

Abstract
The ubiquitinproteasome system is involved in various cellular processes, including transcription, apoptosis, and cell cycle. In vitro, in vivo, and clinical studies suggest the potential use of proteasome inhibitors as anticancer drugs. Cadmium (Cd) is a widespread environmental pollutant that has been classified as a human carcinogen. Recent study in our laboratory suggested that the clinically used anti-alcoholism drug disulfiram (DSF) could form a complex with tumor cellular copper, resulting in inhibition of the proteasomal chymotrypsin-like activity and induction of cancer cell apoptosis. In the current study, we report, for the first time, that DSF is able to convert the carcinogen Cd to a proteasome-inhibitor and cancer cell apoptosis inducer. Although the DSF–Cd complex inhibited the chymotrypsin-like activity of a purified 20S proteasome with an IC50 value of 32 μmol/L, this complex was much more potent in inhibiting the chymotrypsin-like activity of prostate cancer cellular 26S proteasome. Inhibition of cellular proteasome activity by the DSF–Cd complex resulted in the accumulation of ubiquitinated proteins and the natural proteasome substrate p27, which was followed by activation of calpain and induction of apoptosis. Importantly, human breast cancer MCF10DCIS cells were much more sensitive to the DSF–Cd treatment than immortalized but non-tumorigenic human breast MCF-10A cells, demonstrating that the DSF–Cd complex could selectively induce proteasome inhibition and apoptosis in human tumor cells. Our work suggests the potential use of DSF for treatment of cells with accumulated levels of carcinogen Cd.
Keywords: Cadmium, Disulfiram, Proteasome inhibitor, Calpain, Apoptosis
Introduction
Cadmium (Cd) is a widespread environmental pollutant of increasing worldwide concern, which is associated with air and water pollution (Waisberg et al., 2003). For nonoccupationally exposed people, Cd is absorbed into the body from dietary sources and cigarette smoking (Satarug and Moore, 2004; McElroy et al., 2007). Cd and its compounds have been classified as human carcinogens by the International Agency for Research on Cancer (IARC) in 1993, based on high risk of pulmonary tumor related to Cd exposure (Kelley, 1999). Cd has been shown to play a role in carcinogenesis by enhancing DNA mutation rates and to stimulate mitogenic signaling pathways and expression of oncoproteins that control cellular proliferation (Beyersmann and Hechtenberg, 1997). Cd accumulates in the human body with a half-life exceeding 25 years once absorbed (Filipic et al., 2006). Liver and kidney are two main organs that accumulate Cd since they express high levels of metallothionein, a Cd-binding protein (Pandey, 2006). Cd also accumulates in renal cortex, leading to induction of renal cancer (Kolonel, 1976; Il’yasova and Schwartz, 2005; Hu et al., 2002). Furthermore, rat liver epithelial cells could undergo carcinogenic transformation after chronic, low-level Cd exposure, suggesting the involvement of Cd in liver tumor formation (Qu et al., 2005). Cd exposure is also related to prostate, breast, bladder, pancreatic cancers (Goyer et al., 2004; Kellen et al., 2007; Sens et al., 2004; Schwartz and Reis, 2000; McElroy et al., 2006), and gallbladder cancer where Cd is concentrated after secretion by liver (Pandey, 2006; Waalkes, 2003).
The ubiquitin-proteasome pathway is responsible for the degradation of most endogenous proteins related to gene transcription, cell cycle, apoptosis and other major cellular processes (Goldberg, 1995; Dou et al., 2003; Orlowski and Wilk, 2000; Nandi et al., 2006). Since cancer cells are much more dependent on the ubiquitin-proteasome pathway than normal cells, it becomes more important to develop proteasome inhibitors as selective anticancer drugs (Dou et al., 2003; Orlowski and Wilk, 2000; Adams, 2004). Efficacy and tolerance of the first proteasome-inhibitor PS-341 (Bortezomib, Velcade) in clinical trials further encourage researchers to explore proteasome inhibitors for cancer treatment (O’Connor et al., 2005; Orlowski et al., 2005; Papandreou et al., 2004; Adams and Kauffman, 2004).
Disulfiram (DSF) is a member of the dithiocarbamate family that has been approved by the Food and Drug Administration (FDA) for the treatment of alcoholism (Orrenius et al., 1996; Johansson, 1992). It possesses an R1R2NC(S)SR3 functional group, which gives it the ability to complex metals. Previously, we reported that DSF could bind to tumor cellular copper (Cu) and form a DSF–Cu complex that induced apoptotic cell death in human breast cancer cells in vitro and in vivo through protea-some inhibition (Chen et al., 2006).
In the current study, we first screened mixtures of DSF and various metals, including magnesium (Mg), calcium (Ca), Cd, chromium (Cr), manganese (Mn), cobalt (Co), nickel (Ni) and zinc (Zn), for their proteasome-inhibitory activities in human prostate cancer PC-3 cells, and found that the DSF–Cd complex is most potent. Furthermore, DSF–Cd complex selectively inhibited the proteasome activity in human breast cancer cells, but not non-tumorigenic cells and this selective proteasome inhibition is associated with increased sensitivity to apoptosis induction in human breast cancer cells. Our study suggests the potential use of DSF as an agent to convert the carcinogen Cd to a selective anticancer drug through proteasome inhibition. To our knowledge, this is the first report of an effective treatment for chronic Cd intoxication, especially by converting this carcinogen to a specific tumor cell killer.
Materials and methods
Materials
Human prostate cancer PC-3 cells were purchased from American Type Culture Collection (Manassas, VA, USA). Human breast cancer MCF10DCIS (malignant MCF10) and immortalized but non-tumorigenic MCF-10A cells (derived from benign human breast tissue) were provided by Dr. Fred Miller (Karmanos Cancer Institute, Detroit, MI, USA). MG132, Magnesium chloride, calcium chloride, cadmium chloride, chromium chloride, manganese chloride, cobalt chloride, nickel chloride, zinc chloride, copper chloride, tetraethylthiuram disulfide (DSF), dimethylsulfoxide (DMSO), epidermal growth factor, insulin, chelora toxin and hydrocortisone were purchased from Sigma-Aldrich (St. Louis, MO, USA). Both inorganic metals and DSF were dissolved in DMSO at a stock concentration of 50 mmol/L, aliquoted, and stored at −20 °C. Purified rabbit 20S proteasome, fluorogenic peptide substrate Suc-LLVY-AMC (for the proteasomal chymotrypsin-like activity assay) were from Calbiochem (San Diego, CA, USA). Mouse monoclonal antibody against human poly (ADP-ribose) polymerase (PARP) was from Roche Applied Science (Indianapolis, IN, USA). Mouse monoclonal antibodies against human ubiquitin (P4D1), Bax (B-9), p27 (F-8), and goat polyclonal antibody against actin (C-11) were from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). Fetal bovine serum was from Tissue Culture Biologicals (Tulare, CA, USA). RPMI 1640, DMEM/F12, horse serum, penicillin, and streptomycin were from Invitrogen (Carlsbad, CA, USA).
Cell culture and whole cell extract preparation
PC-3 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. MCF10DCIS cells were cultured in 1:1 DMEM/F12 media supplemented with 5% (v/v) horse serum, 0.029 mol/L sodium bicarbonate, 10 mmol/L HEPES buffer solution, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. MCF10A cells were cultured in 1:1 DMEM/F12 media prepared as follows: 500 ml of media was supplemented with 5.26% (v/v) horse serum, 0.029 mol/L sodium bicarbonate, 10 mmol/L HEPES buffer solution, 100 U/ml of penicillin, and 100 μg/ml of streptomycin, 52.55 μg of cholera endotoxin, 5 mg insulin, 10 μg of epidermal growth factor, and 250 μg hydrocortisone. All cell lines were maintained at 37 °C in a humidified incubator with an atmosphere of 5% CO2 and routinely tested for mycoplasma contamination. The cells were subcultured by trypsinization when they reached to 80–90% confluency. Medium was changed when the color was turned to yellowish. A whole cell extract was prepared as described previously (Chen et al., 2005).
Analysis of the proteasome chymotrypsin-like activity in treated cells
PC-3 cells were treated as indicated. Intact cells or cell lysates were incubated with 40 μmol/L of fluorogenic substrate for the proteasomal chymotrypsin-like activity for 2 h at 37 °C in 100 μl of assay buffer (50 μmol/L Tris–HCl, pH 7.5). After incubation, production of hydrolyzed 7-amino-4-methylcoumarin (AMC) groups was measured using a Victor3 Multilabel Counter with an excitation filter of 380 nm and an emission filter of 460 nm (PerkinElmer, Boston, MA, USA). Changes in fluorescence were calculated against non-treated controls and plotted using Microsoft Excel™ software.
In vitro proteasome activity assay
Purified 20S rabbit proteasome (35 ng) was incubated with 20 μmol/L of fluorogenic peptide substrate for chymotrypsin-like activity in 100 μL of assay buffer, with or without Cd, DSF or DSF–Cd mixture at different concentrations for 1 h at 37 °C, followed by the measurement of AMC groups, as described (Li and Dou, 2000).
Western blot and cellular morphology analysis
Cells were treated as indicated in Figure legends, harvested and lysed. Cell lysates (40 μg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane (An et al., 1998), followed by visualization using the enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ, USA). Western blot analysis was performed using specific antibodies as indicated. All microscopic imaging for cellular morphology was taken by using Zeiss Axiovert 25 microscope (Thornwood, NY, USA).
Statistical analysis
T-test was used to compare the difference between immortalized but non-tumorigenic human breast MCF-10A and breast cancer DCIS cells in response to DSF–Cd exposure. SPSS 11.0 software was used to perform ANOVA analysis to compare the proteasome-inhibitory effects by treatment of multiple DSF–metal complexes or by DSF–Cd mixture or each alone in PC-3 cells. Significant differences were set up at p<0.01 and differences were set up at p<0.05.
Results
Treatment with the DSF–Cd complex results in proteasome inhibition and tumor cell apoptosis
Previously, we reported that the clinically used anti-alcoholism drug DSF bound to cellular Cu, forming a complex that possesses proteasome-inhibitory and anti-tumor activities (Chen et al., 2006). To search for new metal-based proteasome inhibitors, we tested the effects of a mixture of DSF and another metal ion, including Mg2+, Ca2+, Cd2+, Cr3+, Mn2+, Co2+, Ni2+ and Zn2+. DSF–Cu complex was used as a control in this experiment. Human prostate cancer PC-3 cells were treated for 12 h with 20 μmol/L of each DSF–metal mixture prepared, followed by measurement of proteasome inhibition and apoptotic cell death (Fig. 1). Results of the proteasomal chymotrypsin-like activity assay using the prepared cell extracts showed that the DSF–Cu complex caused 80% inhibition of cellular proteasome activity (Fig. 1A). Interestingly, DSF–Cd complex caused more than 95% inhibition under the same condition (Fig. 1A), indicating that DSF–Cd is a more potent proteasome inhibitor than DSF–Cu. In a sharp contrast, the mixtures of DSF and other tested metals, Mg2+, Ca2+, Cr3+, Mn2+, Co2+, Ni2+ or Zn2+, failed to inhibit the proteasome activity under the tested experimental concentration (Fig. 1A).
Fig. 1.

Disulfiram–cadmium mixture (DSF–Cd) is a potent proteasome inhibitor and apoptosis inducer in human prostate cancer PC-3 cells. PC-3 cells were treated with indicated DSF–metal mixtures at 20 μM for 12 h, followed by the chymotrypsin-like activity assay (A), Western blot analysis of polyubiquitinated proteins, p27, PARP and Bax (B), and morphological evaluation (C). DMSO and DSF alone at 20 μM were used as controls. Molecular weight of intact PARP and the cleaved PARP fragment were 116 kDa and 65 kDa, respectively. Molecular weight of three forms of Bax, is 18, 21, 36 kDa. β-Actin was used as a loading control. Columns, mean of independent triplicate experiments; bars, SD;**, p<0.01.
Inhibition of the proteasomal activity should result in accumulation of ubiquitinated proteins and target proteins (Li and Dou, 2000). Indeed, the levels of ubiquitinated proteins were accumulated in PC-3 cells treated with DSF–Cd or DSF–Cu, but not with mixtures of DSF and other metals (Fig. 1B). Compared to DSF alone or mixtures of DSF with other metals, DSF–Cd and DSF–Cu increased the protein level of proteasome target protein p27 (Fig. 1B). Interestingly, DSF–Zn complex also slightly increased the p27 protein level (Fig. 1B).
Since inhibition of tumor cellular proteasome activity is associated with apoptosis induction (Nam et al., 2001), we then measured apoptotic cell death by PARP cleavage (Fig. 1B) and cellular apoptotic morphologic changes (Fig. 1C). Cleavage of PARP into a p65 fragment was detected in the cells treated with DSF–Cd, DSF–Cu or DSF–Zn (Fig. 1B). Consistently, apoptotic cells (round-up and condensed) were observed after treatment of DSF–Cd, DSF–Cu or DSF–Zn (Fig. 1C).
It has been shown that calpain cleaves PARP and produces a fragment of 65 kDa (Pink et al., 2000). We and others have also shown that associated with the apoptotic commitment, calpain cleaves Bax protein (p21/Bax) into a p18/Bax fragment, which then forms a homodimer p36/Bax (Gao and Dou, 2000; Wood and Newcomb, 2000; Milacic et al., 2006). Three forms of Bax protein, p21, p18, and p36, were detected in the control cells, with p21/Bax as the major form (Fig. 1B, Bax panel, lane 1). However, the p36/Bax form was accumulated, associated with decreased levels of p21/Bax, mainly by DSF–Cd, DSF–Cu and DSF–Zn while other DSF–metal complexes had much less effect (Fig. 1B). Further studies from our laboratory suggest that organic zinc complexes are weaker proteasome inhibitors than organic copper complexes (unpublished data), explaining why DSF–Zn treatment could increase the proteasome target p27 and p36/Bax (Fig. 1B).
Inhibition of the chymotrypsin-like activity of a purified rabbit 20S proteasome and 26S cellular proteasome in vitro by DSF–Cd complex
To provide direct evidence for the inhibition of proteasomal chymotrypsin-like activity by DSF–Cd complex, we performed a cell-free proteasome activity assay using a purified rabbit 20S proteasome in the presence of Cd, DSF or DSF–Cd complex at various concentrations. As we reported, DSF alone had little inhibitory effect on purified 20S proteasome (Chen et al., 2006). However, DSF–Cd mixture and Cd salt showed proteasome inhibition with IC50 values of 32 and 35 μmol/L, respectively (Fig. 2). At the same concentrations, inhibitory effects to 20S proteasome by DSF–Cd treatment were different from that by DSF or Cd alone treatment (p<0.05). Since DSF–Cd mixture inhibited PC-3 cellular 26S proteasome much more potently (Fig. 1A) than inhibiting the purified 20S proteasome (Fig. 2), we further tested effect of DSF–Cd and Cd on 26S proteasome in a PC-3 cell extract in vitro. The results showed that the IC50 values of DSF–Cd and Cd to 26S proteasome activity in PC-3 cell extract were 3.5 and 9.0 μmol/L, respectively (data not shown). These data suggest that in addition to targeting the 20S proteasomal catalytic subunits, DSF–Cd could also target some other proteasomal components such as 19S (see Discussion).
Fig. 2.
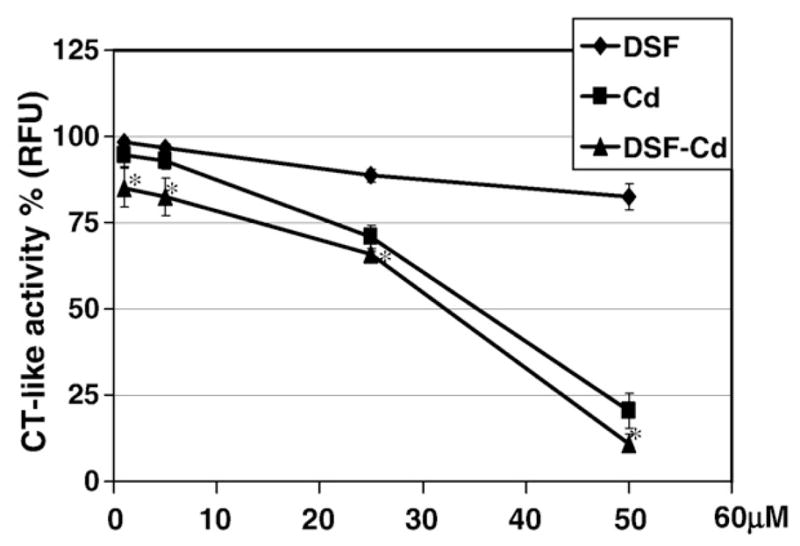
Inhibition of the chymotrypsin-like activity of 20S proteasome by DSF–Cd in vitro. Purified rabbit 20S proteasome (35 ng) was incubated with the peptide substrate for the proteasomal chymotrypsin (CT)-like activity in the presence of DSF, CdCl2 (Cd) and their mixture DSF–Cd at indicated concentrations, as described in Materials and methods. Bars, SD;*, p<0.05.
Comparison on proteasome-inhibitory and cell death-inducing effects of DSF–Cd and MG132
The potency of DSF–Cd on proteasome inhibition was then compared with that of the well-known proteasome-inhibitor MG132. As shown in Fig. 3A, DSF–Cd at 5 to 25 μM inhibited the chymotrypsin-like activity of a purified 20S proteasome by 15 to 33%, and DSF–Cd at 50 μM caused ~88% inhibition (Fig. 3A). In contrast, MG132 at all these concentrations caused around 95% inhibition (Fig. 3A). Therefore, DSF–Cd is a weaker inhibitor than MG132 to purified 20S proteasome.
Fig. 3.

Comparison of proteasome-inhibitory effects between DSF–Cd and MG132. A, Inhibitory effects on purified 20S proteasome by DSF–Cd and MG132 were tested. B, Intact PC-3 cells containing 26S proteasome were incubated with indicated concentrations of DSF–Cd and MG132 for 6 h, followed by a co-incubation for 2 h with the fluorogenic substrate Suc-LLVY-AMC (at 40 μmol/L) for the proteasomal chymotrypsin-like activity. After incubation, production of hydrolyzed AMC groups was measured. C, Morphological changes induced by DSF–Cd and MG132 at different concentrations. Columns, mean of independent triplicate experiments; bars, SD.
We then compared the effect of both proteasome inhibitors in human prostate PC-3 cells. These cells were first treated with different concentrations of DSF–Cd and MG132 for 6 h, followed by additional 2 h incubation with a fluorogenic substrate for the proteasomal chymotrypsin-like activity. Then production of hydrolyzed AMC groups was measured in these cells. By using this assay, we found that DSF–Cd caused a concentration-dependent inhibition on the PC-3 cellular proteasome activity: 5% at 10 μM, 15% at 25 μM and ~25% at 50 μM (Fig. 3B). As a comparison, MG132 at these concentrations induced 13, 22 and 45% inhibition, respectively (Fig. 3B). Therefore, DSF–Cd has comparable potency to MG132 to inhibiting 26S proteasome activity in PC-3 cells. A similar conclusion was obtained from results generated using protein extracts of PC-3 cells treated with DSF–Cd or MG132 at various concentrations (data not shown).
Since DSF–Cd and MG132 caused comparable levels of proteasome inhibition in intact PC-3 cells, we then compared their cell death-inducing effects. As shown in Fig. 3C, DSF–Cd induced apparent cell shrinkage and cell round-up at 2.5–5 μM and further induced extensive cell round-up and condensed morphology at 10 to 25 μM (Fig. 3C). Similarly, MG132 also induced concentration-dependent apoptotic morphological changes (Fig. 3C). These data suggest that DSF–Cd complex has the similar apoptosis-inducing effect to MG132 in PC-3 cells.
Kinetic effect on proteasome inhibition and apoptosis induction by DSF–Cd in PC-3 cells
If inhibition of the proteasome by DSF–Cd is responsible for tumor cell apoptosis induction, we should observe proteasome inhibition prior to cell death. To test this idea, PC-3 cells were treated with 20 μmol/L of DSF–Cd for up to 12 h, followed by measurement of proteasome inhibition and apoptosis (Fig. 4). As shown in Fig. 4A, the proteasomal chymotrypsin-like activity was inhibited by 50–90% by DSF–Cd after 2 h to 12 h treatment. In comparison, DSF had no inhibitory effect and Cd salt had only a transient weak effect (Fig. 4A). Statistical analysis showed that proteasome inhibition by DSF–Cd treatment was significantly different from DSF or Cd treatment (p<0.01). Western blot assay shows that the levels of ubiquitinated proteins accumulated as early as 2 h after the addition of DSF–Cd, and increased gradually in a time-dependent manner (Fig. 4B). Again, in PC-3 cells treated with DSF or Cd alone, ubiquitinated proteins were only slightly accumulated (Fig. 4B).
Fig. 4.

Kinetic studies on proteasome inhibition and apoptosis induction by disulfiram–cadmium in PC-3 cells. Exponentially grown PC-3 cells (0 h) were exposed to 20 μM DSF–Cd, DSF or cadmium chloride (Cd) for the indicated time points, followed by measuring inhibition of the proteasomal chymotrypsin-like activity using Suc-LLVY-AMC (A), Western blot analysis using specific antibodies to ubiquitin, Bax, and PARP (B, D), and apoptotic morphologic changes (C). β-Actin was used as a loading control. Columns, mean of independent triplicate experiments; bars, SD;**, p<0.01.
In the same kinetic experiment, we found that the cleaved p65/PARP fragment appeared 6 h after DSF–Cd treatment, which was apparently increased at 12 h (Fig. 4B). Also, in the cells treated with DSF–Cd, but not DSF or Cd alone, Bax p18 and p36 accumulated as early as 2 h, and further increased afterwards (Fig. 4B). Morphologically, the DSF–Cd-treated cells became rounded after 2 h, while DSF- or Cd-treated cells maintained a normal morphology to the end of treatment (Fig. 4C). In another kinetic experiment, we found that ubiquintinated proteins were accumulated after 15 min treatment with DSF–Cd, several hours before apoptosis induction (Fig. 4D vs B, C). Therefore, DSF–Cd complex induces proteasome inhibition much earlier than apoptosis.
DSF–Cd complex selectively induced proteasome inhibition and apoptosis in human breast cancer, but not in non-tumorigenic cells
It has been shown that various proteasome inhibitors could induce apoptosis selectively in human tumor, but not normal cells (Dou and Li, 1999). To study whether DSF–Cd complex also has a selective activity on tumor cells, we treated human breast cancer DCIS cells and human breast tissue-derived MCF10A (immortalized but non-tumorigenic) cells with DSF–Cd at various concentrations for 10 h, followed by morphological analysis and proteasome activity assay. As shown in Fig. 5A, treatment of breast cancer DCIS cells with DSF–Cd caused concentration-dependent apoptotic morphological changes, which were seen when DSF–Cd was used at as low as 1 μM (Fig. 5A vs. B). In a sharp contrast, the morphological changes in immortalized but non-tumorigenic MCF10A cells were induced only when DSF–Cd was used at 10–20 fold higher concentrations than that used in DCIS (Fig. 5A vs. B). As a comparison, neither DSF nor Cd salt at the tested highest concentration (20 μM) could induce any morphological changes in either DCIS or MCF-10A cell lines (Fig. 5B). Therefore, breast cancer DCIS cells are much more sensitive to DSF–Cd-induced apoptosis than non-tumorigenic MCF-10A cells.
Fig. 5.

Differential effects of DSF–Cd complex on malignant and non-tumorigenic breast cells. Malignant MCF10-DCIS cells (DCIS) and immortalized but non-tumorigenic MCF-10A cells (10A) were treated with DSF–Cd at 1–20 μM, DSF at 20 μM, cadmium chloride (Cd) at 20 μM, or DMSO for 10 h, followed by morphological changes evaluation (A, B) and the CT-like activity assay using whole cell lysates (C). Columns, mean of independent triplicate experiments; bars, SD;**, p<0.01.
To study whether increased sensitivity of DCIS cells to DSF–Cd treatment was due to increased proteasome inhibition, we measured the levels of the proteasomal chymotrypsin-like activity in protein extracts of both cell lines treated with DSF–Cd (Fig. 5C). We found that treatment with DSF–Cd caused a concentration-dependent inhibition of the proteasomal chymotrypsin-like activity in DCIS cells, with 50, 70 and 80% inhibition at 5, 10 and 20 μM, respectively (Fig. 5C). However, the proteasomal chymotrypsin-like activity in MCF10A cells was decreased by only ~20% with DSF–Cd at the same concentrations (Fig. 5B). Statistical analysis showed that proteasome inhibition by 5–20 μM DSF–Cd treatment was significantly different (p <0.01) in DCIS and MCF 10A cell lines. DSF or Cd salt alone had no or much less proteasome–inhibitory activity in both cell lines (Fig. 5C). Therefore, the proteasome activity in immortalized but non-tumorigenic MCF10A cells is not effectively inhibited by DSF–Cd, which may be responsible for lower levels of apoptosis induced in these cells (Fig. 5A). Taken together, our study demonstrates that human breast cancer cells are more sensitive to the novel DSF–Cd proteasome inhibitor than the non-tumorigenic cells.
Discussion
Cd is a ubiquitous environmental pollutant. Cd exposure is associated with extensive health problems including cancers. Unfortunately, so far there was no effective treatment reported to decrease its toxicity (Fotakis and Timbrell, 2006). Here we report that the clinically used anti-alcoholism drug DSF is capable of binding Cd and forming a new complex, which has proteasome-inhibitory and apoptosis-inducing activities in human cancer cells. Furthermore, DSF–Cd complex selectively inhibited proteasome activity and consequently induced apoptosis in human cancer cells, but has less effect on immortalized but non-tumorigenic cells. Our study suggests the potential use of DSF as an agent to convert the carcinogen Cd to a novel proteasome inhibitor and anticancer drug.
DSF, a member of the dithiocarbamate family, has been approved by the FDA for the treatment of alcoholism (Orrenius et al., 1996; Johansson, 1992). Previously, we reported that DSF binding to Cu forms a new complex DSF–Cu that induced apoptosis in human breast cancer via proteasome inhibition (Chen et al., 2006). Since Cd is located close to Cu in the periodic table, it is therefore possible that Cd might favor the similar bioligands to Cu. To test this idea, we determined the biological property of the DSF–Cd complex.
We examined the potential proteasome-inhibitory activity of different organic metal compounds by measuring both cellular proteasome activity and accumulation of ubiquitinated proteins and proteasome target p27. We found that treatment with DSF–Cd mixture significantly reduced chymotrypsin-like activity (Fig. 1A) and resulted in accumulation of ubiquitinated proteins and its natural substrate p27 (Fig. 1B), indicating that indeed proteasome inhibition had occurred. In contrast, ligand DSF alone or other metal mixtures with DSF did not inhibit the proteasome (Fig. 1A). We noticed that DSF–Zn also increased the protein level of p27 (Fig. 1B). We found that some synthetic Zinc complexes are weaker proteasome inhibitors than the corresponding copper complexes (unpublished data), suggesting that DSF–Zn could also slightly inhibit the proteasome activity (Figs. 1A and B). DSF capable of converting Cd to a proteasome inhibitor was further supported by in vitro direct proteasome inhibition. DSF–Cd inhibits the chymotrypsin-like activity of a purified 20S proteasome with an IC50 value of 32 μmol/L and cellular 26S proteasome with an IC50 value of 3.5 μmol/L under cell-free condition (Fig. 2 and data not shown). Compared to its potency to purified 20S proteasome, DSF–Cd showed about 10-fold higher potency to inhibit tumor cellular 26S proteasome. Similar result has been reported showing that 26S proteasome in PC-3 cells was decreased rapidly by 30% to 50% in dose-independent in their three major enzymatic activities following exposure to 1 to 20 Gy, however there was no effect on 20S proteasome (Pervan et al., 2005). The authors claimed that the treatment-sensitive target is located in the 19S cap of the 26S proteasome, rather than in the enzymatically active core (Pervan et al., 2005). We also believe that DSF–Cd has additional target(s) in 19S particle in addition to the 20S proteasome.
The proteasome-inhibitory effect of DSF–Cd was also compared with the authentic proteasome-inhibitor MG132 (Fig. 3). DSF–Cd was less potent to inhibiting a purified 20S proteasome (Fig. 3A). However, DSF–Cd and MG132 had comparable potencies to inhibiting PC-3 cellular 26S proteasome activity (Fig. 3B). Consistently, these two proteasome inhibitors had similar cell death-inducing effects (Fig. 3C), suggesting that inhibition of prostate cancer cellular proteasome activity by DSF–Cd is functional. It appears that DSF–Cd might also target other molecules in cells, in addition to 20S proteasome.
Proteasome inhibition is associated with apoptosis induction (Nam et al., 2001). If inhibition of the proteasome by DSF–Cd is responsible for tumor cell apoptosis, we should observe inhibition of proteasomal activity prior to cell death. Indeed, in the kinetic experiment we found that the proteasomal chymotrypsin-like activity was inhibited at as early as 2 h after addition of DSF–Cd (Fig. 4A). Consistently, ubiquitinated proteins were accumulated at as early as 15 min to 2 h (Figs. 4B, D). However, under the same conditions, apoptosis induction was detected after 4 h as shown by PARP cleavage and morphological changes (Fig. 4). Associated with apoptosis induction, calpain is activated, as shown by the appearance of p65/PARP, p18/Bax and p36/Bax (Fig. 4).
We found that the DSF–Cd complex has minimal effect on immortalized but non-tumorigenic MCF-10A cell line, in sharp contrast to its effect on breast tumor DCIS cells (Fig. 5). We proposed that the toxicity of DSF–Cd to cancer cells was due to their selective proteasome-inhibitory activity, to which non-tumorigenic cells are resistant. This hypothesis was tested by examining the proteasome activity levels in immortalized but non-tumorigenic MCF10A cells compared to malignant-MCF10 DCIS cells after DSF–Cd treatment (Fig. 5). We found that immortalized but non-tumorigenic-MCF 10A cells suffered much less proteasome inhibition when treated with DSF–Cd, although the proteasome activity in malignant-MCF10 cells were significantly inhibited under the same conditions (Fig. 5), further supporting the argument that the DSF–Cd complex is less toxic to normal cells but are toxic to cancer cells through the mechanism of tumor-specific proteasome inhibition.
The data presented here supports the novel concept of using non-toxic organic compounds such as DSF to convert carcinogen Cd into a proteasome inhibitor and an apoptosis inducer for cancer treatment, which should have no or much less effect on normal cells. The model we have presented in this report is limited by comparing immortalized breast cells rather than really normal primary cell lines to tumor cells. Future experiments should examine effect of DSF on Cd-induced tumor in animal model and its apoptosis effect on normal tissues.
Acknowledgments
The authors acknowledge the following grant support: Karmanos Cancer Institute of Wayne State University (to Q.P. Dou), a National Cancer Institute Grant (CA112625 and CA120009 to Q.P. Dou.), and the NCI/NIH Cancer Center Support Grant (to Karmanos Cancer Institute). We also thank Dr. Fred Miller for providing both DCIS and MCF-10A cell lines.
References
- Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Investig. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998;5:1062–1075. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- Beyersmann D, Hechtenberg S. Cadmium, gene regulation, and cellular signaling in mammalian cells. Toxicol Appl Pharmacol. 1997;144:247–261. doi: 10.1006/taap.1997.8125. [DOI] [PubMed] [Google Scholar]
- Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol. 2005;69:1421–1432. doi: 10.1016/j.bcp.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006;66:10425–10433. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- Dou QP, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updat. 1999;2:215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- Dou QP, Smith DM, Daniel KG, Kazi A. Interruption of tumor cell cycle progression through proteasome inhibition: implications for cancer therapy. Prog Cell Cycle Res. 2003;5:441–446. [PubMed] [Google Scholar]
- Filipic M, Fatur T, Vudrag M. Molecular mechanism of cadmium induced mutagenicity. Hum Exp Toxicol. 2006;25:67–77. doi: 10.1191/0960327106ht590oa. [DOI] [PubMed] [Google Scholar]
- Fotakis G, Timbrell JA. Modulation of cadmium chloride toxicity by sulphur amino acids in hepatoma cells. Toxicol In Vitro. 2006;20:641–648. doi: 10.1016/j.tiv.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Gao G, Dou QP. N-terminal cleavage of Bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes Bcl-2-independent cytochrome c release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Functions of the proteasome: the lysis at the end of the tunnel. Science. 1995;268:522–523. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- Goyer RA, Liu J, Waalkes MP. Cadmium and cancer of prostate and testis. BioMetals. 2004;17:555–558. doi: 10.1023/b:biom.0000045738.59708.20. [DOI] [PubMed] [Google Scholar]
- Hu J, Mao Y, White K. Renal cell carcinoma and occupational exposure to chemicals in Canada. Occup Med. 2002;52:157–164. doi: 10.1093/occmed/52.3.157. [DOI] [PubMed] [Google Scholar]
- Il’yasova D, Schwartz GG. Cadmium and renal cancer. Toxicol Appl Pharmacol. 2005;207:179–186. doi: 10.1016/j.taap.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Johansson B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiatr Scand, Suppl. 1992;369:15–26. doi: 10.1111/j.1600-0447.1992.tb03310.x. [DOI] [PubMed] [Google Scholar]
- Kellen E, Zeegers MP, Hond ED. Buntinx F. Blood cadmium may be associated with bladder carcinogenesis: the Belgian case-control study on bladder cancer. Cancer Detect Prev. 2007;31:77–82. doi: 10.1016/j.cdp.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Kelley C. Cadmium therapeutic agents. Curr Pharm Des. 1999;5:229–240. [PubMed] [Google Scholar]
- Kolonel LN. Association of cadmium with renal cancer. Cancer. 1976;37:1782–1787. doi: 10.1002/1097-0142(197604)37:4<1782::aid-cncr2820370424>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Li B, Dou QP. Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A. 2000;97:3850–3855. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006;98:869–873. doi: 10.1093/jnci/djj233. [DOI] [PubMed] [Google Scholar]
- McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Urinary cadmium levels and tobacco smoke exposure in women age 20–69 years in the United States. J Toxicol Environ Health, Part A. 2007;70:1779–1782. doi: 10.1080/15287390600754953. [DOI] [PubMed] [Google Scholar]
- Milacic V, Chen D, Ronconi L, Landis-Piwowar KR, Fregona D, Dou QP. A novel anticancer gold(III) dithiocarbamate compound inhibits the activity of a purified 20S proteasome and 26S proteasome in human breast cancer cell cultures and xenografts. Cancer Res. 2006;66:10478–10486. doi: 10.1158/0008-5472.CAN-06-3017. [DOI] [PubMed] [Google Scholar]
- Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem. 2001;276:13322–13330. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, Esseltine D, Trehu E, Adams J, Schenkein D, Zelenetz A. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2005;23:676–684. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- Orlowski M, Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys. 2000;383:1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105:3058–3065. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Nobel CS, van den Dobbelsteen DJ, Burkitt MJ, Slater AF. Dithiocarbamates and the redox regulation of cell death. Biochem Soc Trans. 1996;24:1032–1038. doi: 10.1042/bst0241032. [DOI] [PubMed] [Google Scholar]
- Pandey M. Environmental pollutants in gallbladder carcinogenesis. J Surg Oncol. 2006;93:640–643. doi: 10.1002/jso.20531. [DOI] [PubMed] [Google Scholar]
- Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- Pervan M, Iwamoto KS, McBride WH. Proteasome structures affected by ionizing radiation. Mol Cancer Res. 2005;3:381–390. doi: 10.1158/1541-7786.MCR-05-0032. [DOI] [PubMed] [Google Scholar]
- Pink JJ, Wuerzberger-Davis S, Tagliarino C, Planchon SM, Yang X, Froelich CJ, Boothman DA. Activation of a cysteine protease in MCF-7 and T47D breast cancer cells during β-Lapachone-mediated apoptosis. Exp Cell Res. 2000;255:144–155. doi: 10.1006/excr.1999.4790. [DOI] [PubMed] [Google Scholar]
- Qu W, Diwan BA, Reece JM, Bortner CD, Pi J, Liu J, Waalkes MP. Cadmium-induced malignant transformation in rat liver cells: role of aberrant oncogene expression and minimal role of oxidative stress. Int J Cancer. 2005;114:346–355. doi: 10.1002/ijc.20736. [DOI] [PubMed] [Google Scholar]
- Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:1099–1103. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz GG, Reis IM. Is cadmium a cause of human pancreatic cancer? Cancer Epidemiol Biomarkers Prev. 2000;9:139–145. [PubMed] [Google Scholar]
- Sens DA, Park S, Gurel V, Sens MA, Garrett SH, Somji S. Inorganic cadmium- and arsenite-induced malignant transformation of human bladder urothelial cells. Toxicol Sci. 2004;79:56–63. doi: 10.1093/toxsci/kfh086. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicol. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- Wood DE, Newcomb EW. Cleavage of Bax enhances its cell death function. Exp Cell Res. 2000;256:375–382. doi: 10.1006/excr.2000.4859. [DOI] [PubMed] [Google Scholar]