

Abstract
Objective
The aim of this study was to determine if a latent variable approach might be useful in identifying shared variance across genetic risk alleles that is associated with antisocial behaviour at age 15 years.
Methods
Using a conventional latent variable approach, we derived an antisocial phenotype in 328 adolescents utilizing data from a 15-year follow-up of a randomized trial of a prenatal and infancy nurse-home visitation program in Elmira, New York. We then investigated, via a novel latent variable approach, 450 informative genetic polymorphisms in 71 genes previously associated with antisocial behaviour, drug use, affiliative behaviours, and stress response in 241 consenting individuals for whom DNA was available. Haplotype and Pathway analyses were also performed.
Results
Eight single-nucleotide polymorphisms (SNPs) from 8 genes contributed to the latent genetic variable that in turn accounted for 16.0% of the variance within the latent antisocial phenotype. The number of risk alleles was linearly related to the latent antisocial variable scores. Haplotypes that included the putative risk alleles for all 8 genes were also associated with higher latent antisocial variable scores. In addition, 33 SNPs from 63 of the remaining genes were also significant when added to the final model. Many of these genes interact on a molecular level, forming molecular networks. The results support a role for genes related to dopamine, norepinephrine, serotonin, glutamate, opioid, and cholinergic signaling as well as stress response pathways in mediating susceptibility to antisocial behaviour.
Conclusions
This preliminary study supports use of relevant behavioural indicators and latent variable approaches to study the potential “co-action” of gene variants associated with antisocial behaviour. It also underscores the cumulative relevance of common genetic variants for understanding the etiology of complex behaviour. If replicated in future studies, this approach may allow the identification of a ‘shared’ variance across genetic risk alleles associated with complex neuropsychiatric dimensional phenotypes using relatively small numbers of well-characterized research participants.
Keywords: Antisocial behaviour, latent variable analysis, shared variance, co-action of gene variants
Genetic factors are integral to the understanding of the etiology of antisocial behaviour, as evidenced by family and twin studies that indicate a heritability of at least 50% (Moffitt, 2005; Viding, Larsson, & Jones, 2008). Quantitative genetic studies indicate that genetic and environmental influences on the emergence of antisocial behaviour appear remarkably similar in their magnitude despite different approaches, assessment methods, age of assessment, or the gender of the participants (Rhee & Waldman, 2002; Rhee & Waldman, 2011; Lahey & Waldman, 2012). In addition, many risk factors that are traditionally thought to be environmental may also reflect genetic vulnerability (Moffitt, 2005).
Over the past decade, it was widely expected that the genetic basis of common disorders would be resolved by genome-wide association studies (GWAS)--large-scale studies in which the entire genome is covered by genetic markers. Although recent GWAS have given us a clearer picture of the allelic architecture of genetic susceptibility for some pediatric-onset disorders including type I diabetes, the bulk of heritable variance remains unexplained for many pediatric-onset neuropsychiatric disorders including antisocial behaviour and substance use disorders.
Information about gene function has led to the identification of a number of relevant candidate systems of genes (CSG) that influence antisocial (delinquent, criminal, and substance use) behaviours. The CSG approach provides the advantages of hypothesis-driven research, while mitigating some limitations of a candidate gene approach that usually targets only one or a small number of loci. Although a CSG approach is a viable alternative to both a candidate gene and GWAS studies, it still requires a very large number of subjects to identify allelic variations that by themselves account for a very small proportion of the phenotypic variance (Maher et al., 2011; Grigorenko et al., 2010; Yrigollen et al., 2008; Greenwood et al., 2011).
While CSG studies examine related families of genes, the analytical approach often focuses on only one allelic variant at a time. Several studies have demonstrated that multiple measurements of genes are needed to fully realize the association between the variance in genes to phenotypic variation (Burt & Mikolajewski, 2008; Grigorenko et al, 2010). Thus, whereas positive CSG studies allow inferences to be made concerning the probable presence of gene-gene interactions, they do not permit an accurate estimation of the phenotypic variance explained since the variance accounted for by gene-gene interactions is unlikely to be simply additive. Indeed, very few approaches, if any, are currently available to assess systematically the presence of a ‘shared’ variance across genetic risk alleles with regard to a particular phenotype of interest.
The application of a latent variable approach to genetic data should facilitate the identification of allelic variants that collectively contribute to particular phenotypic outcome. This approach has been widely used in economics and psychology to study observable behavior that may be influenced by unobservable constructs that are not directly measurable (Avery, 1979; Bollen, 2002). A clear advantage of using a latent variable analysis is that it reduces the dimensionality of data so that a large number of observable variables can be aggregated in a model to represent a single underlying factor when there are strong relationships among variables.
The application of latent variable analyses to genetic data is novel. Because observed variables that have no correlation cannot result in a latent construct, the identification of a latent genetic construct provides a potential path to discover a ‘shared’ variance across genetic risk alleles. This shared variance could be additive, multiplicative or interactive. However, this latent variable approach is limited by its inability to specify the biological nature of the gene-gene interactions that underlie this shared variance. Consequently, we have chosen to refer to the “co-action” of allelic variants (additive, multiplicative, and other) in describing our findings.
The choice of the genetic loci to be assessed is critically important. Several studies have reported associations between antisocial behaviour and drug use and genetic variants associated with dopaminergic and serotoninergic pathways (Chambers et al., 2001; Bierut et al., 2002; Solanto, 2002; Kim-Cohen et al., 2006; Moffitt, 2005; Blum et al., 2012; McCrory et al., 2012; Moul et al., 2013). We also selected genes associated with affiliative behaviors and stress response. The rationale for including in the CSG genetic loci implicated in affiliative behaviours is based, in part, on studies that report strong associations between inconsistent, harsh, or abusive disciplinary practices by parents and child rule-breaking behaviours, delinquency, and aggression (Stouthamer-Loeber et al., 2001; Stanger et al., 2004). Childhood conduct problems have also been associated with a lack of parental involvement, a lack of parental warmth and parent–child conflict (Burt et al., 2005; Caspi et al., 2004; Stanger et al., 2004). Dopaminergic pathways are also critically involved in affiliative (e.g., maternal care) and other reward-related behaviours (Depue & Collins, 1999; Mileva-Seitz et al., 2012). The rationale for including in the CSG genetic loci implicated in stress response is based on studies that have reported an association between cortisol levels and aggression in adolescents (Gao et al., 2009; Poustka et al., 2010; Matthys et al., 2013). In addition, the neural substrates of affiliation (parenting behaviours) are closely interrelated to the stress response, salience, and reward pathways (Leckman & Herman, 2002; Lim & Young, 2006; Gordon et al., 2011).
The goals of this preliminary study were: (1) to construct a latent phenotypic antisocial variable using all, or a subset, of 15 indicators of delinquency, antisocial behaviour and drug use collected as part of the 15 year follow-up of a randomized trial of a prenatal and infancy nurse-home visitation program in Elmira, New York (Olds et al., 1998); (2) to define a novel CSG that includes genetic loci that have been implicated in affiliative behaviours and stress response as delinquency and drug use; (3) to carry out initial univariate analyses of each of the informative single nucleotide polymorphisms (SNPs) within the CSG; (4) to perform a quasi-bootstrapping procedure to construct an initial latent genetic variable; (5) to refine and extend the latent genetic variable by reassessing each of the remaining informative SNPs one at a time leading to a final model where both latent genetic and phenotypic variables are incorporated simultaneously; (6) to determine if there is a continuous relationship between the number of risk alleles and scores from the latent antisocial variable; and (7) to identify biological pathways and processes that are over-represented by all of the genes in the CSG that were implicated in this analysis. A flow chart of these steps and procedures is presented in Figure 1.
Figure 1. Flow Chart.

This diagram presents the step-by-step sequence of the analyses undertaken as part of this study. The Elmira study refers to the phenotypic characterization of the participants in the initial follow-up study of the Nurse Family Partnership conducted in Elmira, NY (Olds et al., 1998). See the text for a more detailed presentation of the measures and the analyses performed. SNP = single nucleotide polymorphism.
METHODS
Participants
Participants were a subset of the first-born offspring of 400 pregnant women who participated in a randomized, controlled study, of the Nurse Family Partnership Program (NFP) based in Elmira, New York (Olds et al., 1998). Although the phenotypic information was collected at the 15-year follow-up, the DNA was obtained in association with the 27-year follow-up. Useable DNA was available from 241 unrelated, primarily Caucasian participants (consenters) who consented to have their DNA genotyped and analyzed. The demographic and clinical characteristics of consenters (n=241) and non-consenters (n=87) were compared. Although the mothers of the consenters, on average, were one year younger than the non-consenters (19.08 vs. 20.29 years, respectively), no other clinically meaningful differences were found between these groups (Table 1).
Table 1.
Demographic Characteristics and Observed Antisocial Indicators for the Consenters vs. non-Consenters
| Maternal background characteristics | N | Consenters (n=241) | N | Non- Consenters (n=87) | P-value |
|---|---|---|---|---|---|
| Maternal age, mean (SD), | 241 | 19.08 (2.93) | 87 | 20.29 (3.69) | 0.002 |
| Maternal education, years completed, mean (SD) | 241 | 11.22 (1.49) | 87 | 11.43 (1,73) | 0.293 |
| Unmarried, % | 241 | 61.83% | 87 | 60.92% | 0.882 |
| Low SES household, % | 241 | 61.41% | 87 | 54.02% | 0.229 |
| White, % | 214 | 88.80% | 76 | 87.36% | 0.719 |
| Adolescent characteristics, % | |||||
| Male, % | 241 | 48.13% | 87 | 56.32% | 0.190 |
| Adolescent antisocial indicators (birth to 15 yrs), mean(SD) | |||||
| No. of times run away | 231 | 0.39 (1.37) | 87 | 0.44 (1.45) | 0.769 |
| No. of times stopped by police | 231 | 1.62 (5.12) | 87 | 1.10 (3.41) | 0.382 |
| No. of times arrested | 234 | 0.15 (0.47) | 87 | 0.06 (0.23) | 0.071 |
| No. of convictions (including probation violations) | 231 | 0.31 (1.81) | 87 | 0.11 (0.52) | 0.319 |
| No. of times sent to youth corrections | 231 | 0.11 (0.49) | 87 | 0.92 (0.50) | 0.794 |
| No. of times booked or charged by police | 231 | 0.34 (0.85) | 87 | 0.16 (0.57) | 0.074 |
| No. of sexual partners | 227 | 1.96 (6.11) | 84 | 1.45 (4.07) | 0.481 |
| Adolescent antisocial indicators (Grades 7–9), mean(SD) | |||||
| No. of short-term school suspensions | 213 | 0.33 (0.96) | 78 | 0.31 (1.08) | 0.874 |
| No. of long-term school suspensions | 213 | 0.02 (0.14) | 78 | 0.08 (0.68) | 0.236 |
| Adolescent antisocial indicators (6 mos prior to interview at 15 yrs), mean(SD) | |||||
| No. of minor delinquency acts | 231 | 3.18 (4.59) | 87 | 2.71 (4.13) | 0.403 |
| No. of major delinquency acts | 231 | 3.41 (5.66) | 87 | 3.09 (5.11) | 0.638 |
| No. of cigarettes/day | 231 | 1.68 (4.27) | 86 | 1.15 (3.02) | 0.293 |
| No. of days drank | 230 | 2.71 (7.81) | 87 | 1.72 (7.56) | 0.314 |
| Alcohol use | 234 | 0.24 (0.98) | 87 | 0.32 (1.34) | 0.568 |
| Days used drugs | 231 | 4.05 (15.8) | 86 | 3.28 (10.9) | 0.677 |
SES indicates socioeconomic status. See text and Olds et al. 1998 for more detailed descriptionsof indicators. P-values in bold are significant at p <.05 level.
Antisocial indicators
Assessments conducted at the15-year follow-up are specified in earlier reports (Olds et al., 1998). Briefly, interviews were conducted with each adolescent in the study group, their biological mothers and their custodial adults, if the biological mother had lost custody. School records were abstracted to assess suspensions. In this study, we initially used all 15 non-dichotomous outcome measures (Table 1). These variables included the lifetime number of times the child/adolescent had run away from home; been stopped by the police, booked and charged, convicted of a crime (including probation violations), and sent to youth correctional facilities; the number of lifetime sexual partners; and number of parent reported youth arrests. For grades 7 through 9, the numbers of short- and long-term school suspensions were included. For the 6 months prior to the interview, additional questions were asked that permitted the estimation of the number of minor and major acts of delinquency. Separate variables were constructed concerning the frequency of using cigarettes per day and the number of days the adolescents had consumed alcohol or used illegal drugs during the 6-month period prior to the interview. The adolescents were asked questions regarding the effect of alcohol on 5 domains of their lives (trouble with parents, trouble at school, problems with friends, problems with someone they were dating, and trouble with police). These data were summarized in an alcohol-use behavioural problem scale (range, 0–5).
Latent antisocial variable
The underlying propensity to display antisocial behaviour was modeled as a latent variable using 15 indicators of delinquency, criminality, and substance use (Table 1). Data from the entire cohort of adolescents (consenters and non-consenters) were used to develop the latent antisocial variable. Each outcome measure was a count variable and upon examination, the variances of each were found to be about twice that of their means, indicating the possibility of over-dispersion. On further screening of the data, we found that on average, more than 70% of study participants had a zero response for an outcome indicating excess zeros. Hence, we developed this latent antisocial construct using a zero-inflated Poisson (ZIP) regression model (Lambert, 1992), because it allows for extra variation (over-dispersion) and an inflated number of zeros. The analyses were performed using the NLMIXED procedure from SAS, version 9.2 (SAS Institute, Gary, NC). The NLMIXED procedure fits non-linear mixed models –models with both fixed and random effects. To fit the 15 outcome measures simultaneously into the NLMIXED procedure, the data were rearranged so that each participant had 15 rows of data with each row containing the response to each of the outcome variables. The 15 available indicators were entered into a one-factor latent variable model. To identify the model, the mean and variance of the antisocial latent variable were set to 0 and 1, respectively. Intercepts and factor loadings could then be estimated.
Candidate genes and genotyping
To define a relevant CSG that may influence antisocial behaviours, the selection of genetic loci was driven by two main factors, specifically the presence of literature supporting the association of a particular gene/genetic variant with antisocial behaviour, drug use, stress response, and/or affiliative behaviours, and the availability of genetic markers that had acceptable distributions of allele frequencies (Maher et al., 2011; Grigorenko et al., 2010; Leckman & Herman, 2002; Yrigollen et al., 2008). A total of 450 SNPs were studied at or near the 71 genes identified from the literature. The number of SNPs studied with each locus ranged from 2 to 31. On average, there was one SNP per 16.77 (SD=30.44) kb for each gene. A complete list of the 71 genes and the 450 informative SNPs included in the CSG is presented in Supplemental Table 1. Supplemental Table 1 includes: RefSNP accession identification numbers (rs numbers; http://www.ncbi.nlm.nih.gov/projects/SNP/), chromosomal locations, gene information, sequence information, and minor allele frequencies for the four HapMap populations as well as a reference to at least one scientific report documenting the potential association with antisocial behaviour. Neither the list of possible utilizable markers nor the list of genes investigated in this research is exhaustive. Yet, to our knowledge, this is one of only a few efforts to investigate multiple polymorphisms nested within multiple genes simultaneously.
For the study sample (n=241), genomic DNA was extracted from lymphocytes (n=218), saliva (n=8) and buccal cells (n=15). Genotyping was performed by the Yale Center for Genome Analysis using Illumina Golden Gate platform. Genotype data were cleaned and selected genotypes were verified. Verification of Hardy-Weinberg equilibrium of genotypes was performed by using the χ2 goodness of fit test and no significant deviations beyond chance levels were observed.
Latent genetic variable
To develop the latent genetic variable, we sought first to identify individual SNPs significantly associated with the latent antisocial variable. We conducted a univariate analysis for each of the 450 informative SNPs within the CSG, using a cumulative logistic regression with two thresholds for 3 possible responses. Autosomal chromosomes were categorized as having 0, 1, or 2 risk alleles for each SNP to reflect heterozygous and homozygous genotypes. This initial analysis identified 26 SNPs that were marginally significantly associated with the latent antisocial variable (data not shown). To ensure that a potentially informative SNP was not overlooked, SNPs with even marginal significance at a trend level with a p-value of ≤ 0.20 were retained.
In constructing our genetic latent variable, we also were interested in identifying potential multicollinearity among these 26 SNPs. To do this, we first identified combinations of 10 SNPs from the initial set of 26 marginally significant SNPs, using a quasi-bootstrapping procedure with replacement. Second, to determine which SNP combinations were collectively predictive of the latent antisocial variable, each combination of 10 was serially entered into a one factor cumulative logistic regression model. The analyses were performed using the NLMIXED procedure from SAS, version 9.2 (SAS Institute, Gary, NC).
An examination of these results led to the serial identification of unique combinations of three to four SNPs that when they were simultaneously present the latent genetic variable was predictive of the latent antisocial construct at a p-value of 0.05. An Initial Model with four SNPs from four genes was formulated that maximized the predictive power of the latent genetic variable. We performed a t-test and a linear regression to determine if there was a significant association between the number risk alleles and the latent antisocial variable scores.
Final model
To ensure that none of the remaining 444 SNPs from the original 450 SNPs in our candidate system of genes (CSG) did not increase the predictive power of our Initial Model, we developed the “Final Model” by adding each of the remaining 444 SNPs individually into the Initial Model that included four risk alleles. Only those SNPs that enhanced the model were retained and included in the Final Model.
Using the “Predict” statement in the NLMIXED procedure, the latent antisocial behaviour scores were derived for each participant using the Final Model. To determine if there was a continuous or a step-wise relationship between the number of risk alleles in the Final Model and the scores of the latent antisocial variable, we examined the distribution of the mean scores for the latent antisocial phenotype according to the number of risk alleles present in each of the participants.
Haplotype analysis
The vulnerability contribution arising from being a carrier of the risk alleles identified in the latent genetic models is not likely to be due to a direct relationship arising from the risk alleles per se unless these can be demonstrated to be mutations that alter the gene expression in some important way. More often it will be genetic factors closely linked to the risk alleles that are the actual causal factor. The extent to which the designated risk alleles provide useful predictive information is a function of how closely associated they are with the actual nearby risk factor(s). If both could be measured we might determine that the risk allele is only associated in a small proportion of the chromosomes carrying a potent nearby risk factor and much of the time it alone does not really add to the predictive value. Or, we might find that the designated risk allele has a high frequency of association with a weak nearby risk factor. Consequently, it is reasonable to expect that multi-SNP haplotypes encompassing the risk SNPs identified in the final model should have the potential for capturing more of the genetic variation underlying the vulnerability to antisocial behaviour. The extent to which multi-SNP haplotypes can capture more of the relevant genetic factor depends not so much on the physical distance as on the degree of linkage disequilibrium between the risk allele and relevant nearby factors. Consequently, haplotypes of 2- to 4-SNPs for each of the genes of interest were examined. To test the predictive value of the haplotypes we rank ordered the 241 individuals according to their antisocial score and created three groupings with roughly equal numbers of individuals in each (low, middle, and top scoring antisocial groups – see Figure 2). If there is no relationship between the haplotypes and the antisocial measure then we would expect to estimate for each gene separately the same frequencies for the same haplotypes (within measurement error) in each of the three groups. If the haplotypes do have predictive value for the antisocial measure then rather different haplotype frequencies should be estimated across the behavioural groupings—especially for the haplotypes carrying the already identified risk alleles.
Figure 2. Distribution of the Latent Antisocial Variable Scores.

This figure presents the frequency distribution of latent antisocial variable scores for the 241 individuals who participated in the Nurse Family Partnership randomized clinical trial and provided DNA (see Olds et al., 1998). Low, Middle and Top refer to the three groups (tertiles) based on their latent antisocial variable scores.
Biological network and pathway analysis
We used the PANTHER Classification System v8.0 (Mi & Thomas, 2009; Mi, Muruganujan, & Thomas, 2013) to investigate all genes implicated in the CSG for enrichment of membership in biological pathways and processes. This tool uses a binomial statistical test to compare classifications of multiple clusters of lists to a reference list to determine over- or under-representation of PANTHER classification categories (Cho & Campbell, 2000).
We employed the online tool GeneMANIA (http://www.genemania.org) to visualize connections among our genes and find other genes related to our set of input genes. The GeneMANIA database consists of genomics and proteomics data from a variety of sources, including data from gene and protein expression profiling studies and primary and curated molecular interaction networks and pathways. Our analysis weighted all network data sources equally (“Equal by network” advanced option) in order to visualize all networks (physical interactions, predicted interactions, pathways, genetic interactions) that connect our input gene list.
RESULTS
Latent antisocial variable
Fourteen of the 15 observed (directly measured) indicators of antisocial behaviour were found to significantly contribute at a p-value of 0.05 to a single latent antisocial variable. Estimates obtained from the zero-inflated Poisson regression model are presented in Supplemental Table 2. The one observed variable that did not significantly contribute to the latent antisocial variable was the number of long-term school suspensions in grades 7–9 and it was omitted from the subsequent analyses. The total number of times the child had run away, had been stopped by the police, and had been sent to a correctional facility from birth to the 15 year follow-up; and the number of days the adolescent had drunk alcohol and used drugs were the phenotypic variables that most robustly contributed to the latent antisocial construct (Supplemental Table 2). Figure 2 presents the distribution of the latent antisocial variable scores.
Latent genetic variable
Using the available DNA from the 241 consenters, we first identified 26 SNPs that were marginally related to the latent antisocial variable at a p-value of <0.20 (data not shown). After identifying combinations of 10 SNPs using the quasi-bootstrapping method, we identified a unique combination of four of these 26 SNPs (Initial Model) from four genes (Table 2). When these four SNPs were entered collectively into the Initial Model, the resulting latent genetic construct was significantly predictive of the latent antisocial construct (β=0.413; p=0.0059) and accounted for 16.0% of the variance of the latent antisocial variable. This Initial Model included: Glutamate receptor, metabotropic, 5 (GRM5, OMIM 604102, Gen-Bank accession NM_000842.1 chromosome 11q14.3, SNP: rs1874946); Dopamine receptor D2 (DRD2, OMIM 126450; Gen-Bank accession NM_016574.2, chromosome 11q23.2, SNP: rs4587762); Arginine vasopressin receptor 1A (AVPR1A, OMIM 600821, Gen-Bank accession NM_000706.3 chromosome12q14-q15, SNP: rs10877970); and Glutamate receptor, ionotropic, N-methy-D-aspartate, subunit 2A (GRIN2A, OMIM 138253, Gen-Bank accession NM_000833.2, chromosome 16p13.2, SNP: rs8047589. The total number of risk alleles was significantly associated with an individual’s latent antisocial variable score ([slope] y= 0.205 x – 0.571, r2 = 0.076, t = 4.44, d.f. = 239, 2-tailed p= 1.381 × 10−5).
Table 2.
Risk alleles contributing to the final latent genetic variable in the Final Model
| Chromosome Location | Gene | Risk SNP (Hets/Hom/#Typed) | Univariate Analysis (p-value) | Final Model (coefficient) | Final Model (p-value) | Haplotyping Results |
|---|---|---|---|---|---|---|
| 4q31.23 | NR3C2 | rs3843413 22/94/121 |
0.065 | −0.524 | 0.007 | L-M* |
| 11q14.3 | GRM5† | rs1874946 100/35/237 |
0.015 | 1.000 | <0.0001 | L-T** |
| 11q23.2 | NCAM1 | rs1545086 127/40/240 |
0.052 | 0.644 | 0.004 | L-M*/L-T** |
| 11q23.2 | DRD2† | rs4587762 107/42/237 |
0.022 | 0.764 | 0.004 | L-M***/M-T***/L-T*** |
| 12q14.2 | AVPR1A† | rs10877970 63/9/236 |
0.051 | 0.699 | 0.003 | M-T* |
| 15q13.3 | CHRNA7 | rs1514246 105/26/236 |
0.076 | 0.515 | 0.005 | L-M**/M-T**/L-T** |
| 15q21.1 | SQRDL | rs626808 75/7/240 |
0.135 | 0.537 | 0.006 | M-T* |
| 16p13.2 | GRIN2A† | rs8047589 99/103/238 |
0.060 | 0.592 | 0.004 | L-M**/M-T**/L-T* |
Loci included in the Initial Model of the Latent genetic variable (see text).
See text for the full names of the genes and Supplemental Table 1 for a complete presentation of the information concerning each risk single nucleotide polymorphism (SNP). The reference SNP ID number (rs#) is provided as is the number of heterozygotes (HETs), the number of homozygotes (HOMs), as well as the total number of individuals typed for the identified risk alleles. L = Low; M = Middle; and T = Top refer to the low, middle and top scores on the latent antisocial variable (see Figure 2). The coefficient for NR3C2 has a negative sign indicating that the more frequently occurring genotype C is associated with an increased risk of antisocial behavior when considered in combination with the other genes.
p <0.05;
p <0.01;
p <0.001
Final model
The serial addition of the remaining 444 informative SNPs found that the Initial model was stable. Four SNPs from four additional genes modestly enhanced the significance of the latent genetic variable yielding the Final model (Table 2, Figure 3). In the Final model, the predictive power of the resulting latent genetic construct that included the eight SNPS was stronger and accounted for slightly more of the total variation of the latent antisocial variable than the Initial model. The four additional genes included in the Final model were: Nuclear receptor subfamily 3, group C, member 2 (NR3C2, OMIM 600983, Gen-Bank accession NG_013350, chromosome 4q31.23, SNP: rs3843413); Neural cell adhesion molecule 1 (NCAM1, OMIM 116930; Gen-Bank accession NG_032036, chromosome 11q23.1, SNP: rs1545086); Cholinergic receptor, nicotinic, alpha 7 (CHRNA7, OMIM 118511; Gen-Bank accession NM_148911.1, chromosome 15q13.3, SNP: rs1514246); and Sulfide quinone reductase-like (yeast) (SQRDL, Gen-Bank accession NM_021199, chromosome 15q13.3, SNP: rs626808).
Figure 3. Final Model.
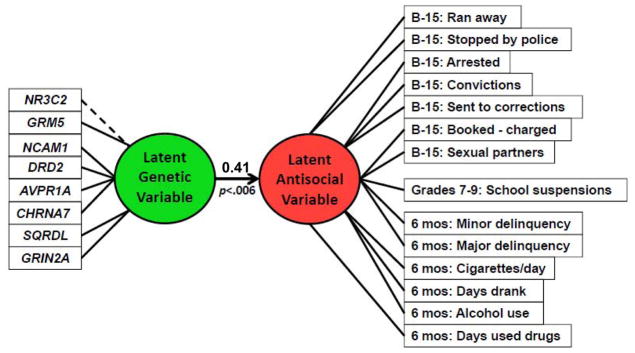
The final model incorporates a latent antisocial outcome variable and a latent genetic predictor variable. The latent genetic variable represents the underlying genetic level manifested by the 8 available genes. The latent antisocial variable represents the antisocial level manifested by 14 available delinquency indicators. The latent genetic variable is significantly predictive of the antisocial construct (β=0.413; p=0.006) and accounts for 16% of the variance in the latent antisocial variable score. The dashed line for NR3C2 indicates that the coefficient for this gene in the Final model has a negative sign (indicating that the more frequently occurring genotype C is associated with an increased risk of antisocial behaviour when considered in combination with the other genes.
We found a continuous linear relationship between the number of risk alleles and the latent antisocial variable (Figure 4; [slope] y= 0.141 x – 0.836, r2 = 0.091, t = 4.889, d.f. = 239, 2-tailed p= 1.861 × 10−6). We also ran the final model after excluding the 27 “non-white” participants (see Table 1) and the results were unchanged (data not shown).
Figure 4. Distribution of the number of risk alleles vs. antisocial latent variable scores in the Final model.

The scatter plot presents the individual data for each of the participants (latent antisocial variable score vs. the total number of the risk alleles (slope y= 0.141 x – 0.836, r2 = 0.091, t = 4.889, d.f. = 239, 2-tailed p= 1.861 × 10−6).
Haplotype analyses
To test the predictive value of the haplotypes we rank-ordered the 241 individuals according to their antisocial score and created three groupings with roughly equal numbers of individuals in each (low, middle, and top scoring antisocial groups – see Figure 2). We found that for all eight of the risk genes the overall haplotype frequency estimates were statistically different for some or all of behavioural subgroups when compared two at a time (see Table 2). A more detailed explanation of these haplotype analyses can be found in the supplemental materials. An examination of the results shows somewhat complex patterns in that the risk alleles are found on a number of different haplotype backgrounds at each gene and some of these show patterns where a particular risk allele haplotype systematically increases in frequency from the low to middle to top (high) antisocial groups. Figure S1 (in the supplement) is a bar-graph illustration of the results with Figure S1a displaying the results for the 3 genes (DRD2 > CHRNA7 > GRIN2A) with the strongest and most powerful statistical differences comparing the 3 behavioural groupings. The haplotyping results for the remaining five genes (NCAM1 > AVPR1A = SQRD = NR3C2 > GRM5) also show significant differences between at least two of the antisocial groups in the predicted direction (Figure S1b).
Network and Pathway Analysis
In addition, 33 SNPs from 63 of the remaining genes were also significant at a univariate level when added to the final model one at a time. In an effort to identify biological pathways and processes that may contribute to the latent anti-social phenotype, we completed this analysis for 41 implicated genes (eight in the Final model plus the additional 33 that were minimally significant). The most prominent over-represented biological pathways and processes included G protein-coupled dopaminergic, serotoninergic, and opioid receptors that activate an intracellular second messenger cascades to produce excitatory or inhibitory responses in the CNS (Table 3). Furthermore, there is evidence for genetic, physical and pathway interactions among many of the input implicated gene list (Supplemental Figure 2).
Table 3.
Overrepresentation of biological pathways and processes
| PANTHER Pathways | Genes # | RefSeq Gene IDs | Expected # | P-value1 |
|---|---|---|---|---|
| Heterotrimeric G-protein signaling pathway-Gi alpha and Gs alpha mediated pathway | 9 | OPRM1, CREB1, DRD1, DRD2, GRM5, HTR1B, HTR7, HTR4, HTR2A | 0.38 | 2.5 × 10−8 |
| Dopamine receptor mediated signaling | 7 | SLC6A3, COMT, DRD1, DDC, DRD2, MAOA, TH | 0.17 | 8.1 × 10−8 |
| Adrenaline and noradrenaline biosynthesis | 5 | SLC6A3, COMT, DDC, MAOA, TH | 0.07 | 1.8 × 10−6 |
| Huntington disease | 5 | GRIN2B, JUN, FOS, BDNF, GRIN2A | 0.34 | 3.9 × 10−3 |
| Gonadotropin releasing hormone receptor | 6 | JUN, FOS, CREB1, HSPA8, DRD2, PRLR | 0.58 | 4.1 × 10−3 |
| Metabotropic glutamate receptor group I | 3 | GRIN2B, GRM5, GRIN2A | 0.06 | 6.1 × 10−3 |
| 5-Hydroxytryptamine biosynthesis | 2 | DDC, TH | 0.01 | 0.013 |
| Apoptosis signaling | 4 | JUN, FOS, CREB1, HSPA8 | 0.27 | 0.028 |
| Heterotrimeric G-protein signaling- Gq alpha and Go alpha mediated | 4 | OPRM1, DRD1, DRD2, GRM5 | 0.31 | 0.048 |
| Nicotine pharmacodynamics | 3 | DDC, DRD2, TH | 0.13 | 0.049 |
| Neurological system processes | 26 | OXTR, GRIN2B, CNTNAP2, SEMA6D, OPRM1, SLC6A3, COMT, FOS, CREB1, NR3C2, CHRNA7, DRD1, ASPM, DRD2, SLC6A4, BDNF, HTR1B, GRIN2A, ESR1, GRM5, AVPR1A, HTR4, FLNB, NCAM1, HTR7, HTR2A | 4.45 | 1.8 × 10−13 |
| Signal transduction | 29 | OXTR, GRIN2B, CNTNAP2, SEMA6D, JUN, OPRM1, SLC6A3, CREB1, STAT5B, NR3C2, CHRNA7, DRD1, ASPM, DRD2, SLC6A4, PTEN, BDNF, TH, HTR1B, GRIN2A, ESR1, GRM5, AVPR1A, HTR4, FLNB, NCAM1, PRLR, HTR7, HTR2A | 8.8 | 3.5 × 10−9 |
| Cell-cell signaling | 17 | GRIN2B, CNTNAP2, OPRM1, SLC6A3, CHRNA7, DRD1, DRD2, SLC6A4, BDNF, GRIN2A, HTR1B, FLNB, NCAM1, PRLR, HTR7, HTR4, HTR2A | 2.96 | 1.9 × 10−7 |
| Cellular processes | 32 | OXTR, GRIN2B, CNTNAP2, SEMA6D, JUN, OPRM1, SLC6A3, FOS, CREB1, STAT5B, NR3C2, CHRNA7, DRD1, ASPM, DRD2, SLC6A4, PTEN, BDNF, MAOA, TH, HTR1B, GRIN2A, ESR1, HDAC5, GRM5, AVPR1A, HTR4, FLNB, NCAM1, PRLR, HTR7, HTR2A | 13.12 | 3.1 × 10−7 |
| Cell surface receptor linked signal transduction | 18 | OXTR, GRIN2B, SEMA6D, CNTNAP2, OPRM1, CHRNA7, DRD1, DRD2, BDNF, GRIN2A, HTR1B, GRM5, AVPR1A, NCAM1, PRLR, HTR7, HTR4, HTR2A | 4.58 | 2.0 × 10−5 |
| Synaptic transmission | 11 | GRIN2B, CNTNAP2, OPRM1, SLC6A3, CHRNA7, BDNF, DRD2, SLC6A4, GRIN2A, FLNB, HTR4 | 1.55 | 4.2 × 10−5 |
| Nerve-nerve synaptic transmission | 5 | GRIN2B, CHRNA7, BDNF, DRD2, GRIN2A | 0.25 | 9.0 × 10−4 |
| G-protein coupled receptor protein signaling pathway | 11 | OXTR, OPRM1, CHRNA7, DRD1, DRD2, GRM5, AVPR1A, HTR1B, HTR7, HTR4, HTR2A | 2.14 | 9.6 × 10−3 |
| Blood circulation | 6 | OXTR, DRD2, AVPR1A, HTR1B, HTR7, HTR2A | 0.5 | 1.8 × 10−3 |
| Muscle contraction | 8 | OXTR, CHRNA7, ASPM, DRD2, AVPR1A, NCAM, FLNB, HTR4 | 1.26 | 5.1 × 10−3 |
| Apoptosis | 9 | FOS, STAT5B, BDNF, DRD2, MAOA, PTEN, HDAC5, NCAM, HTR4 | 2.22 | 0.049 |
Assessment of relative representation is compared to the entire genome, using the PANTHER Classification System (pantherdb.org).
P-values use Bonferroni correction for multiple testing.
DISCUSSION
The genetic, epigenetic and neurobiological mechanisms by which childhood adversity increases vulnerability to psychopathology including antisocial outcomes and a heightened risk of substance abuse remain poorly understood. Efforts to identify susceptibility genes using GWAS have largely failed, in part because they evaluate potential risk alleles one-by-one rather than in combination with one another. Latent variable constructs using multiple risk alleles may provide a useful approach to identify susceptibility genes that in combination contribute to antisocial outcomes. Knowledge of individual differences in risk genotypes may contribute to a deeper understanding of the complex interaction between environmental experiences (such as abuse) and antisocial outcomes and has the potential to identify the molecular pathways that influence the neurobiological circuitry underpinning psychological and emotional development.
In this study, a novel latent variable approach identified eight genes, from a predefined CSG of 71 genes, which collectively accounted for 16% of the variance in a latent antisocial phenotype. Notably, we found a linear relationship between the number of risk alleles and the individual’s latent antisocial variable score (Figure 4). The results of the haplotyping analyses were also consistent with each of these loci being adjacent to a functional genetic variant. If replicated, this is a remarkably high proportion of the phenotypic variance to be explained by genetic factors. In our estimation these results suggest both additive non-additive effects. Specifically, the amount of variance explained by the 8 gene final model is slightly above 16%, but when we simply do a regression analysis looking at the association between an individual’s total number of risk alleles and their antisocial latent variable the amount of variance explained is just 7.8% (Figure 4). This difference (16% vs. 9.1%) may well be due to various “non-additive” effects that involve a more complex “co-action” of some of these risk alleles.
To put this finding in a clinical perspective, well-designed and well-executed GWAS have provided a glimpse into the complex genetic architecture of medical disorders such as a type-2 diabetes mellitus (T2DM). Specifically, at least 36 diabetes-associated genes have been identified that increase the risk of T2DM, but these loci collectively account for only a small fraction (5–10%) of the genetic contribution to T2DM (Stolerman & Florez, 2009). Interestingly, most of the discovered gene variants have been linked to beta-cell dysfunction rather than insulin resistance. This finding has challenged the established thinking of T2DM as being simply a disorder of insulin action.
There are several weaknesses of this study. First, the sample of consenting subjects was primarily Caucasian of European descent limiting the generalizability of these findings to other ethnic groups. Second, a larger dataset from a single ethnic background would be desirable especially in order to establish which particular risk allele haplotype backgrounds are most and least associated with the latent antisocial variable score. Third, the decision to use a single latent antisocial and a single genetic variable can also be questioned. It is possible that a multifactorial antisocial variable could account for more of the phenotypic variance. Likewise this analytic approach does not permit the characterization of the biological processes that contribute to this shared variance. However, twin data strongly suggests that a common set of genetic risks are associated with four disorders reflecting antisocial behaviour (conduct disorder and antisocial personality disorder) and substance use disorders (alcohol and drug abuse or dependence) (Kendler et al., 2011; Lahey & Waldman, 2012). Fourth, our selection of genetic loci and the selection of SNPs is somewhat dated. Additional genes, particularly nodal genes identified by the pathway analysis (see Supplemental Figure 2) G-protein-coupled kinase 5 [GRK5]; Adenylate cyclase 6 [ADCY6]; BCL2-associated athanogene 1 [BAG1]; DISCS large, drosophila, homolog of, 2 [DLG2]; 5-Hydroxytryptamine receptor 1D [HTR1D]; Adenosine A2 receptor [ADORA2A]; Neurotrophic tyrosine kinase, receptor, type 2 [NTRK2]; and Signal transducer and activator of transcription 5A [STAT5A]) should be included in future efforts to replicate these findings (see Supplemental Figure S2). Given the likely importance of epigenetic modification in altering levels of gene expression including SNPs from DNA methyltransferase 3 –like protein [DNMT3L], along with DNMT3B, will also be of interest.
In the current study the strongest contributors included genes over-representing dopamine, norepinephrine, serotonin, and glutamate signaling (Table 3). This is largely congruent with our understanding of the neurobiology of antisocial behaviour (Gao et al., 2009), although finding an enrichment for these signaling processes may be biased by the CSG selection processes and tendency for prior studies to focus on genes in neurotransmitter systems. We also found a highly significant result with regard to CHRNA7 and glucocorticoid receptor signaling which is consistent with Raine’s (2002) theory that the role of prefrontal deficits, low autonomic arousal, and early health factors acting in part through cholinergic mechanisms affecting cardiac functioning may play an important role in the development of antisocial and aggressive behaviour in children.
Consistent with the importance of glucocorticoid receptor signaling, we also found that NR3C2, in the Final model, had a “negative” coefficient indicating that the major allele was associated with an increased risk of antisocial behaviour when considered in combination with the seven other risk genes. NR3C2 encodes the mineralocorticoid receptor (MR). NR3C2 encodes the mineralocorticoid receptor (MR). MR belongs to the nuclear receptor superfamily and functions as a ligand-dependent transcription factor that mediates the effects of glucocorticoids as well as aldosterone on a variety of target tissues including the central nervous system. While the MR has a very high affinity for glucocorticoids, the glucocorticoid receptor (GR) has a somewhat lower affinity. The glucocorticoids are known to produce profound and diverse effects in the brain in relation to cognition and behavior (de Kloet et al., 2005). Although the GR has received a great deal of attention, it is now clear that the MR also plays an important role in the coordination of normal circadian activity and the reactivity of the hypothalamus- pituitary- adrenal (HPA) axis in response and adaptation to stress (Conway-Campbell et al., 2012; Zalachoras, Houtman, & Meijer, 2013).
However, for this latent variable modeling approach to have true clinical significance, environmental risk and protective variables also need to be included in these models. This in turn has the potential to clarify interactions between genotypes and response to preventive interventions including the Nurse Family Partnership (Olds et al., 1998; 2010) as well as other interventions including lifestyle changes and cognitive-behavioural interventions over the course of development (Lochman et al., 2011; Rutter, 2013).
Supplementary Material
Supplementary Table S1. Candidate system of genes (CSG)
Supplementary Table S2. Estimates from the zero-Inflated Poisson Regression Model
Supplemenatal Figure S1. Haplotype frequencies within 3 subgroups
Supplementary Figure S2. Biological network
Key Points.
The genetic, epigenetic and neurobiological mechanisms by which childhood adversity increases vulnerability to psychopathology including antisocial outcomes and a heightened risk of substance abuse remain poorly understood.
Efforts to identify susceptibility genes using Genome-Wide Association Studies have largely failed, in part because they evaluate potential risk alleles one by one rather than in combination with one another.
Latent variable analyses have been widely used in economics and psychology. Its application to genetics is novel. Our preliminary data suggests that this may be a useful approach to identify susceptibility genes that in multiple combinations contribute to a shared vulnerability to develop antisocial outcomes.
Knowledge of individual differences in risk genotypes may contribute to a deeper understanding of the complex interaction between environmental experiences (such as abuse) and antisocial outcomes, and has the potential to identify the molecular pathways that influence the neurobiological circuitry underpinning psychological and emotional development and that are responsive to preventive interventions as well as lifestyle changes and cognitive-behavioural treatments.
Acknowledgments
Dr. David Olds receives no personal remuneration from the licensing of Nurse Family Partnership program. However, the University of Colorado Denver receives funds from the licensing and some of these funds go to the Prevention Research Center for Family and Child Health (PRC). Dr. Olds is the Director of the PRC that conducts research on the program for improvements. While the PRC staff provides consultation regarding program implementation and evaluation, their consultancy provides no personal remuneration to PRC staff members beyond their university salaries. Dr. Leckman has received research support from Grifols, NIH, and the Associates of the Child Study Center, and book royalty payments from a number of publishing companies. He has received travel funding for invited presentations at departmental conferences, CME events, and other clinical or scientific venues. The other authors report no financial relationships with commercial interests.
This research was supported in part by NIH grants; R01DA021624 (Olds), R01DA216240 (Olds), R01DA021624 (Olds), and T32MH018268 (JFL) and donations from the Associates of the Yale Child Study Center.
Footnotes
Conflict of interest: Potential conflict of interest disclosed in acknowledgments.
- Substantial contributions to conception and design (MJB, HL, ELG, JFL), acquisition of data (DLO, ML, CMY, ELG), or analysis and interpretation of data (MJB, HL, TVF, ML, AJP, LK, ELG, JFL).
- Drafting the article or revising it critically for important intellectual content (MJB, HL, TVF, AJP, ELG, JFL)
- Final approval of the version to be published (all authors).
Additional supporting information is provided along with the online version of this article.
Please note that Wiley-Blackwell Publishing are not responsible for the content or functionality of any supporting materials supplied by the authors (although this material was peer reviewed by JCPP referees and Editors along with the main article). Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Avery RB. Modeling monetary policy as an unobserved variable. Journal of Econometrics. 1979;10:291–311. [Google Scholar]
- Bierut LJ, Rice JP, Edenberg HJ, Goate A, Foroud T, Cloninger CR, Reich T. Family-based study of the association of the dopamine D2 receptor gene (DRD2) with habitual smoking. American Journal of Medical Genetics. 2002;90:299–302. doi: 10.1002/(sici)1096-8628(20000214)90:4<299::aid-ajmg7>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Blum K, Chen AL, Giordano J, Borsten J, Chen TJ, Hauser M, Barh D. The addictive brain: all roads lead to dopamine. Journal of Psychoactive Drugs. 2012;44(2):134–143. doi: 10.1080/02791072.2012.685407. [DOI] [PubMed] [Google Scholar]
- Bollen KA. Latent variables in psychology and the social sciences. Annual Review of Psychology. 2002;53:605–634. doi: 10.1146/annurev.psych.53.100901.135239. [DOI] [PubMed] [Google Scholar]
- Bornovalova MA, Blazei R, Malone SH, McGue M, Iacono WG. Disentangling the relative contribution of parental antisociality and family discord to child disruptive disorders. Personality Disorders. 2012 Aug 13; doi: 10.1037/a0028607. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, McGue M, Krueger RF, Iacono WG. Sources of covariation among the child-externalizing disorders: Informant effects and the shared environment. Psychological Medicine. 2005;35:1133–1144. doi: 10.1017/S0033291705004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt SA, Mikolajewski AJ. Preliminary evidence that specific candidate genes are associated with adolescent-onset antisocial behavior. Aggressive Behavior. 2008;34(4):437–45. doi: 10.1002/ab.20251. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Morgan J, Rutter M, Taylor A, Arseneault L, Polo-Tomas M. Maternal expressed emotion predicts children’s antisocial behavior problems: Using monozygotic-twin differences to identify environmental effects on behavioral development. Developmental Psychology. 2004;40:149–161. doi: 10.1037/0012-1649.40.2.149. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Krystal JH, Self DW. A neurobiological basis for substance abuse comorbidity in schizophrenia. Biological Psychiatry. 2001;50(2):71–83. doi: 10.1016/s0006-3223(01)01134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ. Transcription, genomes, function. Trends in Genetics. 2000;16(9):409–415. doi: 10.1016/s0168-9525(00)02065-5. [DOI] [PubMed] [Google Scholar]
- Conway-Campbell BL, Pooley JR, Hager GL, Lightman SL. Molecular dynamics of ultradian glucocorticoid receptor action. Molecular and Cellular Endocrinology. 2012;348:383–393. doi: 10.1016/j.mce.2011.08.014. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Fitzsimons CP, Datson NA, Meijer OC, Vreugdenhil E. Glucocorticoid signaling and stress-related limbic susceptibility pathway: about receptors, transcription machinery and microRNA. Brain Research. 2009;1293:129–141. doi: 10.1016/j.brainres.2009.03.039. [DOI] [PubMed] [Google Scholar]
- Depue RA, Collins PF. Neurobiology of the structure of personality: dopamine, facilitation of incentive motivation, and extraversion. Behavioral and Brain Sciences. 1999;22(3):491–517. doi: 10.1017/s0140525x99002046. [DOI] [PubMed] [Google Scholar]
- Gao Y, Glenn AL, Schug RA, Yang Y, Raine A. The neurobiology of psychopathy: a neurodevelopmental perspective. Canadian Journal of Psychiatry. 2009;54(12):813–83. doi: 10.1177/070674370905401204. [DOI] [PubMed] [Google Scholar]
- Gordon I, Martin C, Feldman R, Leckman JF. Oxytocin and social motivation. Developmental and Cognitive Neuroscience. 2011;1(4):471–493. doi: 10.1016/j.dcn.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, Braff DL. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia. American Journal of Psychiatry. 2011;168:930–946. doi: 10.1176/appi.ajp.2011.10050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorenko EL, De Young CG, Eastman M, Getchell M, Haeffel GJ, Klinteberg B, Yrigollen CM. Aggressive behavior, related conduct problems, and variation in genes affecting dopamine turnover. Aggressive Behavior. 2010;36:158–176. doi: 10.1002/ab.20339. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Aggen SH, Knudsen GP, Roysamb E, Neale MC, Reichborn-Kjennerud T. The structure of genetic and environmental risk factors for syndromal and subsyndromal common DSM-IV axis I and all axis II disorders. American Journal of Psychiatry. 2011;168:29–39. doi: 10.1176/appi.ajp.2010.10030340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE. MAOA, maltreatment, and gene-environment interaction predicting children’s mental health: new evidence and a meta-analysis. Molecular Psychiatry. 2006;11(10):903–13. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Lahey BB, Piacentini JC, McBurnett K, Stone P, Hartdagen S, Hynd G. Psychopathology in the parents of children with conduct disorder and hyperactivity. Journal of the American Academy of Child & Adolescent Psychiatry. 1988;27:163–170. doi: 10.1097/00004583-198803000-00005. [DOI] [PubMed] [Google Scholar]
- Lahey BB, Waldman ID. Annual research review: phenotypic and causal structure of conduct disorder in the broader context of prevalent forms of psychopathology. Journal of Child Psychology and Psychiatry. 2012;53(5):536–557. doi: 10.1111/j.1469-7610.2011.02509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert D. Zero-inflated poisson regression, with an application to defects in manufacturing. Technometrics. 1992;34:1–14. [Google Scholar]
- Leckman JF, Herman AE. Maternal behavior and developmental psychopathology. Biological Psychiatry. 2002;51:27–43. doi: 10.1016/s0006-3223(01)01277-x. [DOI] [PubMed] [Google Scholar]
- Lim MM, Young LJ. Neuropeptidergic regulation of affiliative behavior and social bonding in animals. Hormones and Behavior. 2006;50:506–517. doi: 10.1016/j.yhbeh.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Lochman JE, Powell NP, Boxmeyer CL, Jimenez-Camargo L. Cognitive-behavioral therapy for externalizing disorders in children and adolescents. Child Adolescent Psychiatric Clinics of North America. 2011;20(2):305–318. doi: 10.1016/j.chc.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Maher BS, Vladimirov VI, Latendresse SJ, Thiselton DL, McNamee RF, Kang M, Vanyukov MM. The AVPR1A gene and substance use disorders: Association, replication, and functional evidence. Biological Psychiatry. 2011;70:519–527. doi: 10.1016/j.biopsych.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthys W, Vanderschuren LJ, Schutter DJ. The neurobiology of oppositional defiant disorder and conduct disorder: Altered functioning in three mental domains. Developmental Psychopathology. 2013;25(1):193–207. doi: 10.1017/S0954579412000272. [DOI] [PubMed] [Google Scholar]
- McCrory E, De Brito SA, Viding E. The link between child abuse and psychopathology: a review of neurobiological and genetic research. Journal of the Royal Society of Medicine. 2012;105(4):151–156. doi: 10.1258/jrsm.2011.110222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Molecular Biology. 2009;563:123–40. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Research. 2013;41(Database issue):D377–386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileva-Seitz V, Fleming AS, Meaney MJ, Mastroianni A, Sinnwell JP, Steiner M, Sokolowski MB. Dopamine receptors D1 and D2 are related to observed maternal behavior. Genes, Brain and Behavior. 2012;11(6):684–694. doi: 10.1111/j.1601-183X.2012.00804.x. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. The new look of behavioral genetics in developmental psychopathology: Gene-environment interplay in antisocial behaviors. Psychological Bulletin. 2005;131:533–554. doi: 10.1037/0033-2909.131.4.533. [DOI] [PubMed] [Google Scholar]
- Moul C, Dobson-Stone C, Brennan J, Hawes D, Dadds M. An exploration of the serotonin system in antisocial boys with high levels of callous-unemotional traits. PLoS One. 2013;8(2):e56619. doi: 10.1371/journal.pone.0056619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olds D, Henderson CR, Jr, Cole R, Eckenrode JF, Kitzman HF, Luckey DF, Powers J. Long-term effects of nurse home visitation on children’s criminal and antisocial behavior: 15-year follow-up of a randomized controlled trial. Journal of the American Medical Association. 1998;280:1238–1244. doi: 10.1001/jama.280.14.1238. [DOI] [PubMed] [Google Scholar]
- Olds DL, Kitzman HJ, Cole RE, Hanks CA, Arcoleo KJ, Anson EA, Stevenson AJ. Enduring effects of prenatal and infancy home visiting by nurses on maternal life course and government spending: follow-up of a randomized trial among children at age 12 years. Archives of Pediatric and Adolescent Medicine. 2010;164(5):419–424. doi: 10.1001/archpediatrics.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poustka L, Maras A, Hohm E, Fellinger J, Holtmann M, Banaschewski T, Laucht M. Negative association between plasma cortisol levels and aggression in a high-risk community sample of adolescents. Journal of Neural Transmission. 2010;117(5):621–627. doi: 10.1007/s00702-010-0386-7. [DOI] [PubMed] [Google Scholar]
- Raine A. Annotation: the role of prefrontal deficits, low autonomic arousal, and early health factors in the development of antisocial and aggressive behavior in children. Journal of Child Psychology and Psychiatry. 2002;43(4):417–434. doi: 10.1111/1469-7610.00034. [DOI] [PubMed] [Google Scholar]
- Rhee SH, Waldman ID. Genetic and environmental influences on aggression. In: Shaver MPR, editor. Human aggression and violence: Causes, manifestations and consequences. Washington, DC: American Psychological Association; 2011. pp. 143–163. [Google Scholar]
- Rhee SH, Waldman ID. Genetic and environmental influences on antisocial behavior: A meta-analysis of twin and adoption studies. Psychological Bulletin. 2002;128:490–529. [PubMed] [Google Scholar]
- Rutter M. Resilience-clinical implications. Journal of Child Psychology and Psychiatry. 2013;54(4):474–487. doi: 10.1111/j.1469-7610.2012.02615.x. [DOI] [PubMed] [Google Scholar]
- Solanto MV. Dopamine dysfunction in ADHD: integrating clinical and basic neuroscience research. Behavior and Brain Research. 2002;130(1–2):65–71. doi: 10.1016/s0166-4328(01)00431-4. [DOI] [PubMed] [Google Scholar]
- Stanger C, Dumenci L, Kamon J, Burstein M. Parenting and children’s externalizing problems in substance-abusing families. Journal of Clinical Child and Adolescent Psychology. 2004;33:590–600. doi: 10.1207/s15374424jccp3303_16. [DOI] [PubMed] [Google Scholar]
- Stolerman ES, Florez JC. Genomics of type 2 diabetes mellitus: implications for the clinician. Nature Reviews Endocrinology. 2009;5(8):429–436. doi: 10.1038/nrendo.2009.129. [DOI] [PubMed] [Google Scholar]
- Stouthamer-Loeber M, Loeber R, Homish DL, Wei E. Maltreatment of boys and the development of disruptive and delinquent behavior. Developmental Psychopathology. 2001;13(4):941–955. [PubMed] [Google Scholar]
- Viding EF, Larsson H, Jones AP. Quantitative genetic studies of antisocial behavior. Philosophical Transactions of the Royal Society B. 2008;363:2519–2527. doi: 10.1098/rstb.2008.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrigollen CM, Han SS, Kochetkova AF, Babitz T, Chang JT, Volkmar FR, Grigorenko EL. Genes controlling affiliative behavior as candidate genes for autism. Biological Psychiatry. 2008;63:911–916. doi: 10.1016/j.biopsych.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalachoras I, Houtman R, Meijer OC. Understanding stress-effects in the brain via transcriptional signal transduction pathways. Neuroscience 2013. 2013 Mar 29; doi: 10.1016/j.neuroscience.2013.03.038. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Candidate system of genes (CSG)
Supplementary Table S2. Estimates from the zero-Inflated Poisson Regression Model
Supplemenatal Figure S1. Haplotype frequencies within 3 subgroups
Supplementary Figure S2. Biological network