

SUMMARY
Research advancing our understanding of Mycobacterium tuberculosis (Mtb) biology and complex host-Mtb interactions requires consistent and precise quantitative measurements of Mtb proteins. We describe the generation and validation of a compendium of assays to quantify 97% of the 4,012 annotated Mtb proteins by the targeted mass spectrometric method selected reaction monitoring (SRM). Furthermore, we estimate the absolute abundance for 55% of all Mtb proteins, revealing a dynamic range within the Mtb proteome of over four orders of magnitude, and identify previously un-annotated proteins. As an example of the assay library utility, we monitored the entire Mtb dormancy survival regulon (DosR), which is linked to anaerobic survival and Mtb persistence, and show its dynamic protein-level regulation during hypoxia. In conclusion, we present a publicly available research resource that supports the sensitive, precise, and reproducible quantification of virtually any Mtb protein by a robust and widely accessible mass spectrometric method.
INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), one of the most devastating infectious diseases. One-third of the world’s population is latently infected with Mtb, and ~1.4 million people die from the disease each year (WHO, 2012). In immunocompetent individuals, Mtb is contained in granulomatous lesions in the lungs by the host immune system, and thus Mtb infection is controlled in a clinically silent state, termed latency (Gengenbacher and Kaufmann, 2012). When the host immune system is compromised, dormant bacilli are resuscitated and cause active TB. Latent Mtb infections are difficult to treat and can persist lifelong. In vitro culture systems mimicking aspects of the complex host environment, such as exposure to hypoxia and nitric oxide, led to the identification of a phenotypically drug-tolerant, nonreplicating form of Mtb and the underlying genetic program controlled by the dormancy survival regulon (DosR) (Boon and Dick, 2002; Park et al., 2003; Voskuil et al., 2003). Several studies have shown that DosR is required for survival during anaerobic dormancy in vitro and that it is induced during infection of macrophages, in animal models and in humans, during active and latent Mtb infection (Boon and Dick, 2012). Because of its implication in Mtb persistence and latent TB, the DosR regulon has been widely studied, but most studies to date have focused on transcriptional regulation, and only limited quantitative data are available for the respective proteins.
To advance the understanding of Mtb biology during infection, survival, and persistence, the reproducible quantification of the proteins that catalyze and control these processes is critically important. To date, two predominant strategies have been used to measure Mtb proteins; the first is based on affinity reagents such as antibodies and includes methods like western blotting and ELISA. While these methods are robust and well established, affinity reagents are only available for a small subset of Mtb proteins, and their development and validation are expensive, time-consuming, and challenging to scale to high throughput. The high effort to generate these affinity reagent-based protein assays has moreover led to a bias in research activity toward where there are such high-quality tools available, leaving less-accessible research areas mostly unexplored (Edwards et al., 2011). The second, more exploratory strategy is mass spectrometry (MS)-based proteome discovery. Some of the studies using this technique have reached remarkably high levels of qualitative proteome coverage and improved Mtb genome annotation (de Souza et al., 2011; Jungblut et al., 2001; Kelkar et al., 2011). However, in contrast to the quantitative analysis of the Mtb transcriptome, quantitative proteomic measurements have not yet become routine and, due to technical limitations, still lag behind genomic techniques in number, depth, and consistency. The aim of this study, therefore, was to generate a publicly accessible research resource that makes essentially any protein of the Mtb proteome quantifiable in complex samples via a robust and widely accessible MS technique, thus supporting unbiased protein-based research in Mtb, beyond the restrictions imposed by the limited availability of antibodies.
Today MS offers a range of mature methods for the identification and quantification of proteins and proteomes. They have in common that proteins are enzymatically digested into peptides, which are then separated in time by reverse-phase liquid chromatography, ionized, and injected into the mass spectrometer (Aebersold and Mann, 2003). To date, discovery-driven MS, also known as shotgun MS, has been most widely used for qualitative and quantitative measurements. This technique generally aims at maximizing proteome coverage, but its focus on scale generates some trade-offs related to the technical complexity of the method, its reproducibility, quantitative accuracy, and the number of samples that can be measured with reasonable effort. In contrast, the more recently developed targeted MS techniques are focused on the highly accurate and reproducible measurement of predetermined sets of proteins across many samples and therefore optimally interface with the requirements of hypothesis-based research projects. Currently, the gold standard targeting method is selected reaction monitoring (SRM), also known as multiple reaction monitoring (MRM). In SRM mode the instrument is instructed to monitor predefined combinations of precursor and fragment ions (transitions) continuously over time. These MS coordinates, together with the chromatographic retention time of the peptides, constitute a definitive assay for the detection and quantification of the target peptide in a sample and have to be determined prior to the actual experiment for each protein of interest. Thanks to its high selectivity and sensitivity, SRM allows accurate and highly reproducible measurements of proteins covering a large dynamic range (Cima et al., 2011; Picotti et al., 2009). In recent years, major advances have been accomplished in many aspects of SRM-based targeted proteomics, including improvements in MS instrumentation, advances in the generation of SRM assays (Picotti et al., 2010), and the development of computational tools to support SRM method preparation and data analysis (Chang et al., 2012; MacLean et al., 2010a; Reiter et al., 2011).
To eliminate the need for tedious SRM assay generation before each experiment, a strategy based on high-throughput synthesis of peptides representing every protein of a proteome has recently been presented for the model organism yeast (Picotti et al., 2013). Here we apply this approach to an organism of high clinical relevance and expand it by validating every SRM assay in an unfractionated mycobacterial lysate. The resulting Mtb Proteome Library represents a public resource of definitive assays for the quantification of Mtb proteins in complex biological samples. It can be applied by every researcher with access to appropriate MS instrumentation and greatly enhances the range of Mtb proteins amenable to sensitive and reproducible quantitative analysis.
The Mtb Proteome Library contains SRM assays for 97% of all annotated proteins of Mtb; for 72% of the proteome, the generated SRM assays could be validated. Furthermore, absolute protein abundances were estimated for 55% of all theoretical Mtb proteins, revealing a dynamic range of over four orders of magnitude. Finally, we deployed the Mtb Proteome Library to obtain protein-level insights into the dynamics of the DosR regulon in an in vitro model of dormancy.
RESULTS
Generation of the Mtb Proteome Library followed a three-phase workflow as outlined in Figure 1.
Figure 1. The Mtb Proteome Library Workflow in Three Phases.

(A) Phase I, Proteome Mapping: After harvesting bacterial cultures, proteins are extracted and digested with the proteolytic enzyme trypsin. The resulting peptides are separated into 24 fractions using off-gel isoelectric focusing to reduce sample complexity, and each fraction is analyzed by discovery-driven MS. Peptide identifications can then be used to infer proteins that were present in the sample. The peptide and protein identifications, as well as the corresponding spectra, can be browsed interactively in the PeptideAtlas database (http://www.PeptideAtlas.org).
(B) Phase II, Proteome Library Generation: From the collected data, the most MS-suited, unique peptides are selected for every annotated protein of Mtb. For proteins that have never been observed previously, representative peptides are predicted. The peptides are synthesized, pooled in mixes of 96, and analyzed in SRM-triggered MS2 mode (SRM-MS2). From the resulting spectra the most intense fragment ions, as well as the chromatographic retention times, can be extracted. These mass spectrometric coordinates, called SRM assays, constitute the synthetic Mtb Proteome Library and can be downloaded from the SRMAtlas database (http://www.SRMAtlas.org).
(C) Phase III, Proteome Library Validation: The SRM assays in the synthetic Mtb Proteome Library are validated for the detection of proteins in unfractionated mycobacterial lysates by SRM. The resulting quantitative SRM traces and statistical scores can be viewed in the PASSEL database (http://www.PeptideAtlas.org/passel).
Mapping the MS-Accessible Proteome of Mtb by Discovery MS
As a first step toward the Mtb Proteome Library, we defined the MS-observable fraction of the annotated Mtb proteome (Phase I, Figure 1A). To capture a broad spectrum of physiological states, we harvested Mtb H37Rv cultures at exponential and stationary growth. A peptide fractionation step prior to MS analysis was applied to increase identification rates by reducing sample complexity. This strategy allowed us to assign 144,175 fragment ion spectra to 36,924 peptides mapping to 3,074 (77%) of the annotated 4,012 proteins of Mtb at a protein false discovery rate (FDR) of <1% (Figure 2A).
Figure 2. Defining the MS-Accessible Proteome of Mtb by Discovery MS.
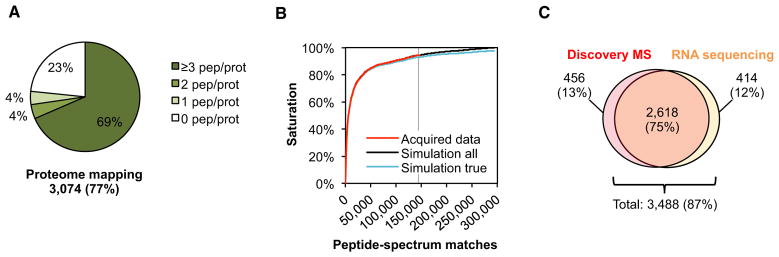
(A) Proteome coverage and distribution of the peptides detected by discovery MS in an extensively fractionated lysate of Mtb during exponential and stationary growth.
(B) Proteome saturation analysis showing the progression of protein identifications as a function of identified spectra. The red line represents data acquired from the fractionated Mtb lysate; the black and blue lines represent the simulated progressions of all identifications and the true positive identifications at an FDR of 1%. The vertical gray line marks the size of our data set in terms of acquired peptide-spectrum matches. One hundred percent proteome saturation is defined at 300,000 peptide-spectrum matches.
(C) Comparison showing the overlap of the protein identifications with the transcriptome determined by RNA sequencing (Arnvig et al., 2011). The numbers represent the total identifications in exponential and stationary phase cultures for both technologies.
Due to the stochastic nature of discovery MS, only a fraction of a complex proteome is identified in a single run. Thus, repeated analyses on the same sample result in different sets of identified proteins and lead to an increase in proteome coverage, the true progression and saturation of which are nontrivial to quantify. Recently, we developed a statistical approach to estimate the saturation level of peptide and protein identifications in such MS experiments (Claassen et al., 2011). Applying this analysis to the data acquired on the 24 Mtb fractions confirmed that 95% of the MS-observable proteome was reached (Figure 2B).
Complete proteome coverage in a biological sample is inherently limited by actual gene expression in a cell. In-depth transcriptome analyses based on RNA deep sequencing of exponential- and stationary-phase Mtb cultures have shown that <80% of the annotated genome is actually expressed in these two cellular states (Arnvig et al., 2011). Our proteome data set covers 86% of these RNA sequencing data (Figure 2C).
By means of extensive discovery-driven MS, we have thus created a comprehensive protein database that covers nearly the entire expressed Mtb proteome. These data were combined with other available MS data on Mtb and made publicly accessible in the PeptideAtlas database (http://www.PeptideAtlas.org), where it can be browsed on the protein, peptide, and individual spectrum levels through a web interface (Deutsch, 2010).
Mtb Proteome Library Generation Based on Synthetic Peptides
In a second step, we generated SRM assays for all annotated proteins of Mtb using synthetic peptides (Phase II, Figure 1B). We thus selected MS-suitable proteotypic peptides, representing the entire Mtb proteome, for chemical synthesis as described earlier (Picotti et al., 2010). Peptides that have previously been detected in the proteome mapping phase were prioritized, but if no or insufficient empirical evidence was available for a given protein, a combination of prediction algorithms was used to determine its best-suited peptides. In total, 17,463 peptides covering 3,930 proteins were selected and synthesized (see Table S1 online). The remaining 2% of the annotated Mtb proteins do not have unique peptides meeting the applied selection criteria (see the Experimental Procedures). The synthetic peptides were pooled into samples of 96 and analyzed on a Qtrap mass spectrometer. We thus acquired highly specific and minimally interfered spectra for 15,679 peptides covering 3,894 (97%) of the annotated Mtb proteins (Figure 3A and Figures S1A–S1C). Importantly, the majority of the proteins are represented by three or more peptides. The synthetic peptides were analyzed likewise on two additional instrument types, an Orbi-trap and a TripleTOF mass spectrometer, to support all conceivable MS workflows which require MS coordinates. For each peptide precursor ion in the resulting spectral libraries, we extracted the most intense peptide fragment ions, their relative intensities, and the chromatographic retention time, which together constitute an SRM assay. These MS coordinates of all three instrument platforms are compiled in the SRMAtlas database (http://www.srmatlas.org), where they can be browsed and downloaded through a web interface (Picotti et al., 2008).
Figure 3. Generation and Validation of the Mtb Proteome Library.
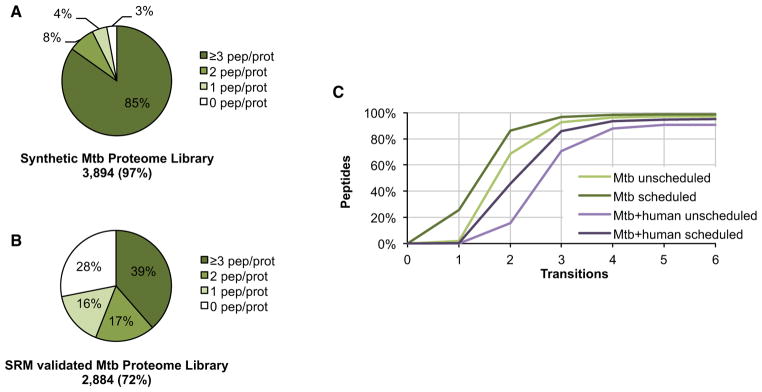
(A) Proteome coverage and distribution of the 15,679 synthetic peptides for which a fragment ion spectrum, and thus an SRM assay, could be obtained.
(B) Proteome coverage and distribution of the 7,094 peptides for which the synthetic SRM assay could be validated by SRM in a mixed unfractionated Mtb lysate of exponential and stationary phase cultures.
(C) Theoretical specificity of SRM assays determined by the SRMCollider algorithm is shown as a cumulative plot of the number of peptides which can be uniquely identified with a given number of transitions. Transitions were selected with decreasing intensity. Scheduled indicates that only background peptides with a retention time close to the target peptide are taken into consideration as interfering background.
See also Figures S1 and S2 and Table S1.
Mtb Proteome Library Validation in Whole-Cell Lysates
The performance of the SRM assays in a biological sample was validated in an unfractionated mixed lysate of Mtb H37Rv cultures grown to exponential and stationary phase, respectively (Phase III, Figure 1C). If available, six transitions were measured per peptide precursor. This led to the identification of 7,094 (41%) of the targeted mycobacterial peptides covering 2,884 (72% of 4,012) proteins at an FDR of 1% (Figure 3B and Table S1). This stringent cutoff resulted in a low number of false positives but came at the price of false negatives (Figures S1D and S1E). The comparison of the proteome coverage reached by SRM and discovery proteomics shows that 88% of the MS-observable proteome defined by discovery MS of extensively fractionated Mtb lysates was amenable to SRM analysis even without prior fractionation, and that 172 proteins were exclusively detectable by SRM (Figures S2A and S2B). Proteins identified by only one of the two MS techniques are generally less abundant. While SRM is more sensitive, the reduced complexity of the fractionated lysate could explain identification of the 362 proteins solely detected by discovery MS. We could confirm that there was no bias against any subcellular localization, even though we did not apply specific protocols to extract or enrich for specific protein classes (Figure S2C). Note that failed validation of an SRM assay does not necessarily mean that it is not functional. The absence of a validation signal is more likely to indicate minute or absent protein expression under the culture conditions used. The SRM traces, together with the statistical confidence score, can be browsed interactively in the PeptideAtlas SRM Experiment Library (PASSEL, http://www.peptideatlas.org/passel) (Farrah et al., 2012).
Theoretical Assessment of SRM Assay Specificity
To compute the number of transitions needed to unambiguously identify an Mtb peptide in a bacterial extract, we applied the SRMCollider (Röst et al., 2012). This algorithm computes potentially interfering transitions from the in silico digested and fragmented proteome and can thus estimate the specificity of an SRM assay. For Mtb whole-cell lysates the three most intense transitions per peptide are in >90% of the cases sufficient to identify a peptide in unscheduled as well as scheduled acquisition mode (Figure 3C). However, in order to target Mtb proteins in a sample also containing a human proteome, e.g., in samples from infected macrophages, at least five of the most intense transitions are needed to unambiguously identify a peptide in >90% of the cases. These theoretical considerations also reveal that the addition of retention time information generally increases the specificity of an SRM assay to an extent similar to that of an additional transition. If measurement time and sample size are sufficient, we recommend acquiring several peptides per protein and a few extra transitions per peptide rather than the minimal numbers suggested by the SRMCollider. On the one hand, this is because the algorithm makes certain assumptions which cannot, in all cases, precisely reflect true conditions; on the other hand, for quantitative SRM studies it is advantageous to exceed the minimal number of transitions actually needed for identification to be able to select, after the measurement, a subset of transition signals that does not contain any interferences—an essential requirement for accurate quantification.
Proteome-wide Absolute Abundance Estimates for Mtb
It has been shown recently that SRM signal intensities of the best flying peptides per protein correlate linearly with absolute protein concentrations (Ludwig et al., 2012). To estimate the concentrations of all proteins identified in the SRM data set, we first selected 34 anchor proteins spanning a wide abundance range of the Mtb proteome and accurately determined their absolute concentrations using isotopically labeled synthetic reference peptides. We then used these anchor proteins to identify and optimize the linear correlation of the ion current signal and the absolute protein abundance in fmol/μg cellular protein extract (Figures S3A–S3C). As proteins represented by fewer than two peptides showed significantly lower quantification accuracy, they were excluded from further analyses. The here-described strategy allowed us to estimate absolute concentrations for 2,195 (55%) of the annotated Mtb proteins with an estimated mean fold error of 2.1 ± 0.6 (Table S2). Mtb protein abundances span four orders of magnitude (Figure 4A). There are only a few very highly abundant proteins, while the majority is within a range of two orders of magnitude. By far the most abundant protein is the chaperonine GroEL2, followed by its close homolog GroEL1 and cognate GroES (Figure 4B).
Figure 4. Proteome-wide Absolute Abundance Estimates for Mtb.

SRM-based absolute label-free abundance estimates for every protein identified by SRM with two or more peptides (2,195 proteins). The abundance estimate has a mean fold error of 2.1 ± 0.6.
(A) Abundance distribution of all quantified proteins.
(B) Absolute concentrations of the ten most abundant proteins in Mtb.
(C) Absolute concentrations mapped on selected protein classes and the metabolic network of Mtb (http://pathways.embl.de/iTuby). Colors correspond to the ones in (A).
(D) Abundance distribution among the functional classes of Mtb as defined in TubercuList. The first and second rows show the distribution of genes in the genome and of the quantifiable proteins, respectively. The third row shows the relative protein concentration for each functional class.
Abbreviations are as follows: Met, metabolism; TCA, tricarboxylic acid cycle; CH, carbohydrate; AA, amino acid; and Vit, vitamine and cofactor. See also Figure S3 and Table S2.
From the pathway map iTuby (http://pathways.embl.de/iTuby), it becomes apparent that the central carbon metabolism, including the TCA cycle, as well as lipid and amino acid metabolism, are in the upper abundance range, whereas components of the nucleotide, vitamin, and cofactor metabolism are less abundant (Figure 4C). As expected, most ribosomal proteins are highly abundant, while signaling pathways, such as the two-component systems, are generally of low abundance. Also ABC transporters and components of the bacterial secretion system are in the mid- to low-abundance range of the Mtb proteome. Figure 4D shows the distribution of proteins according to the ten functional classes as defined in TubercuList and recently extended (Doerks et al., 2012). The distribution of functional classes among the proteins that could be quantified shows homogenous representation of all classes except for members of the PE/PPE family and insertion sequences and phages. The absolute concentrations of proteins multiplied by their length can be used as a proxy for the fraction of the total cellular synthesis budget spent on each functional class (Malmström et al., 2009). This analysis shows that Mtb generally invests a large synthesis budget into information pathways as well as virulence, adaptation, and detoxification strategies. Conversely, cell wall and cell processes, conserved hypotheticals, and regulatory proteins have a below-average synthesis budget.
Time-Resolved Protein Level Regulation of the DosR Regulon under Hypoxic Stress
To assess the utility of the Mtb Proteome Library, we interrogated the protein level regulation of the DosR regulon in response to hypoxic conditions in a standing culture model using the closely related but attenuated strain M. bovis BCG. Figure S5 summarizes the recommended procedure to progress from a list of proteins of interest to a refined transition list. We gathered SRM assays for 51 out of 52 members of the DosR regulon from the Mtb Proteome Library. Additionally, the sensor histidine kinase DosT, which is also part of the DosR/S two-component system but not part of the DosR regulon, was included in the target list. Every target protein was represented by up to three peptides with five transitions per peptide (Table S3). In total, 45 out of 53 proteins (85%) could be confidently quantified over the hypoxic time course (Figures 5A and 5B). Of these, nine proteins were not previously validated because they were expressed below detection limits in exponential- and stationary-phase cultures.
Figure 5. Time-Resolved Protein Level Regulation of the DosR Regulon under Hypoxic Stress.

(A) Growth curve of three exponentially growing M. bovis BCG cultures subjected to hypoxic conditions in a standing culture model. Agitation of shake-flask cultures was stopped at day 0 and resumed after 6 days (reaeration). (A)–(C) represent biological replicates.
(B) Summary table of the DosR regulon study.
(C and D) Fold changes versus day 0 as determined by SRM using heavy isotope-labeled reference peptides for each protein. The data were subjected to hierarchical clustering, and each of the ten clusters was colored differently (corresponding colors in C and D).
(E) For two operons of the DosR regulon the protein fold changes after 4 days of exposure to hypoxia are shown. Error bars represent the standard error.
See also Figures S4 and S5 and Table S3.
The SRM data revealed that many proteins were already induced 6 hr after stopping agitation (Figure 5C). Protein levels reached close to the maximal induction after day 1 and then remained stable over 5 days until reagitation of the cultures. The proteins were clustered according to the dynamics and intensity of regulation (Figure 5D). Following reaeration, a drop in protein levels coinciding with the resumption of growth could be observed. Clearly, the notorious chaperonine HspX was most strongly induced (340-fold), followed by Hrp1 (153-fold), with protein levels peaking at day 4. Interestingly, the transcription factor DosR itself was upregulated later than other members of the DosR regulon, indicating regulation based on mechanisms other than mere gene expression. The sensor histidine kinase DosT remained unchanged, while DosS expression correlated tightly with the induction of DosR. Most DosR regulon members are located in nine discrete genomic locations where they are arranged singly or in operons (Figure S4) (Chauhan et al., 2011). Figure 5E depicts two operons showing the typical staircase gene expression pattern with decreasing levels the further away from the promoter a gene is located. This pattern is indicative of a predominantly transcriptional control in the DosR regulon induction.
In conclusion, we could identify and quantify almost the complete DosR regulon using the Mtb Proteome Library, thereby facilitating straightforward SRM experiments by providing high-quality and ready-to-use quantitative protein assays together with information on detectability of each peptide in standard cultures.
Previously Unannotated Proteins Identified by Proteogenomics
The current version of the Mtb Proteome Library is based on the Mtb H37Rv genome annotation (TubercuList version 2.3). It is conceivable that the proteomic data sets generated contain fragment ion spectra that could add to or correct protein-coding genes in Mtb. We therefore translated the Mtb genome in the six reading frames and searched the discovery MS data set obtained in the proteome mapping phase against this database. As the FDR is more difficult to control if such a large database is used, only proteins with at least two peptide hits at an FDR of <1% were considered. We identified 29 proteins that have not yet been annotated in the Mtb strain H37Rv (Table S4). These proteins were included in the Mtb Proteome Library workflow, and 22 (76%) of them could be validated by SRM in an unfractionated mycobacterial lysate. In Figure 6A, one of the unannotated proteins is shown which was detected by eight peptides. Our results emphasize that the genome annotation of Mtb H37Rv is still incomplete and that it is worthwhile to consider expanding the search database by these previously unannotated proteins.
Figure 6. Proteogenomics Combined with the Mtb Proteome Library Reveals Previously Unannotated Proteins.
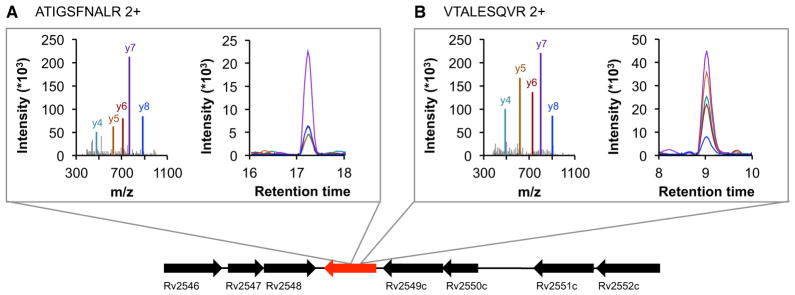
(A) and (B) exemplify two peptides that belong to a protein, identified by proteogenomics, which has so far not been annotated in Mtb H37Rv. Spectra were acquired from synthetic peptides, whereas SRM traces were acquired from endogenous peptides in a whole-cell lysate.
See also Table S4.
Transferability of the Mtb Proteome Library to Other Mycobacterial Strains
The Mtb Proteome Library was developed for the laboratory strain Mtb H37Rv, but it can also be used to investigate proteins in clinical Mtb strains and closely related members of the Mtb complex, such as the vaccine strain M. bovis BCG or M. africanum (Table 1). The proteome of strains belonging to the Mtb complex are well covered by the assays (94%), whereas the proteomes of more distantly related strains are only partially covered.
Table 1.
Transferability of the Mtb Proteome Library to Other Mycobacterial Strains
| Strain (Number of Annotated ORFs) | Peptides | Proteins | Proteome Coverage (%) |
|---|---|---|---|
| M. tuberculosis H37Rv (4012)a,b | 15,679 | 3,894 | 97 |
| M. tuberculosis H37Rv (3989)b | 15,609 | 3,834 | 96 |
| M. africanum (3830)b | 14,733 | 3,672 | 96 |
| M. bovis (3920)b | 15,007 | 3,762 | 96 |
| M. bovis BCG (3952)b | 14,884 | 3,722 | 94 |
| M. tuberculosis CDC1551 (4189)b | 14,680 | 3,642 | 87 |
| M. leprae (1602) | 951 | 639 | 40 |
| M. ulcerans (4241) | 1,901 | 1,249 | 30 |
| M. marinum (5452) | 2,245 | 1,466 | 27 |
| M. smegmatis (6716) | 978 | 709 | 11 |
Table showing for several mycobacterial strains the number of peptides and proteins that are covered in the Mtb Proteome Library, as well as the corresponding proteome coverage. Abbreviations are as follows: ORF, open reading frame; M., Mycobacterium.
The first entry of Mtb H37Rv represents the genome annotation in Tuber-cuList, which was used throughout this study. For all other strains, the NCBI RefSeq genome annotation was used.
Members of the Mtb complex.
DISCUSSION
More efficient intervention measures are urgently needed for better TB control, notably with respect to increasing incidences of drug-resistant strains. However, their development is in part hampered by the incomplete understanding of the molecular mechanisms underlying infection, survival, and persistence of Mtb in the human host. Systems-biological approaches promise to fill knowledge gaps by capturing emergent properties of systems that are not readily apparent from studying isolated components. Proteins are critical players in such systems, as they constitute the functional link between genotype and phenotype, and are attractive drug targets. The consistent quantification of proteins over a large range of differentially perturbed states is a fundamental component of any systems-biology strategy. An attractive tool facilitating such measurements is the targeted MS technique SRM, because it allows accurate quantification of a defined set of proteins with high sensitivity and reproducibility over many samples.
Here we generated the Mtb Proteome Library containing high-quality SRM assays for 97% of all annotated Mtb proteins. Given access to an SRM-capable mass spectrometer, these SRM assays can be directly applied to many proteins in parallel and to any number of samples without the need to invest in the time-consuming and costly development of antibodies.
We were able to detect more than 80% of the annotated Mtb proteome by MS (discovery MS and SRM combined). A recent proteomic study has reported a similarly high coverage of the Mtb proteome, and the overlap between the two is very high (2,948 proteins) (Kelkar et al., 2011). Approximately 20% of the annotated proteins in Mtb could be detected neither by discovery MS nor by SRM. There are three main reasons for the absence of protein-level evidence for predicted coding sequences. First, some proteins are intrinsically not detectable by MS due to their sequence properties; i.e., they do not give rise to unique tryptic peptides of MS-suitable length and hydrophobicity or are not extracted from the cell lysate. Second, as we and others have shown, the genome annotation of Mtb H37Rv is still incomplete, and it is therefore possible that several annotated coding sequences are in fact pseudogenes or annotated in the wrong reading frame (Arnvig et al., 2011; de Souza et al., 2011; Kelkar et al., 2011). Third, certain genes are only transcribed or translated in response to specific in vivo stimuli and are not induced during normoxic exponential and stationary growth in rich medium—the conditions used for the discovery MS and SRM validation measurements. This latter reason was the case for eight proteins of the DosR regulon, which only reached detectable levels under the hypoxic conditions obtained in the standing culture model.
Overall, we could show that the Mtb proteome coverage approaches not only technical limits but also biological (i.e., gene expression) limits, as it closely resembles the expressed fraction of the transcriptome as determined by RNA deep sequencing (Arnvig et al., 2011). Gene products detected exclusively on RNA level are enriched for low abundant ones and membrane proteins, which are often difficult to extract and prevent from precipitation. Gene products detected only by MS may result from unstable RNAs and very stable proteins.
The absolute label-free quantification strategy applied to the extensive SRM data set allowed us to estimate absolute protein concentrations for a large fraction of the Mtb proteome, giving insight into the average composition of the protein content of the cell and its distribution over different functional classes. It is important to note that in particular for mycobacteria, the complete extraction of proteins from the membrane and cell wall is difficult, and consequently the concentrations for these types of proteins could be underestimated compared to soluble proteins.
Absolute protein abundance estimates of a protein extract can be converted into copies per cell using the total cellular protein content, which has been determined for the closely related M. bovis BCG to be within 38 and 72 femtograms (Beste et al., 2005). However, this conversion is only valid if both exhaustive protein extraction and absence of any protein or peptide loss during sample preparation can be ensured. Making these assumptions, the protein copy numbers per cell in Mtb span a range from a few to more than 20,000 copies per cell, correlating well with numbers determined recently for other bacteria. For instance, the ubiquitous chaperonine GroEL in Leptospira interrogans was determined to be present with 19,000 copies, whereas for Mtb 21,000 copies per cell are estimated using above assumptions (GroEL2) (Schmidt et al., 2011). Moreover, the total protein content of a cell when compared to its volume is similar between Mtb (400,000 proteins per cell) and the larger Leptospira interrogans (1,100,000 proteins per cell) (Beck et al., 2009; Cook et al., 2009; Schmidt et al., 2011).
To assess the potential and applicability of the Mtb Proteome Library, we set out to characterize the dynamics and degree of DosR regulon protein expression in a standing culture model of growth arrest. Apart from a few highly abundant proteins, such as HspX and Hrp-1, the DosR regulon has, to the best of our knowledge, never been monitored in full on the protein level. It should be noted, however, that in vitro models can rarely precisely mimic true conditions, and it will thus be important to investigate whether the DosR regulon dynamics observed here reflect the situation in human latent Mtb infection and active TB disease.
Although the genome of Mtb was one of the first to be sequenced, its annotation is still challenging due to its high GC content and low sequence conservation compared to other prokaryotes. Our proteogenomic approach, combined with the Mtb Proteome Library pipeline, revealed 29 proteins not annotated to date, 13 of which have also been identified in an earlier extensive proteogenomic study (Kelkar et al., 2011). Unexpectedly, we found that most of these proteins were already annotated in other Mtb strains, such as CDC1551. The protein-level evidence we provide here for a major part of the annotated Mtb H37Rv proteome, as well as the previously unannotated proteins, will be incorporated into ongoing Mtb H37Rv genome annotation efforts (TubercuList; Lew et al., 2011).
Conclusion and Perspectives
Here we present the Mtb Proteome Library, a resource of validated, high-quality protein assays for targeted, hypothesis-driven studies, in which sensitivity and reproducibility are needed. Due to its selective nature, SRM is affected to a lower degree by sample complexity and background, and we believe that it will allow identification and quantification of Mtb proteins not only in mycobacterial cultures, as shown for the DosR regulon under hypoxic conditions, but also in infected cells, animal models, and potentially even clinical samples, such as lung resections, blood, or sputum (Domon and Aebersold, 2010; Mehaffy et al., 2012).
The synthetic peptide library generated for the Mtb Proteome Library was acquired on three different instrument types, and hence the usability of the MS coordinates is not restricted to SRM-based studies. Other proteomic strategies and techniques which require reference MS coordinates, such as the emerging SWATH technology, will profit from the Mtb Proteome Library as well (Gillet et al., 2012).
In sum, the Mtb Proteome Library is a unique resource which facilitates the generation of high quality SRM data sets, also by nonspecialized proteomics laboratories. These may lead to new mechanistic insights into the complex life cycle of Mtb on the protein level and thus catalyze research toward new treatments, diagnostics, biomarkers, and vaccines for the benefit of the numerous TB patients.
EXPERIMENTAL PROCEDURES
Detailed descriptions are given in the Supplemental Information.
Proteome Definition
The complete Mtb H37Rv proteome was defined as annotated in the Tubercu-List database and contained 4,012 proteins (TubercuList v2.3, April 2011) (Lew et al., 2011).
Organisms and Culture Conditions
Mtb H37Rv and M. bovis BCG (BCG) were grown to early exponential phase, late exponential phase, and stationary phase. To obtain hypoxic conditions and induce the DosR regulon, we employed a standing culture model, where agitation was stopped once cultures had reached exponential growth phase (Kendall et al., 2004).
Sample Preparation
Bacterial cell pellets were dissolved in lysis buffer containing 8 M Urea and Rapi-Gest (Waters) and were disrupted either by sonication (Mtb) or glass bead beating (BCG).Proteinswere reduced andalkylated, followed by a trypticdigest. The peptide solution was desalted by C18 reverse-phase chromatography, dried under vacuum, and resolubilized to a final concentration of 1 mg/ml. For the peptide fractionation through off-gel electrophoresis, peptides from exponential and stationary growth phases of Mtb were pooled and separated on a 3100 OFFGEL Fractionator (Agilent Technologies) using an immobilized pH gradient strip (pH 3–10, GE Healthcare). The resulting 24 peptide fractions were desalted again on C18 reverse-phase columns before MS analysis.
Discovery MS Data Acquisition and Analysis
One microgram of each peptide sample was analyzed on an LTQ Orbitrap XL (Thermo Fisher Scientific). The acquired MS2 spectra were searched against an Mtb H37Rv protein database. Only peptides at an FDR of <1% were considered for further analysis.
Proteome Coverage Prediction
The PeptideProphet output from above was processed with the software MAYU (Reiter et al., 2009). Peptide-spectrum matches were selected at an FDR of 0.16% to obtain a protein FDR of 1%. Proteome coverage prediction was performed as described by Claassen et al. (2011).
Selection and Preparation of Synthetic Peptides
For each protein, several proteotypic peptides were selected using a recently developed algorithm (our unpublished data). Only fully tryptic peptides without missed cleavages and a length between 6 and 21 amino acids were allowed. Furthermore, peptides had to be unique for a particular protein and were not allowed to map to any human protein. Highest priority was given to peptides with the highest number of previous observations in the PeptideAtlas database. If no or insufficient peptides had been observed previously, the best MS-suited peptides for a protein were predicted. A total of 17,463 synthetic peptides were purchased in unpurified form (JPT Peptide Technologies). The peptides were pooled in mixes of 96, desalted using reverse-phase C18 columns, and resolubilized at a concentration of 1 pmol/peptide/μl.
Generation of the Mtb Proteome Library
The synthetic peptide mixes were analyzed on a 4000 QTRAP mass spectrometer (AB Sciex) in SRM-triggered MS2 mode, and in data-dependent acquisition mode on an LTQ Orbitrap XL (Thermo Fisher Scientific), and a TripleTOF mass spectrometer (AB Sciex). All spectra identified with an FDR <1% were considered as true hits and were used to generate the synthetic Mtb Proteome Library. Retention times were extracted from the spectra and converted into a system-independent retention time (iRT) using spiked-in calibration peptides (Biognosys) (Escher et al., 2012).
Validation of the Mtb Proteome Library by SRM
If available, the six highest fragment ion peaks belonging to the y-ion series of the 2+ and 3+ precursors were extracted from the synthetic Mtb Proteome Library. The transition groups were measured in unfractionated Mtb H37Rv ly-sates (1:1 mixture of exponential and stationary phase cultures) in scheduled (±2 min) SRM acquisition mode on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific). Up to 450 transitions per run were acquired with a cycle time of 2 s and a dwell time of at least 20 ms. Transition groups were detected and assigned a statistical confidence score using the mQuest/mProphet software. The resulting data set was filtered at an FDR cutoff of 1%.
Prediction of SRM Assay Specificity
Unique ion signatures were calculated with the SRMCollider tool (Röst et al., 2012). The minimal number of transitions necessary to uniquely identify a precursor species was defined as the minimal n for which no other precursor existed in the background whose ions contained all n query ions.
Proteome-wide Absolute Label-free Quantification
For absolute quantification, 34 anchor proteins were selected covering a wide abundance range of the Mtb proteome. Each anchor protein was accurately quantified by SRM using heavy isotope-labeled reference peptides in defined concentrations determined by amino acid analysis (Thermo Fisher Scientific). All anchor proteins were represented in the Mtb Proteome Library by at least two peptides. The optimal model to combine SRM intensities of best flying peptides and most intense transitions to a single MS signal was determined by Monte Carlo crossvalidation and used to estimate proteome-wide concentrations from the SRM signal intensities of the validation data set (Ludwig et al., 2012). The data set was normalized using the signals of three endogenous peptides which were measured as positive controls in each run.
SRM Analysis of the DosR Regulon
For each protein of the DosR regulon, SRM assays were selected from the Mtb Proteome Library. If possible, three peptides with five y-ion transitions were chosen. For each target peptide, heavy isotope-labeled, synthetic peptides (JPT Peptide Technologies) were added in adjusted concentrations to every sample. For three proteins, no heavy isotope-labeled reference peptides were available (Rv0573, Rv1735, Rv1998c). SRM analysis was done as described above. Undetectable peptides and interfered transitions were manually removed using Skyline, and the resulting transition intensities were subjected to statistical analysis using linear mixed models with the SRMstats R package (Chang et al., 2012; MacLean et al., 2010b).
Proteogenomic Analysis
The genomic sequence of Mtb was translated in all six reading frames using the Bacterial, Archaeal, and Plant Plastid Code provided by NCBI. The MS2 spectra from the 24 OGE fractions were searched against this six-frame translated genome database. The Proteogenomic Mapping Tool (Sanders et al., 2011) was used to map peptides back to their genome location. A protein was only considered if there were at least two peptides with an FDR <1% mapping to the same region of the genome.
Databases
All resources described in this study are publicly accessible for interactive browsing and downloading in the following databases: discovery MS data from fractionated lysates as well as the spectra of all synthetic peptides, and Peptide Atlas (http://www.PeptideAtlas.org); SRM assays based on synthetic peptides, SRMAtlas (http://www.SRMAtlas.org); SRM validation data in Mtb whole-cell lysates and DosR regulon SRM data set, PASSEL (http://www.PeptideAtlas.org/passel).
Supplementary Material
Acknowledgments
We thank Mariette Matondo, Andreas Frei, Simon Hauri, Ludovic Gillet, and Samuel Bader for instrument maintenance and support with the MS measurements, and Ariel Bensimon and Mary-Louise Grossman for critically discussing and reading the manuscript. We also thank Douglas Young for discussions about the proteogenomics data, and Takuji Yamada for development of the pathway map iTuby. This work has been financially supported by the SysteMTb Collaborative Project (grant number 241587), which is funded by the Framework Programme 7 of the European Commission; the ERC advanced grant “Proteomics v3.0” (grant number 233226); the UBS Promedica Foundation; the Swiss Federal Commission for Technology and Innovation (grant number 13539.1); in part by American Recovery and Reinvestment Act funds (grant number R01 HG005805 to R.L.M.) from the National Institutes of Health, National Human Genome Research Institute; and the Bill and Melinda Gates Foundation (grant number OPP1039684 to R.L.M.). P.K.A. was the recipient of EMBO and SNF fellowships.
Footnotes
Supplemental Information includes five figures, four tables, Supplemental Experimental Procedures, and Supplemental References and can be found with this article at http://dx.doi.org/10.1016/j.chom.2013.04.008.
References
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, Croucher NJ, Rose G, Perkins TT, Parkhill J, Dougan G, Young DB. Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002342. doi: 10.1371/journal.ppat.1002342. http://dx.doi.org/10.1371/journal.ppat.1002342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Malmström JA, Lange V, Schmidt A, Deutsch EW, Aebersold R. Visual proteomics of the human pathogen Leptospira interrogans. Nat Methods. 2009;6:817–823. doi: 10.1038/nmeth.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beste DJV, Peters J, Hooper T, Avignone-Rossa C, Bushell ME, McFadden J. Compiling a molecular inventory for Mycobacterium bovis BCG at two growth rates: evidence for growth rate-mediated regulation of ribosome biosynthesis and lipid metabolism. J Bacteriol. 2005;187:1677–1684. doi: 10.1128/JB.187.5.1677-1684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon C, Dick T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J Bacteriol. 2002;184:6760–6767. doi: 10.1128/JB.184.24.6760-6767.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon C, Dick T. How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later. Future Microbiol. 2012;7:513–518. doi: 10.2217/fmb.12.14. [DOI] [PubMed] [Google Scholar]
- Chang CY, Picotti P, Hüttenhain R, Heinzelmann-Schwarz V, Jovanovic M, Aebersold R, Vitek O. Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol Cell Proteom. 2012;11:M111 014662. doi: 10.1074/mcp.M111.014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Sharma D, Singh A, Surolia A, Tyagi JS. Comprehensive insights into Mycobacterium tuberculosis DevR (DosR) regulon activation switch. Nucleic Acids Res. 2011;39:7400–7414. doi: 10.1093/nar/gkr375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cima I, Schiess R, Wild P, Kaelin M, Schüffler P, Lange V, Picotti P, Ossola R, Templeton A, Schubert O, et al. Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc Natl Acad Sci USA. 2011;108:3342–3347. doi: 10.1073/pnas.1013699108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen M, Aebersold R, Buhmann JM. Proteome coverage prediction for integrated proteomics datasets. J Comput Biol. 2011;18:283–293. doi: 10.1089/cmb.2010.0261. [DOI] [PubMed] [Google Scholar]
- Cook GM, Berney M, Gebhard S, Heinemann M, Cox RA, Danilchanka O, Niederweis M. Physiology of mycobacteria. Adv Microb Physiol. 2009;55:81–182. 318–319. doi: 10.1016/S0065-2911(09)05502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza GA, Arntzen MØ, Fortuin S, Schürch AC, Målen H, McEvoy CRE, van Soolingen D, Thiede B, Warren RM, Wiker HG. Proteogenomic analysis of polymorphisms and gene annotation divergences in prokaryotes using a clustered mass spectrometry-friendly database. Mol Cell Proteom. 2011;10:M110 002527. doi: 10.1074/mcp.M110.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW. The PeptideAtlas Project. Methods Mol Biol. 2010;604:285–296. doi: 10.1007/978-1-60761-444-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerks T, van Noort V, Minguez P, Bork P. Annotation of the M. tuberculosis hypothetical orfeome: adding functional information to more than half of the uncharacterized proteins. PLoS ONE. 2012;7:e34302. doi: 10.1371/journal.pone.0034302. http://dx.doi.org/10.1371/journal.pone.0034302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol. 2010;28:710–721. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011;470:163–165. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
- Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrah T, Deutsch EW, Kreisberg R, Sun Z, Campbell DS, Mendoza L, Kusebauch U, Brusniak MY, Hüttenhain R, Schiess R, et al. PASSEL: the PeptideAtlas SRMexperiment library. Proteomics. 2012;12:1170–1175. doi: 10.1002/pmic.201100515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev. 2012;36:514–532. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst HL, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteom. 2012;11:O111 016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut PR, Müller EC, Mattow J, Kaufmann SHE. Proteomics reveals open reading frames in Mycobacterium tuberculosis H37Rv not predicted by genomics. Infect Immun. 2001;69:5905–5907. doi: 10.1128/IAI.69.9.5905-5907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar DS, Kumar D, Kumar P, Balakrishnan L, Muthusamy B, Yadav AK, Shrivastava P, Marimuthu A, Anand S, Sundaram H, et al. Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol Cell Proteom. 2011;10:M111 011445. doi: 10.1074/mcp.M111.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall SL, Movahedzadeh F, Rison SCG, Wernisch L, Parish T, Duncan K, Betts JC, Stoker NG. The Mycobacterium tuberculosis dosRS two-component system is induced by multiple stresses. Tuberculosis (Edinb) 2004;84:247–255. doi: 10.1016/j.tube.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Lew JM, Kapopoulou A, Jones LM, Cole ST. TubercuList—10 years after. Tuberculosis (Edinb) 2011;91:1–7. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Ludwig C, Claassen M, Schmidt A, Aebersold R. Estimation of absolute protein quantities of unlabeled samples by selected reaction monitoring mass spectrometry. Mol Cell Proteom. 2012;11:M111 013987. doi: 10.1074/mcp.M111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 2010a;82:10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010b;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmström JA, Beck M, Schmidt A, Lange V, Deutsch EW, Aebersold R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature. 2009;460:762–765. doi: 10.1038/nature08184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehaffy MC, Kruh-Garcia NA, Dobos KM. Prospective on Mycobacterium tuberculosis proteomics. J Proteome Res. 2012;11:17–25. doi: 10.1021/pr2008658. [DOI] [PubMed] [Google Scholar]
- Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, Schoolnik GK, Sherman DR. Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol Microbiol. 2003;48:833–843. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Lam H, Campbell DS, Deutsch EW, Mirzaei H, Ranish J, Domon B, Aebersold R. A database of mass spectrometric assays for the yeast proteome. Nat Methods. 2008;5:913–914. doi: 10.1038/nmeth1108-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009;138:795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Rinner O, Stallmach R, Dautel F, Farrah T, Domon B, Wenschuh H, Aebersold R. High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat Methods. 2010;7:43–46. doi: 10.1038/nmeth.1408. [DOI] [PubMed] [Google Scholar]
- Picotti P, Clément-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Röst HL, Sun Z, Rinner O, Reiter L, et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L, Claassen M, Schrimpf SP, Jovanovic M, Schmidt A, Buhmann JM, Hengartner MO, Aebersold R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–2417. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L, Rinner O, Picotti P, Hüttenhain R, Beck M, Brusniak MY, Hengartner MO, Aebersold R. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat Methods. 2011;8:430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- Röst HL, Malmström L, Aebersold R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol Cell Proteomics. 2012;11:540–549. doi: 10.1074/mcp.M111.013045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders WS, Wang N, Bridges SM, Malone BM, Dandass YS, McCarthy FM, Nanduri B, Lawrence ML, Burgess SC. The proteogenomic mapping tool. BMC Bioinformatics. 2011;12:115. doi: 10.1186/1471-2105-12-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Beck M, Malmström JA, Lam H, Claassen M, Campbell DS, Aebersold R. Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol Syst Biol. 2011;7:510. doi: 10.1038/msb.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Global Tuberculosis Report 2012. World Health Organization; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.