protein Sci. 2003;12:1567–1572. doi: 10.1110/ps.0301403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gehring H, Christen P. A diagonal procedure for isolating sulfhydryl peptides alkylated with N-ethylmaleimide. Anal Biochem. 1980;107:358–361. doi: 10.1016/0003-2697(80)90396-6. [DOI] [PubMed] [Google Scholar]
- 24.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 25.Gorin G, Matic PA, Doughty G. Kinetics of reaction of N-ethylmaleimide with cysteine and some congeners. Arch Biochem Biophys. 1966;115:593–597. doi: 10.1016/0003-9861(66)90079-8. [DOI] [PubMed] [Google Scholar]
- 26.Lindorff-Larsen K, Winther JR. Thiol alkylation below neutral pH. Anal Biochem. 2000;286:308–310. doi: 10.1006/abio.2000.4807. [DOI] [PubMed] [Google Scholar]
- 27.Wakselman M, Guibejampel E, Raoult A, Busse WD. 1-cyano-4-dimethylamino-pyridinium salts - New water-soluble reagents for cyanylation of protein sulfhydryl groups. J Chem Soc Chem Com. 1976:21–22. [Google Scholar]
- 28.Wu J, Watson JT. A novel methodology for assignment of disulfide bond pairings in proteins. Protein Sci. 1997;6:391–398. doi: 10.1002/pro.5560060215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Chou YT, Husain R, Watson JT. Integration of hydrogen/deuterium exchange and cyanylation-based methodology for conformational studies of cystinyl proteins. Anal Biochem. 2004;331:130–137. doi: 10.1016/j.ab.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 30.Pipes GD, Kosky AA, Abel J, Zhang Y, Treuheit MJ, Kleemann GR. Optimization and applications of CDAP labeling for the assignment of cysteines. Pharmaceut Res. 2005;22:1059–1068. doi: 10.1007/s11095-005-5643-3. [DOI] [PubMed] [Google Scholar]
- 31.Barbirz S, Happersberger HP, Przybylski M, Glocker MO. Selective cyanylation of cysteinyl residues as an approach for the mass spectrometric determination of protein structures. Eur Mass Spec. 1999;5:123–131. [Google Scholar]
- 32.Wu J, Yang Y, Watson JT. Trapping of intermediates during the refolding of recombinant human epidermal growth factor (hEGF) by cyanylation, and subsequent structural elucidation by mass spectrometry. Protein Sci. 1998;7:1017–1028. doi: 10.1002/pro.5560070419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat Chem Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. A comparison between the sulfhydryl reductants tris(2- carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- 36.Cline DJ, Redding SE, Brohawn SG, Psathas JN, Schneider JP, Thorpe C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: Reactivity and membrane permeability. Biochemistry. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- 37.Daithankar VN, Wang WZ, Trujillo JR, Thorpe C. Flavin-linked Erv-family sulfhydryl oxidases release superoxide anion during catalytic turnover. Biochemistry. 2012;51:265–272. doi: 10.1021/bi201672h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jocelyn PC. Standard redox potential of cysteine-cystine from thiol-disulphide exchange reaction with glutathione and lipoic acid. Eur J Biochem. 1967;2:327–331. doi: 10.1111/j.1432-1033.1967.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 39.Chau MH, Nelson JW. Direct measurement of the equilibrium between glutathione and dithiothreitol by high-performance liquid-chromatography. Febs Lett. 1991;291:296–298. doi: 10.1016/0014-5793(91)81305-r. [DOI] [PubMed] [Google Scholar]
- 40.Singh R, Lamoureux GV, Lees WJ, Whitesides GM. Reagents for rapid reduction of disulfide bonds. Methods Enzymol. 1995;251:167–173. doi: 10.1016/0076-6879(95)51119-9. [DOI] [PubMed] [Google Scholar]
- 41.Lees WJ, Whitesides GM. Equilibrium-constants for thiol disulfide interchange reactions - A coherent, corrected set. J Org Chem. 1993;58:642–647. [Google Scholar]
- 42.Lukesh JC, Palte MJ, Raines RT. A potent, versatile disulfide-reducing agent from aspartic Acid. J Am Chem Soc. 2012;134:4057–4059. doi: 10.1021/ja211931f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown WD. Reduction of protein disulfide bonds by sodium borohydride. Biochim Biophys Acta. 1960;44:365–367. [Google Scholar]
- 44.Hansen RE, Ostergaard H, Norgaard P, Winther JR. Quantification of protein thiols and dithiols in the picomolar range using sodium borohydride and 4,4′-dithiodipyridine. Anal Biochem. 2007;363:77–82. doi: 10.1016/j.ab.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Erlandsson M, Hallbrink M. Metallic zinc reduction of disulfide bonds between cysteine residues in peptides and proteins. Int J Pept Res Ther. 2005;11:261–265. [Google Scholar]
- 46.Cecil R, Weitzman PD. The electroreduction of the disulphide bonds of insulin and other proteins. Biochem J. 1964;93:1–10. doi: 10.1042/bj0930001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leach SJ, Meschers A, Swanepoel OA. Electrolytic reduction of proteins. Biochemistry. 1965;4:23–27. doi: 10.1021/bi00877a005. [DOI] [PubMed] [Google Scholar]
- 48.Torchinskii IM, Metzler DE. Sulfur in Proteins. Pergamon. 1981 [Google Scholar]
- 49.Kruusma J, Benham AM, Williams JAG, Kataky R. An introduction to thiol redox proteins in the endoplasmic reticulum and a review of current electrochemical methods of detection of thiols. Analyst. 2006;131:459–473. doi: 10.1039/b515874e. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Cui WD, Zhang H, Dewald HD, Chen H. Electrochemistry-assisted top-down characterization of disulfide-containing proteins. Anal Chem. 2012;84:3838–3842. doi: 10.1021/ac300106y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harfield JC, Batchelor-McAuley C, Compton RG. Electrochemical determination of glutathione: a review. Analyst. 2012;137:2285–2296. doi: 10.1039/c2an35090d. [DOI] [PubMed] [Google Scholar]
- 52.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 53.Riddles PW, Blakeley RL, Zerner B. Reassessment of Ellman's reagent. Methods Enzymol. 1983;91:49–60. doi: 10.1016/s0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 54.Zhu JG, Dhimitruka I, Pei D. 5-(2-aminoethyl)dithio-2-nitrobenzoate as a more base-stable alternative to Ellman's reagent. Org Lett. 2004;6:3809–3812. doi: 10.1021/ol048404+. [DOI] [PubMed] [Google Scholar]
- 55.Humphrey RE, Ward MH, Hinze W. Spectrophotometric determination of sulfite with 4,4′-dithiodipyridine and 5,5′-dithiobis-(2-nitrobenzoic acid) Anal Chem. 1970;42:698–702. [Google Scholar]
- 56.Tyagarajan K, Pretzer E, Wiktorowicz JE. Thiol-reactive dyes for fluorescence labeling of proteomic samples. Electrophoresis. 2003;24:2348–2358. doi: 10.1002/elps.200305478. [DOI] [PubMed] [Google Scholar]
- 57.Chen X, Zhou Y, Peng XJ, Yoon J. Fluorescent and colorimetric probes for detection of thiols. Chem Soc Rev. 2010;39:2120–2135. doi: 10.1039/b925092a. [DOI] [PubMed] [Google Scholar]
- 58.Kosower NS, Kosower EM, Newton GL, Ranney HM. Bimane fluorescent labels: Labeling of normal human red cells under physiological conditions. Proc Nat Acad Sci U S A. 1979;76:3382–3386. doi: 10.1073/pnas.76.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Imai K, Toyooka T, Watanabe Y. A novel fluorogenic reagent for thiols - ammonium 7-fluorobenzo-2-oxa-1,3-diazole-4-sulfonate. Anal Biochem. 1983;128:471–473. doi: 10.1016/0003-2697(83)90404-9. [DOI] [PubMed] [Google Scholar]
- 60.Toyo'oka T, Imai K. Isolation and characterization of cysteine-containing regions of proteins using 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole and high-performance liquid chromatography. Anal Chem. 1985;57:1931–1937. doi: 10.1021/ac00286a032. [DOI] [PubMed] [Google Scholar]
- 61.Chin CC, Wold F. The use of tributylphosphine and 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole in the study of protein sulfhydryls and disulfides. Anal Biochem. 1993;214:128–134. doi: 10.1006/abio.1993.1466. [DOI] [PubMed] [Google Scholar]
- 62.Husted LB, Sorensen ES, Sottrup-Jensen L. 4-(Aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole is not specific for labeling of sulfhydryl groups in proteins as it may also react with phenolic hydroxyl groups and amino groups. Anal Biochem. 2003;314:166–168. doi: 10.1016/s0003-2697(02)00650-4. [DOI] [PubMed] [Google Scholar]
- 63.Treuheit MJ, Kirley TL. Reversibility of cysteine labeling by 4-(aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole. Anal Biochem. 1993;212:138–142. doi: 10.1006/abio.1993.1303. [DOI] [PubMed] [Google Scholar]
- 64.Abukhalaf IK, Silvestrov NA, Menter JM, von Deutsch DA, Bayorh MA, Socci RR, Ganafa AA. High performance liquid chromatographic assay for the quantitation of total glutathione in plasma. J Pharm Biomed Anal. 2002;28:637–643. doi: 10.1016/s0731-7085(01)00658-6. [DOI] [PubMed] [Google Scholar]
- 65.Li YH, Yang Y, Guan XM. Benzofurazan sulfides for thiol imaging and quantification in live cells through fluorescence microscopy. Anal Chem. 2012;84:6877–6883. doi: 10.1021/ac301306s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: The two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- 67.Joly JC, Swartz J. In vitro and in vivo redox states of the Escherichia coli periplasmic oxidoreductases DsbA and DsbC. Biochemistry. 1997;36:10067–10072. doi: 10.1021/bi9707739. [DOI] [PubMed] [Google Scholar]
- 68.Goodson RJ, Katre NV. Site-directed pegylation of recombinant interleukin-2 at its glycosylation site. Bio-Technol. 1990;8:343–346. doi: 10.1038/nbt0490-343. [DOI] [PubMed] [Google Scholar]
- 69.Odom OW, Kudlicki W, Kramer G, Hardesty B. An effect of polyethylene glycol 8000 on protein mobility in sodium dodecyl sulfate-polyacrylamide gel electrophoresis and a method for eliminating this effect. Anal Biochem. 1997;245:249–252. doi: 10.1006/abio.1996.9993. [DOI] [PubMed] [Google Scholar]
- 70.Karala AR, Ruddock LW. Does S-methyl methanethiosulfonate trap the thiol-disulfide state of proteins? Antioxid Redox Signal. 2007;9:527–531. doi: 10.1089/ars.2006.1473. [DOI] [PubMed] [Google Scholar]
- 71.Nielsen JW, Jensen KS, Hansen RE, Gotfredsen CH, Winther JR. A fluorescent probe which allows highly specific thiol labeling at low pH. Anal Biochem. 2012;421:115–120. doi: 10.1016/j.ab.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 72.Smyth DG, Konigsberg W, Blumenfeld OO. Reactions of N-ethylmaleimide with peptides and amino acids. Biochem J. 1964;91:589–595. doi: 10.1042/bj0910589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharpless NE, Flavin M. The reactions of amines and amino acids with maleimides. Structure of the reaction products deduced from infrared and nuclear magnetic resonance spectroscopy. Biochemistry. 1966;5:2963–2971. doi: 10.1021/bi00873a028. [DOI] [PubMed] [Google Scholar]
- 74.Brewer CF, Riehm JP. Evidence for possible nonspecific reactions between N-ethylmaleimide and proteins. Anal Biochem. 1967;18:248–255. [Google Scholar]
- 75.Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006;7:271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monostori P, Wittmann G, Karg E, Turi S. Determination of glutathione and glutathione disulfide in biological samples: An in-depth review. J Chromatogr B. 2009;877:3331–3346. doi: 10.1016/j.jchromb.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 78.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione - Applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 79.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione-reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 80.Shaik IH, Mehvar R. Rapid determination of reduced and oxidized glutathione levels using a new thiol-masking reagent and the enzymatic recycling method: application to the rat liver and bile samples. Anal Bioana Chem. 2006;385:105–113. doi: 10.1007/s00216-006-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Melnyk S, Pogribna M, Pogribny I, Hine RJ, James SJ. A new HPLC method for the simultaneous determination of oxidized and reduced plasma aminothiols using coulometric electrochemical detection. J Nutr Biochem. 1999;10:490–497. doi: 10.1016/s0955-2863(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 82.Diopan V, Shestivska V, Zitka O, Galiova M, Adam V, Kaiser J, Horna A, Novotny K, Liska M, Havel L, Zehnalek J, Kizek R. Determination of plant thiols by liquid chromatography coupled with coulometric and amperometric detection in lettuce treated by lead(II) Ions. Electroanal. 2010;22:1248–1259. [Google Scholar]
- 83.Sun YP, Smith DL, Shoup RE. Simultaneous detection of thiol-containing and disulfide-containing peptides by electrochemical high-performance liquid-chromatography with identification by mass-spectrometry. Anal Biochem. 1991;197:69–76. doi: 10.1016/0003-2697(91)90357-y. [DOI] [PubMed] [Google Scholar]
- 84.Ostergaard H, Henriksen A, Hansen FG, Winther JR. Shedding light on disulfide bond formation: engineering a redox switch in green fluorescent protein. EMBO J. 2001;20:5853–5862. doi: 10.1093/emboj/20.21.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 86.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid Redox Signal. 2010;13:621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 87.Ostergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bjornberg O, Ostergaard H, Winther JR. Mechanistic insight provided by glutaredoxin within a fusion to redox-sensitive yellow fluorescent protein. Biochemistry. 2006;45:2362–2371. doi: 10.1021/bi0522495. [DOI] [PubMed] [Google Scholar]
- 89.Gutsche M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, Dick TP. Proximity-based Protein Thiol Oxidation by H2O2-scavenging Peroxidases. J Biol Chem. 2009;284:31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bjornberg O, Ostergaard H, Winther JR. Measuring Intracellular Redox Conditions Using GFP-Based Sensors. Antioxid Redox Signal. 2006;8:354–361. doi: 10.1089/ars.2006.8.354. [DOI] [PubMed] [Google Scholar]
- 91.Morgan B, Ezerina D, Amoako TN, Riemer J, Seedorf M, Dick TP. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat Chem Biol. 2012;9:119–125. doi: 10.1038/nchembio.1142. [DOI] [PubMed] [Google Scholar]
- 92.Grauschopf U, Winther JR, Korber P, Zander T, Dallinger P, Bardwell JC. Why is DsbA such an oxidizing disulfide catalyst? Cell. 1995;83:947–955. doi: 10.1016/0092-8674(95)90210-4. [DOI] [PubMed] [Google Scholar]
- 93.Hatahet F, Ruddock LW. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 94.Wunderlich M, Glockshuber R. Redox properties of protein disulfide isomerase (DsbA) from Escherichia coli. Protein Sci. 1993;2:717–726. doi: 10.1002/pro.5560020503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bagiyan GA, Koroleva IK, Soroka NV, Ufimtsev AV. Oxidation of thiol compounds by molecular oxygen in aqueous solutions. Russ Chem Bull. 2003;52:1135–1141. [Google Scholar]
- 96.Munday R, Munday CM, Winterbourn CC. Inhibition of copper-catalyzed cysteine oxidation by nanomolar concentrations of iron salts. Free Radical Bio Med. 2004;36:757–764. doi: 10.1016/j.freeradbiomed.2003.12.015. [DOI] [PubMed] [Google Scholar]