

Abstract
Background
Expression of the circadian gene, Npas2, is altered in fetal life with maternal high fat diet exposure by virtue of alterations in the fetal histone code. We postulated that these disruptions would persist postnatally.
Methods
Pregnant macaques were fed a control (CTR) or high fat (HF) diet and delivered at term. When offspring were weaned, they were placed on either CTR or HF diet for a period of 5 months to yield four exposure models (in utero diet/postweaning diet: CTR/CTR n = 5; CTR/HF n = 4; HF/CTR n = 4; HF/HF n = 5). Liver specimens were obtained at necropsy at one year of age.
Results
Hepatic trimethylation of lysine 4 of histone H3 is decreased, (CTR/HF 0.87-fold, P = 0.038; HF/CTR 0.84-fold, P = 0.038) while hepatic methyltransferase activity increased by virtue of diet exposure (HF/HF 1.3-fold, P = 0.019). Using chromatin immunoprecipitation to determine Npas2 promoter occupancy, we found alterations of both repressive and permissive histone modifications specifically with postweaning high fat diet exposure.
Conclusion
We find altered Npas2 expression corresponds with a change in the histone code within the Npas2 promoter.
INTRODUCTION
Studies on the molecular underpinnings of the developmental origins of health and disease investigate how perturbations in the in utero environment can predispose an individual to disease in adulthood (1). Individuals exposed in the womb to various maternal constraints such as a low protein diet, maternal caloric restriction or maternal high fat diet show a higher susceptibility to the adult onset of metabolic syndrome than unexposed offspring (2). In light of the current obesity epidemic, study of the long lasting effects of in utero exposure to a maternal HF diet and maternal obesity are extremely important. How this memory of in utero exposure is maintained remains an unanswered question in the field, and epigenetic modifications in the offspring have logically emerged as potential molecular mediators.
DNA methylation and the removal and addition of post-translational modifications to the histone proteins are mechanisms the cell utilizes to regulate gene transcription. Although the “histone code” has not been specifically defined, in general, certain modifications are associated with gene expression (permissive modifications) while others are associated with gene repression (repressive modifications) (Figure 1A). Acetylated histones, such as H3K14ac, are traditionally associated with gene expression (3). Trimethylation of lysine 4 of histone H3 (H3K4me3) is also found in the promoter region of active genes (4). However, histone lysine trimethylation does not necessarily correspond with gene activation. Both H3K9me3 and H3K27me3 are enriched in promoters of inactive genes (5) (Figure 1A). Histone post-translational modifications are not permanent. Enzymes such as histone acetyltransferases (HATs) and histone deacetylases (HDACs) add and remove acetyl moieties from histone lysine residues (6) (Figure 1B). Histone methyltransferases add mono-, di-, or tri-methyl groups to histones (7) (Figure 1B). These modifications work in coordination with transcriptional activators and repressors to ensure proper regulation of gene expression.
Figure 1. Histone modifications are associated with different chromatin states.

(A) The histone modifications H3K14ac and H3K4me3 are both associated with actively transcribed, open euchromatin. Both H3K9me3 and H3K27me3 are associated with transcriptionally repressed, closed heterochromatin. Green diamond = H3K4me3; red triangle = H3K14ac; yellow pentagon = H3K9me3; orange octagon = H3K27me3. (B) Addition of an acetyl group to the N-terminal histone domain is achieved through the enzymatic activity of histone acetyltransferases (HATs). These modifications can be removed by histone deacetylases (HDACs). Similarly, the enzymatic activity of histone methyltransferases (HMTases) can add mono-, di- or tri-methyl groups to the histone proteins, and these modifications can be removed by histone demethylases (HDMases).
In recent years, several investigators have shown that epigenetic changes are modifiable with maternal diet constraints (2). Changes in DNA methylation and histone modifications have been reported with exposure to maternal calorie and protein restriction using rat model systems (8,9). In humans, in utero exposure to famine results in changes in DNA methylation which persist into adulthood (10). Using a mouse model, it has been shown that exposure to maternal methyl supplements results in changes in DNA methylation at specific loci(11). Interestingly, in each of these studies, the in utero constraint was associated with an increased susceptibility to hypertension and obesity in the offspring in adulthood.
We have previously reported epigenetic, metabolomic, circadian and pathological changes associated with maternal high fat diet exposure in a non-human primate model of excess nutrition (12–18). Specifically, we have previously demonstrated that H3K14ac is increased in the fetal liver with high fat diet exposure, while HDAC activity is decrease (12). Further characterization of the fetal liver revealed alterations in the expression of Npas2, an important regulator of circadian genes in peripheral organs such as the liver. This altered expression was accompanied by alterations in the histone code in the promoter of the Npas2 gene (15). The Npas2 promoter contains a transcriptional DNA binding element (tandem RORE response elements) essential for gene expression (19). In the late gestation fetus, Npas2 expression is increased with HF diet exposure and in both temporal and spatial association H3K14ac is enriched within the RORE.
Study in our macaque model system has revealed effects of the in utero diet exposure which are manifested postnatally. Specifically, offspring show alterations the seritonergic system and increased anxiety(20). Because we and others have found that epigenetic modifications are modifiable through environmental interactions (21,22), we set out to investigate whether the changes we found in fetal liver would persist postnatally in 1 year old Japanese macaques. Offspring were exposed to maternal CTR or HF diet in utero and postnatally until weaning at approximately 7 months of age. At weaning, offspring were either assigned to a CTR or a HF diet, yielding four different exposure models (Figure 2, in utero diet/postweaning diet: CTR/CTR; CTR/HF; HF/CTR; and HF/HF). We hypothesized that animals exposed to a high fat diet in utero would have an altered histone code, potentially showing an increase in H3K14ac levels similar to fetal life. We also sought to determine if activity of the histone modifying enzymes differed by virtue of diet exposure. Finally, we sought to interrogate the Npas2 promoter in the juvenile animals. We have previously shown that Npas2 expression is increased with postweaning HF diet exposure (15). We sought to determine if the increase in expression was associated with altered promoter occupancy in our juvenile animals.
Figure 2. Model of in utero and postweaning diet exposure in Japanese macaques.
Animals were exposed to either a control or high fat diet in utero. Animals were delivered at term, and were placed on one of two postweaning diets, either control or high fat. This model yielded four cohorts of diet exposure, depending on the combination of in utero and postweaning diet exposures; designated as in utero/ postweaning.
MATERIALS AND METHODS
Study Design
All animal procedures were approved by the ONPRC Institutional Animal Care and Use Committee and conformed to NIH guidelines on the ethical use of animals. Briefly, weight and age matched adult female Japanese macaques (macaca fuscata), were fed a control (CTR) or high fat (HF) for up to 4 years. The CTR diet (Fiber Balanced Monkey Diet 5000, Purina Lab Diet, St. Louis, MO) provided 13% of calories from fat and the HF (Test Diet, 5LOP, Purina Lab Diet) provided 32% of calories from fat and was supplemented with calorically-dense treats. The HF represents a typical Western diet in regards to the saturated fat content. Monkeys were housed in indoor/outdoor pens in groups of approximately 12 individuals (male: female ratio 2:10). Monkeys had ad libitum access to food and water.
In order to generate juvenile offspring (1 year of life), a cohort of dams (n = 18) progressed to term (e167). Dams had no more than 2 prior pregnancies before delivering the subjects of this study. To evaluate the effects of the postweaning diet, juveniles were grouped by diet exposure in utero and by postweaning diet to yield four offspring exposure models (Figure 2, in utero diet/postweaning diet: i.e., CTR/CTR, n = 5; CTR/HF, n = 4; HF/CTR, n = 4; HF/HF, n = 5). Juvenile liver samples were obtained at 5 months postweaning when animals were sacrificed at approximately 1 year of age. Briefly, animals are fasted for approximately 4 hours prior to necropsy and are sedated with 15–25 mg/kg of ketamine IM. The animals are deeply anesthetized with a surgical dose of sodium pentobarbital (25–30 mg/kg IV) by veterinary staff and then exsanguinated. Liver specimens are then snap frozen in liquid nitrogen and stored at −80°C until use. All procedures involving animals have undergone an extensive IACUC review process at OHSU and BCM.
Western blotting
Histones were extracted as previously described (12). Samples were run on 18% Tris-Glycine gels (Invitrogen, Carlsbad, CA), transferred to PVDF (BioRad, Hercules, CA) and incubated overnight in primary antibody (Millipore, Billerica, MA) at a dilution of 1:1000. The blot was incubated for 45 minutes in secondary antibody (anti-rabbit IgG, Cell Signaling, Beverly, MA) and visualized using Chemiluminescence (Perkin-Elmer, Waltham, MA). Blots were stripped with stripping buffer (Pierce, Rockford, IL) and reprobed with GAPDH antibody (Abcam, Cambridge, MA) in order to normalize loading. Of note, for the Western blot experiments, the HF/CTR group had an n of 3.
HDAC and HMTase assays
Nuclear extracts were prepared and HDAC assays (Biomol, Farmingdale, NY) were performed as previously described (12). For HMTase assays, 80 μg of hepatic lysate was used and assay was performed according to manufacturer conditions (Epigentek, Farmingdale, NY).
Chromatin Immunoprecipitation
ChIP was performed as previously described15. Briefly, 100 μg of chromatin was incubated overnight with histone modification specific antibodies (Millipore). Ten percent of total chromatin used was set aside as the “input” control. Bound chromatin was precipitated using Protein A Dynal beads (Invitrogen), eluted with 10% SDS and purified with QIAquick columns (Qiagen, Valencia, CA). PCR reactions were set up using primers as described(15).
Analysis
A Student’s t-test was used for statistical significance for all assays. To determine percent IP, samples were first normalized to the input for each sample. Percent IP was calculated as 100*2^(Input-IP).
RESULTS
H3K4me3 is altered by virtue of diet exposure
We have previously reported that the fetal histone code at H3K14ac is upregulated in association with exposure to maternal high fat diet in the fetal liver at gestational day 130 (term 167 days in the Japanese macaque) (12). This change in histone H3K14ac is associated with hepatic markers of non-alcoholic fatty liver disease in the fetus (13). We have also shown that the increased fetal hepatic triglycerides persisted to postnatal day 180 (13). We sought to determine if the upregulation in altered histone acetylation also persists postnatally. Using Western blotting on acid extracted histones to detect alterations in the juvenile hepatic histone code, we interrogated the abundance of two permissive (H3K14ac and H3K4me3) and two repressive (H3K9me3 and H3K27me3) histone modifications in our four cohorts of offspring. We found that levels of H3K4me3 changed significantly with differing diet exposures both in utero and postweaning (Figure 3A). Comparing each group to the CTR/CTR (i.e., maternal control diet and control diet postweaning cohort we found a significant decrease in H3K4me3 in the CTR/HF group (0.87-fold, P = 0.038). Furthermore, there was a decrease (0.84-fold, P = 0.038) in H3K4me3 in the HF/CTR. However, in the group chronically exposed to HF diet (HF/HF) there was no significant difference in H3K4me3 levels. While we have found that fetal H3K14ac levels are altered in utero with HF diet exposure, we do not find a significant change in any of our postweaning exposure cohorts at one year of age (Figure 3B). Neither H3K9me3 (Figure 3C) nor H3K27me3 (Figure 3D) were significantly altered in any of the cohorts.
Figure 3. H3K4me3 methylation in the juvenile liver is altered by virtue of diet exposure.

Using Western blotting of extracted hepatic histones, we found that H3K4me3 (A) is decreased in two exposure models (CTR/HF (p=0.038) and HF/CTR (p=0.038)). Levels of H3K14ac (B), H3K9me3 (C) and H3K27me3 (D) do not change in any of the groups tested. Horizontal bars indicate the fold-change compared to CTR/CTR for each group analyzed.
Histone H3 lysine 4 methyltransferase activity is altered with chronic HF diet exposure
After determining that hepatic H3K4me3 levels were altered in our model system, we sought to determine if there is a change in H3K4 histone methyltransferase (HMTase) activity in hepatic lysates from each of the four cohorts. Using a commercially available kit which specifically measures only H3K4 methyltransferase activity, we found that indeed, chronic exposure to a maternal and postweaning high fat diet (HF/HF) is associated with increased HMTase activity (1.34-fold, P = 0.019) (Figure 4A). There were no significant changes in the other cohorts. Because we have previously reported and consistently observe that fetal HDAC activity is altered in utero with maternal HF diet exposure (12), we sought to determine if this alteration persists postnatally. Consistent with our observations that the juvenile histone code is altered by a maternal high fat diet on H3K4me3 but not on H3K14ac, we did not observe significant changes in HDAC activity in our model system (Figure 4B).
Figure 4. Juvenile hepatic histone methyltransferase activity is increased in HF/HF exposed animals.
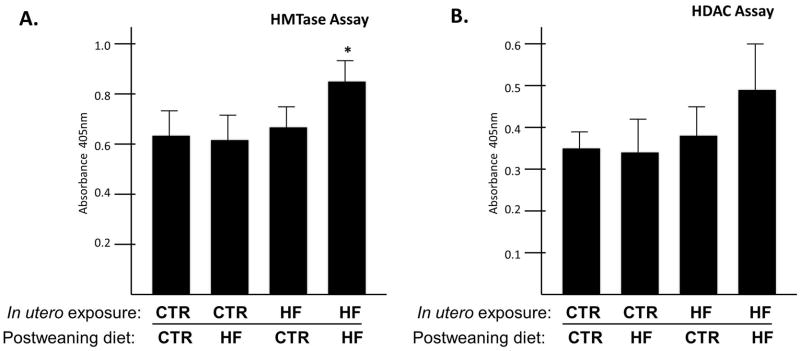
Using a commercially available kit, we measured histone H3 K4 trimethylase activity. (A) We found that HMTase activity is increased in the HF/HF exposed group (p=0.019). (B) HDAC activity in juvenile animals did not change by virtue of diet exposure.
Promoter occupancy of the RORE transcriptional element of Npas2 is altered with diet exposure
We have previously published that H3K14ac is enriched in the RORE of Npas2 in fetal life with high fat diet exposure (15). This increase in acetylation, a modification associated with gene expression, was concomitant with an increase in Npas2 gene expression and demonstrated significant correlation between alterations in promoter occupancy and exon transcription. In juvenile liver, exposure to a postweaning high fat diet leads to significant increases in Npas2 expression(15) Since juvenile Npas2 expression levels are increased in both the CTR/HF and HF/HF offspring(15), we sought to interrogate the Npas2 promoter in the liver of juvenile animals to determine if promoter occupancy is altered in a manner akin to that observed in the fetus.
Using antibodies specific for the permissive H3K4me3, we found this modification was enriched in the HF/HF group (7.7% vs. 15.0%, P = 0.016), compared with CTR/CTR (Figure 5A). This change corresponds with the increase in HMTase activity we found in this cohort (Figure 4A). Using an antibody specific for H3K14ac we found that there were no significant changes in any juvenile cohort interrogated (Figure 5B). Interestingly, both repressive modifications showed depletion in the postweaning HF diet groups. Using an antibody to determine enrichment of H3K9me3 at the RORE, we found that the modification is depleted in the cohorts on a postweaning HF diet, CTR/HF (2.7% vs. 0.7%, P = 0.001) and HF/HF (2.7% vs 0.6%, P < 0.01) (Figure 5C). We found similar results with H3K27me3 (Figure 5D). The modification is depleted in CTR/HF (4.6% vs. 3.0%, P = 0.021) and HF/HF (4.6% vs. 1.9%, P < 0.01) compared with CTR/CTR.
Figure 5. The RORE of Npas2 is depleted for repressive modifications with postweaning exposure to a HF diet.

Using ChIP for various histone modifications, we interrogated the occupancy of the RORE of Npas2 in juvenile liver. (A) We found that H3K4me3 is enriched in the RORE of the HF/HF exposed animals (p=0.016). (B) H3K14ac levels are not altered with any diet condition. H3K9me3 (C) is depleted in CTR/HF (p=0.001) and HF/HF (p<0.01). H3K27me3 (D) is similarly depleted in CTR/HF (p=0.021) and HF/HF (p<0.01). Horizontal bars indicate the fold change for each group compared with CTR/CTR.
DISCUSSION
How a caloric dense high fat maternal diet ultimately affects the phenotypic outcome of the offspring is unclear. However, studies using non human primates demonstrate that without intervention the consequences are significant. High fat exposure during the prenatal period predisposes offspring to be more susceptible to increased fat mass and insulin resistance (23), cardiovascular reactivity(24), liver dysfunction(25), and anxiety(20) later in life. The molecular mechanisms behind the development of these phenotypes in adulthood are largely ill-defined. However, we believe that our primate model, examining both in utero and postnatal diet exposures, will yield information as to how these phenotypes emerge. Epigenetic changes which occur in utero may provide a mechanism to maintain a long lasting, stable memory of the in utero experience. While we have reported epigenetic changes in the fetal liver in our non-human primate model system (12,15,17,18), whether these alterations persist and how they parlay into later phenotypic alterations remains to be elucidated.
Our previous epigenetic characterizations of the fetal primate liver exposed to maternal HF diet revealed that H3K14ac is increased compared with CTR diet exposed animals (12). We have also determined that this modification is enriched in the promoter of the circadian transcription factor, Npas2, specifically in the fetal livers of HF diet exposed animals (15). This change in promoter occupancy was also accompanied by disrupted expression of Npas2 in the HF diet exposed fetal liver (15). Analysis of gene expression in the juvenile liver showed that Npas2 regulation is disrupted only in the animals who consumed a HF diet postweaning, regardless of their in utero exposure.
While it has been shown that HF diet feeding disrupts circadian rhythm and gene expression in the mouse (26), we were the first to show that in utero exposure to HF diet feeding disrupts the peripheral circadian rhythm in the fetus. Many elegant studies in animal models have demonstrated a cyclicity of histone modifications in the promoter regions of these circadian regulated genes (27). Over a 24 hour period, such promoters see a change in histone acetylation, due to the interaction of NPAS2 with the histone acetyltransferase p300 (28). Abundance of H3K4 methylation in specific promoters also changes over a 24 hour period due to interactions of the circadian genes with the histone methyltransferase MLL (29). In our present study, we wanted to determine if the HF diet dependent changes in both histone modification abundance, and localization to the Npas2 promoter that we observed in fetal life were maintained postnatally in our juvenile cohort.
We hypothesized that animals exposed in utero to a HF diet may maintain the increased H3K14 acetylation postnatally. Using histone modification specific antibodies, we measured H3K14ac abundance in juvenile liver. Consistent with our prior observations in the fetal liver, we observed that the histone code in juvenile animals is modified by both maternal and postweaning high fat diet. Of interest, however, were the site and nature of these histone modifications. Specifically, we found that in the juvenile, H3K14 acetylation did not differ significantly with maternal or postnatal high fat diet exposure compared with CTR/CTR (Figure 3B). However, another permissive modification, H3K4me3, was altered by virtue of diet exposure. Specifically, we found that levels were slightly but significantly decreased in both the CTR/HF and the HF/CTR groups compared with CTR/CTR (Figure 3A). Because H3K4me3 is altered with both CTR and HF in utero exposures, we cannot conclude that the in utero environment is responsible for the reprogramming of the juvenile hepatic histone code. Nor can chronic exposure to a HF diet be implicated, as the HF/HF group did not show a difference in abundance of this modification. Potentially, a disconnect between in utero and postnatal environments may contribute to the alterations in H3K4me3. Interrogation of the abundance of two repressive histone modifications, H3K9me3 and H3K27me3, revealed no difference (Figure 3C and D). Taken together and in consideration of our prior observations, these findings suggest that the fetal histone code is modified at H3K14 in association with maternal high fat diet intake while the juvenile code is modified at H3K4me3. We find it of significance that H3K14 appears to be modifiable specifically in fetal life; this finding is consistent with others observations regarding developmental specificity to the histone code(30).
Our unique model system gives us insights into the potential mechanisms which may lie behind the discrepancy we observe between our fetal and juvenile cohorts. We have recently published that the abundance and activity of the histone deacetylase, SIRT1, is decreased in the fetal liver with HF diet exposure (17). We also found that SIRT1 deacetylates H3K14ac in vitro. While we believe that SIRT1 may play a role in the increased H3K14ac seen in fetal life, we have not tested the abundance and activity of SIRT1 in the juvenile liver. It would be of interest to test whether these changes in SIRT1 level and activity are stable in the juvenile liver, and if not, which specific HMTases are responsible for the increase in H3K4me3 observed in the juvenile liver.
Differences between the fetal and postnatal characteristics may also play a role in changes in the epigenetic milieu of the liver during development. In our NHP model system, we have found that fetuses exposed to a maternal HF diet weigh 10% less at the beginning of the third trimester than those exposed to a CTR diet (13). This is likely due to lower lean body mass. However, at both postnatal day 30 and 180, these animals weigh the same as control diet exposed animals but have almost twice the body fat, as determined by DEXA, compared with controls. In these juvenile animals we have also found an increased plasma insulin level and an abnormal cardiovascular morphology (31). Perhaps the accumulation of white adipose tissue and the disparity in body composition from the third trimester to juvenile life in these animals is helping to orchestrate the alterations we observe in the hepatic histone code.
We sought to determine the mechanism behind this decrease in H3K4me3 levels, and hypothesized that this decrease could be due to lower levels of HMTase activity (32). Using a commercially available kit, we specifically measured histone H3 lysine 4 trimethyltransferase activity from hepatic lysates. Rather than finding decreased HMTase activity in the CTR/HF and HF/CTR groups which would account for the decreased trimethylation, we found that activity was significantly increased in the HF/HF group (Figure 4A). This could potentially explain why this cohort did not exhibit a decrease in H3K4me3 abundance by Western blot as the increased activity may be resultantly affecting the measurement of histone methyl group stability. We have also previously shown that HDAC activity is decreased in HF diet exposed fetal liver, compared with CTR (12). We sought to determine if this decrease in activity persists postnatally with in utero HF diet exposure. We found that HDAC activity did not change in any group tested compared with CTR/CTR which is consistent with our observations regarding acetylation (Figure 4B).
Based on our previous findings that the permissive modification, H3K14ac, was enriched in the promoter of Npas2, in the HF diet exposed fetal liver, we had hypothesized that such a permissive modification may be similarly enriched in the juvenile cohort. Due to our findings in this study that the abundance of H3K4me3 is altered while H3K14ac is not, we hypothesized that perhaps H3K4me3 would be enriched in the Npas2 promoter. Because we have found that expression of Npas2 in juvenile animals is increased with a postweaning HF diet, and not by in utero HF diet exposure, (15) we believed that H3K4me3 would be enriched specifically in these two groups. However, we only found increased enrichment only in the HF/HF group (Figure 5A). Our ChIP analysis utilizing an antibody for H3K14ac did not reveal the differential promoter occupancy we observed in fetal life. However, using antibodies against two repressive modifications demonstrated differential enrichment in the RORE of Npas2. Both H3K9me3 and H3K27me3 are depleted in the groups which also show increased expression, CTR/HF and HF/HF (Figure 5C and D). Although these results were unexpected, the depletion of repressive modifications in these two groups correlates with the higher expression of Npas2.
We have previously characterized epigenetic changes to the fetal hepatic histone code. We found that H3K14ac is an important molecular mediator in the fetal liver exposed to maternal HF diet. However, given our current data, we speculate that this modification may be specific for fetal life. In our four postweaning diet exposure groups, we find that only H3K4me3 abundance is altered by virtue of diet exposure in juvenile liver. Furthermore, the change in HDAC activity we reported in fetal life does not persist, but instead we find an increase in H3K4 trimethylase activity. Finally, we find that rather than the Npas2 RORE being enriched for permissive histone modifications in the cohorts which have an increased Npas2 expression, we find it is depleted for repressive modifications.
Our data reveal that postweaning diet influences the epigenomic signature of the individual. The epigenetic changes we have reported in fetal life, namely increased H3K14ac abundance and decreased HDAC activity with HF diet exposure, do not persist postnatally. Instead, we report that novel histone modifications are altered by virtue of postweaning diet exposure. We find alterations in the abundance of H3K4me3 and in HMTase activity, and observe differential promoter occupancy of repressive histone modifications. Because we cannot conclude that site specific epigenetic alterations which occur during fetal life maintain the memory of the in utero exposure, we must determine how epigenetic changes in fetal life influence the alterations postnatally.
While we have not studied the circadian behavior of our juvenile cohort, we have found that the histone occupancy of the promoter region, and the expression level of Npas2 is altered with HF diet exposure in fetal and juvenile life. We believe these alterations to an important circadian rhythm transcription factor warrant further investigation into the transcriptional, epigenetic, and perhaps even behavioral consequences of HF diet consumption during pregnancy. The findings herein lay the foundation for future investigations, and suggest that the while the histone code is developmental stage specific, our initial overarching observations persist. In sum, a maternal high fat diet modifies both the fetal and postnatal histone code and postweaning exposure to a high fat diet similarly modifies histones in a type- and site-specific manner.
Acknowledgments
This work was supported by the National Institutes of Health (NIH, Bethesda, MD) Director New Innovator Award (DP2120OD001500-01 K.A.), NICHD/NIDDK #R01DK080558-01 (K.A.), and the NIH REACH IRACDA K12 GM084897 (M.S.).
Footnotes
The authors have neither conflicts of interest nor financial disclosures.
Presented at the 31st Annual Meeting of the Society for Maternal Fetal Medicine, San Francisco, California, February 10, 2011.
References
- 1.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suter MA, Aagaard-Tillery KM. Environmental influences on epigenetic profiles. Semin Reprod Med. 2009;27:380–90. doi: 10.1055/s-0029-1237426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–3. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 4.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127–34. doi: 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011;2011:371832. doi: 10.1155/2011/371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kota SK, Feil R. Epigenetic transitions in germ cell development and meiosis. Dev Cell. 2010;19:675–86. doi: 10.1016/j.devcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–6. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 9.Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–73. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tobi EW, Lumey LH, Talens RP, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 12.Aagaard-Tillery KM, Grove K, Bishop J, et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008;41:91–102. doi: 10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119:323–35. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox J, Williams S, Grove K, Lane RH, Aagaard-Tillery KM. A maternal high-fat diet is accompanied by alterations in the fetal primate metabolome. Am J Obstet Gynecol. 2009;201:281, e1–9. doi: 10.1016/j.ajog.2009.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suter M, Bocock P, Showalter L, et al. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J. 2011;25:714–26. doi: 10.1096/fj.10-172080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frias AE, Morgan TK, Evans AE, et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology. 2011;152:2456–64. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suter MA, Chen A, Burdine MS, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012 doi: 10.1096/fj.12-212878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suter MA, Sangi-Haghpeykar H, Showalter L, et al. Maternal High-Fat Diet Modulates the Fetal Thyroid Axis and Thyroid Gene Expression in a Nonhuman Primate Model. Mol Endocrinol. 2012 doi: 10.1210/me.2012-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto T, Nakahata Y, Soma H, Akashi M, Mamine T, Takumi T. Transcriptional oscillation of canonical clock genes in mouse peripheral tissues. BMC Mol Biol. 2004;5:18. doi: 10.1186/1471-2199-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan EL, Grayson B, Takahashi D, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010;30:3826–30. doi: 10.1523/JNEUROSCI.5560-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–8. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 22.Lillycrop KA, Burdge GC. Epigenetic changes in early life and future risk of obesity. Int J Obes (Lond) 2011;35:72–83. doi: 10.1038/ijo.2010.122. [DOI] [PubMed] [Google Scholar]
- 23.Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2008;294:R528–38. doi: 10.1152/ajpregu.00316.2007. [DOI] [PubMed] [Google Scholar]
- 24.Rudyk O, Makra P, Jansen E, Shattock MJ, Poston L, Taylor PD. Increased cardiovascular reactivity to acute stress and salt-loading in adult male offspring of fat fed non-obese rats. PLoS One. 2011;6:e25250. doi: 10.1371/journal.pone.0025250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Strakovsky R, Zhou D, Zhang Y, Pan YX. A maternal high-fat diet represses the expression of antioxidant defense genes and induces the cellular senescence pathway in the liver of male offspring rats. J Nutr. 2011;141:1254–9. doi: 10.3945/jn.111.139576. [DOI] [PubMed] [Google Scholar]
- 26.Kohsaka A, Laposky AD, Ramsey KM, et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell metabolism. 2007;6:414–21. doi: 10.1016/j.cmet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Feng D, Lazar MA. Clocks, metabolism, and the epigenome. Molecular cell. 2012;47:158–67. doi: 10.1016/j.molcel.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature. 2003;421:177–82. doi: 10.1038/nature01314. [DOI] [PubMed] [Google Scholar]
- 29.Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. 2010;17:1414–21. doi: 10.1038/nsmb.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab. 2010;21:223–9. doi: 10.1016/j.tem.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan L, Lindsley SR, Comstock SM, et al. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. International journal of obesity. 2012 doi: 10.1038/ijo.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teperino R, Schoonjans K, Auwerx J. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 2010;12:321–7. doi: 10.1016/j.cmet.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]