

Abstract
Drug design studies targeting one of the primary toxic agents in Alzheimer’s Disease, soluble oligomers of amyloid β-protein (Aβi), have been complicated by the rapid, heterogeneous aggregation of Aβ and the resulting difficulty to structurally characterize the peptide. To address this, we have developed [Nle35, D-Pro37]Aβ42, a substituted peptide inspired from molecular dynamics simulations which forms structures stable enough to be analyzed by NMR. We report herein that [Nle35, D-Pro37]Aβ42 stabilizes the trimer, and prevents mature fibril and β-sheet formation. Further, [Nle35, D-Pro37]Aβ42 interacts with WT Aβ42 and reduces aggregation levels and fibril formation in mixtures. Using ligand-based drug design based on [Nle35, D-Pro37]Aβ42, a lead compound was identified with effects on inhibition similar to the peptide. The ability of [Nle35, D-Pro37]Aβ42 and the compound to inhibit the aggregation of Aβ42 provides a novel tool to study the structure of Aβ oligomers. More broadly, our data demonstrate how molecular dynamics simulation can guide experiment for further research into AD.
Introduction
Alzheimer’s Disease (AD) is a degenerative brain disorder and a major cause of dementia for which no cure is known. Currently, >35 million people worldwide are believed to suffer from the disease and with the aging of the population this number is expected to increase by a factor of 3-4 over the next 40 years.1 AD is characterized by two pathological hallmarks – extracellular deposits composed primarily of amyloid β-protein (Aβ) fibrils and intracellular, hyperphosphorylated tau fibrils. While the identity of the molecular species responsible for the initiation of AD pathology has been debated historically, genetic evidence from early-onset familial AD,2-5 and experimentally observed neurotoxicity of Aβ,6-11 have indicated that Aβ assembly into β–sheet rich aggregates is a crucial, early-stage, neurotoxic element in AD.
Although Aβ is an important neurotoxin in the etiology of AD, the heterogeneous nature of Aβ assemblies has made it difficult to study particular toxic Aβ oligomers individually. An early hypothesis of Aβ toxicity stated that the energetically stable, insoluble Aβ fibrils were the primary pathologic species.12 However, a large body of evidence demonstrated poor correlation between fibril content and disease severity, thus potentially contradicting the original amyloid fibril hypothesis.13-16 Instead, evidence connecting synaptotoxicity and early cognitive deficits to soluble Aβ oligomers has emerged.9, 17, 18 Still, due to the rapid aggregation kinetics of Aβ and the existence of Aβ oligomers in dynamically changing mixtures, it has been difficult to elucidate the exact mechanism of assembly and the relationship between specific oligomer structures and their toxic activity. Bitan et al. have used Photo-Induced Cross-Linking of Unmodified Proteins (PICUP) to demonstrate the rapid equilibration of monomers and oligomers in freshly prepared Aβ, with distinct oligomer size distributions for Aβ40 and Aβ4219 Recently, the relationship between oligomer order and toxicity was elucidated for Aβ40 monomer through tetramer using PICUP-stabilized oligomers.20 Nonetheless, testing the biological activity of single oligomeric Aβ species has been highly challenging.
An additional consequence of the heterogeneous composition of Aβ assemblies is the difficulty determining the structure of soluble Aβ oligomers under physiological conditions. Traditional high-resolution methods, such as X-ray crystallography and solution-state NMR spectroscopy have been unsuccessful at determining the structure of the pathologically and physiologically relevant soluble Aβ oligomers. Efforts have been made to stabilize low molecular weight oligomers, but to date, high-resolution structures of Aβ oligomers are not available. To gain insight into the structural features of Aβ oligomers and the mechanisms by which they cause neurotoxicity, more information is needed.
Towards this end, we have previously reported the results of a Markov State Model Molecular Dynamics (MSM-MD) simulation of four C-terminal fragments of Aβ42 at physiological concentrations.21 Due to the strong dependence of aggregation kinetics on the exact amino acid sequence of the C-terminus, C-terminal fragments of Aβ21-43 were used in the simulation. The study revealed that Aβ trimers were a semi-stable state in the aggregation pathway, in which monomers contained a β-turn in the C-terminus. Similar structural motifs containing a C-terminal β-turn had been previously predicted in coarse grained simulations.22 Based on these results, a Gly37→D-Pro substitution was predicted to stabilize the β-turn. Given that this turn is not seen in any proposed structure of Aβ fibrils, it was hypothesized that the Gly37→D-Pro substitution would stabilize soluble forms of Aβ42.
Recently, we have reported experimental studies of the structure and behavior of an Aβ analogue containing both Gly37→D-Pro and Met35→Nle (norleucine) substitutions ([Nle35,D-Pro37]Aβ42.23 The Nle substitution was included to preclude oxidation of Met35, which can impact assembly kinetics and toxicity.24 Solution-state NMR studies of [Nle35,D-Pro37]Aβ42 showed that this analogue indeed formed a C-terminus β-turn centered around residues 37–39. Additional evidence from atomic force microscopy (AFM) and circular dichroism (CD) spectroscopy suggested that [Nle35,D-Pro37]Aβ42 did not form fibrils, and instead was stable in soluble oligomeric forms comprising 3-7 monomers. Here, we report further characterization of the behavior of [Nle35,D-Pro37]Aβ42 and its effect on the aggregation of WT Aβ42. In addition, intrigued by the results of these studies, we used ligand-based drug design to identify a small molecule capable of recapitulating the behavior of [Nle35,D-Pro37]Aβ42. In doing so, we have identified and characterized novel modulators of the Aβ42 aggregation pathway, capable of stabilizing Aβ42 oligomers that may be used for detailed structural and mechanistic investigations of their interactions with their cellular targets. Our studies demonstrate the utilization of molecular simulations to guide experiment in developing small molecule inhibitors targeting toxic amyloidogenic protein oligomers.
Results
[Nle35,D-Pro37]Aβ42 forms stable oligomers, not fibrils, in solution
In a previous computational study, Kelley et al. hypothesized that a turn-promoting substitution, such as Gly37→D-Pro would stabilize trimeric Aβ oligomers.21 To test this hypothesis, an Aβ42 analogue with the Gly37→D-Pro substitution was synthesized. An additional Met35→Nle substitution was introduced to avoid oxidation of the native methionine at position 35 during assay. Thioflavin T experiments, in which fluorescence of the dye is proportional to the extent of β-sheet character of the protein aggregates, were performed to assess the extent of Aβ aggregation (Figure 1). Following four days of incubation, the [Nle35,D-Pro37]Aβ42 samples showed negligible fluorescence whereas substantial fluorescence was measured for WT Aβ42, indicating negligible β-sheet character in the substituted peptide. This result is consistent with our predictions that a Gly37→D-Pro substitution stabilizes oligomers.
Figure 1.
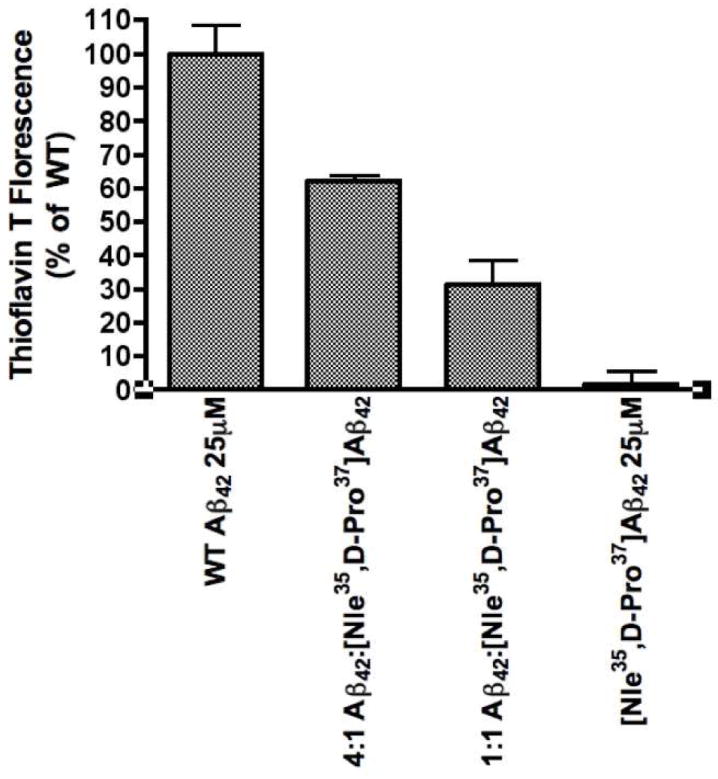
ThT fluorescence experiments of mixtures incubated for four days containing [Nle35,D-Pro37]Aβ42 and Aβ42. Mixtures contained 25 μM Aβ42 with [Nle35,D-Pro37]Aβ42 at 6.25 μM in the 4:1 mixture, and 25 μM in the 1:1 mixture. Samples were incubated at 37°C for the duration of the experiment.
To further evaluate the effect of the Gly37→D-Pro substitution on fibril formation, peptide preparations were imaged using transmission electron microscopy (TEM) at the same time point as the ThT fluorescence measurement. Aβ42 showed extensive fibril formation with co-deposition of amorphous aggregates (Figure 2A,B). In contrast, in identically prepared solutions of [Nle35,D-Pro37]Aβ42, we observed only amorphous aggregates and no fibrils (Figure 2C,D). Additionally, [Nle35,D-Pro37]Aβ42 samples aggregated for up to 6 months showed no fibril formation (data not shown).
Figure 2.
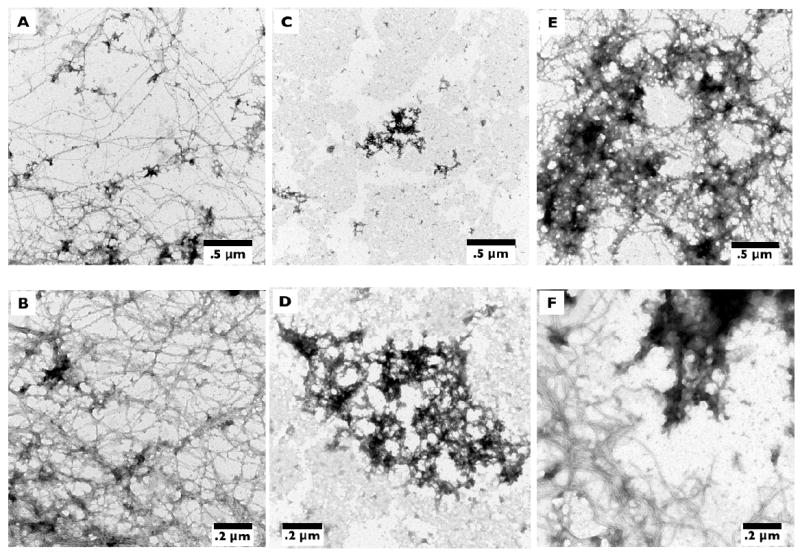
Morphological analysis of mixtures of [Nle35,D-Pro37]Aβ42 and Aβ42 after a four day incubation by TEM. Solutions containing 25 μM Aβ42, (A) and (B), 25 μM [Nle35,D-Pro37]Aβ42, (C) and (D), or a 1:1 mixture of 25 μM [Nle35,D-Pro37]Aβ42 and 25 μM Aβ42, (E) and (F), were incubated at 37°C until immediately prior to imaging. After incubation, samples were transferred to carbon-coated Formvar grids and stained with uranyl acetate.
Our previous simulations had indicated that, apart from the C-terminal turn, [Nle35,D-Pro37]Aβ42 is largely disordered in solution, and should not form stable secondary structure elements. To assess this, [Nle35,D-Pro37]Aβ42 was analyzed by circular dichroism (CD). At time zero, both WT Aβ42 and [Nle35,D-Pro37]Aβ42 exhibited spectra characteristic of a statistical coil, with a minimum at 197 nm (Figure 3). The spectrum of WT Aβ42 changed gradually over the course of the five-day experiment to develop a mimimum at 217 nm and a maximum at 196 nm, indicative of β-sheet. In contrast, the spectrum of [Nle35,D-Pro37]Aβ42 exhibited almost no change over five days.
Figure 3.
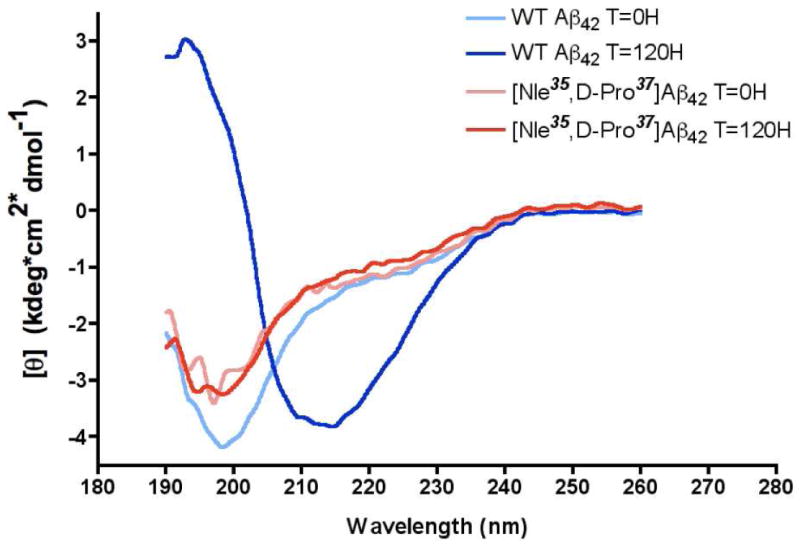
Secondary structure analysis of [Nle35,D-Pro37]Aβ42 and Aβ42 by CD spectroscopy. CD spectra of [Nle35,D-Pro37]Aβ42 and Aβ42 taken immediately after dissolution of the monomer (light blue and pink, respectively), and after five day incubation (dark blue and red, respectively). 25 μM samples of either [Nle35,D-Pro37]Aβ42 or Aβ42 were incubated at 37°C with continuous shaking for the duration of the experiment.
Finally, to investigate the effect of [Nle35,D-Pro37]Aβ42 on equilibrium mixtures of freshly prepared Aβ42, we conducted PICUP experiments, fractionated the cross-linked oligomers using SDS-PAGE, and visualized them by silver staining as previously reported (Figure 4A).25 Quantitative analysis of the gels with ImageJ indicated a 70% increase in the relative abundance of the trimer state for [Nle35,D-Pro37]Aβ42 compared with WT Aβ42 (Figure 4B). Concomitant with the increase in trimer abundance, we observed a decrease in monomer and tetramer in [Nle35,D-Pro37]Aβ42 relative to WT Aβ42, consistent the prediction that the Gly37→D-Pro substitution stabilizes Aβ42 trimer.
Figure 4.
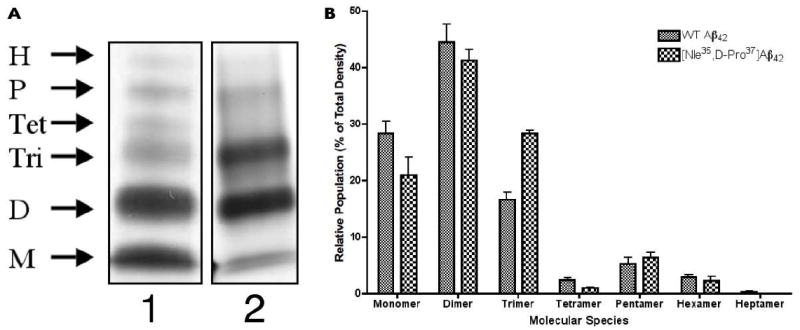
PICUP reveals distinct oligomer size distributions for [Nle35,D-Pro37]Aβ42 and Aβ42 following cross-linking immediately after preparation of fresh monomer. (A) SDS-PAGE of cross-linked samples of 25 μM Aβ42 and [Nle35,D-Pro37]Aβ42 (1 and 2, respectively), following silver staining. Arrows to the left of the gel images indicate the identity of oligomer bands, as deduced from the molecular weight marker. (B) Densiometric analysis of [Nle35,D-Pro37]Aβ42 and Aβ42 created using ImageJ. The data are the result of three independent experiments, each of which is well represented by the images in (A).
As a control, we examined an analogue with an identical amino acid composition to [Nle35,D-Pro37]Aβ42, but with sequence positions randomly assigned (Aβscramble). In both turbidity and Thioflavin T experiments, Aβscramble aggregated substantially more rapidly than WT Aβ42 (Supplementary Figure 3). This result demonstrates that any D-Proline substitution alone is not sufficient to replicate the effects of the Gly37→D-Pro substitution on aggregation. Additionally, a selenomethionine substituted peptide [Sem35,D-Pro37]Aβ42 was created for attempted x-ray studies. This peptide gave similar ThT and PICUP results as [Nle35,D-Pro37]Aβ42, thus demonstrating that the Met35→Nle substitution is not required to reproduce the effects of [Nle35,D-Pro37]Aβ42(Supplementary Figure 4). These results agree with other PICUP studies which demonstrate that a Met35→Nle substitution slightly destabilizes the trimer state, and is otherwise dissimilar from the distribution of [Nle35,D-Pro37]Aβ42.26
[Nle35,D-Pro37]Aβ42 reduces aggregation in mixtures with WT Aβ42 in a dose dependent manner
Next, we asked whether [Nle35,D-Pro37]Aβ42 could interact with and affect the aggregation of WT Aβ42. To answer this question, we measured ThT fluorescence in solutions of 25 μM WT Aβ42 mixed with 25 μM or 6.25 μM [Nle35,D-Pro37]Aβ42 (1:1 or 4:1 Aβ42:[Nle35,D-Pro37]Aβ42 concentration ratio, respectively). Following four day incubation experiments, the fluorescence of the mixtures was intermediate between the values observed for each peptide alone and inversely proportional to [Nle35,D-Pro37]Aβ42 concentration (Figure 1). The fluorescence signal of the 1:1 and 4:1 mixtures were 31.4±12.5% and 62.2±3.3% that of WT Aβ42 alone, respectively.
Morphological examination by TEM of the [Nle35,D-Pro37]Aβ42:Aβ42 mixtures supported the ThT data (Figure 2E,F). At a 1:1 molar ratio, the extent of mature fibril formation was reduced and an increase in amorphous aggregated was observed. Taken together with ThT data, the results suggest that [Nle35,D-Pro37]Aβ42 interacts with WT Aβ42, decreases formation of β-sheet-rich Aβ42 fibrils, and increases the relative abundance of non-fibrillar assemblies.
Identification of a small molecule Aβ42 aggregation inhibitor
Intrigued by the effects of [Nle35,D-Pro37]Aβ42 on aggregation, we conducted a ligand-based screen to identify small molecules capable of recapitulating structural and chemical motifs of the substituted peptide. The previously reported NMR structure of [Nle35,D-Pro37]Aβ42 was stripped of side chain atoms, except for the D-Pro, and used as the query for the screen (Figure 5A). In doing so, molecules mimicking the β-turn structure and recapitulating hydrogen-bonding interactions were selected. The top 100 scoring hits from the Maybridge Screening Database were purchased and investigated for their ability to reduce Aβ42 aggregation.
Figure 5.
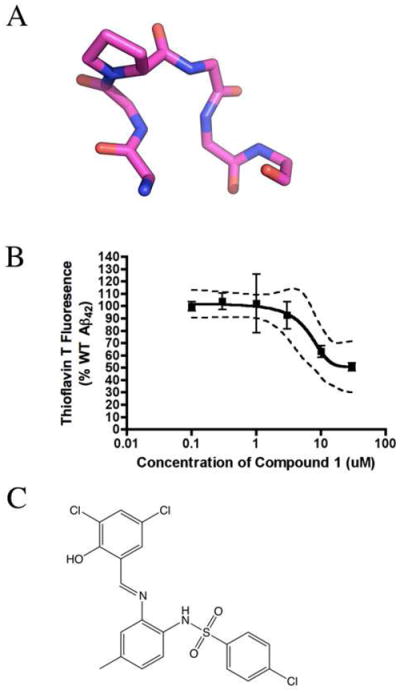
(A) The query used in the ligand-based drug design screen to identify 1, as visualized in PyMol. Residues 35-40 of the NMR structure of [Nle35,D-Pro37]Aβ42 were stripped of side chain atoms, except for the D-Pro residue. Carbons are shown in purple, oxygen in red, and nitrogen in blue. (B) Dose–response effect of 1 on Aβ42 aggregation as measured by ThT fluorescence. Aβ42 was incubated at a concentration of 25 μM in the presence of the indicated concentrations of 1. (C) and Structure of 1.
The compounds were screened in a single-dose ThT aggregation assay (data not shown). Compounds identified as potential hits were further evaluated in a dose–response ThT aggregation assay. Of these compounds, 4-chloro-N-[2-[(E)-(3,5-dichloro-2-hydroxyphenyl)methylideneamino]-4-methylphenyl] benzenesulfonamide (1) demonstrated reproducible, dose-dependent decreases in fluorescence (Figure 5B,C). 1 was found to inhibit WT Aβ42 aggregation as read by ThT fluorescence with an IC50 of 13 μM.
Characterization of the effects of 1 on WT Aβ42 aggregation
We then evaluated the effect of 1 on the secondary structure of aggregated Aβ42 by CD. In initial scans, identically prepared solutions of WT Aβ42 and a 1:1 ratio of drug:Aβ42 showed a minimum absorbance around 197 nm and were otherwise characteristic of a statistical coil conformation (Figure 6A). Over the course of the 5 day aggregation, the WT Aβ42 spectra shifted to have an absorbance maximum around 196 nm and minimum at 217 nm, indicating the transition of the WT peptide from a statistical coil to β–sheet rich conformation. In comparison, the CD spectrum of the 1:1 solution did not change over the course of the experiment and remained consistent with a random coil structure.
Figure 6.
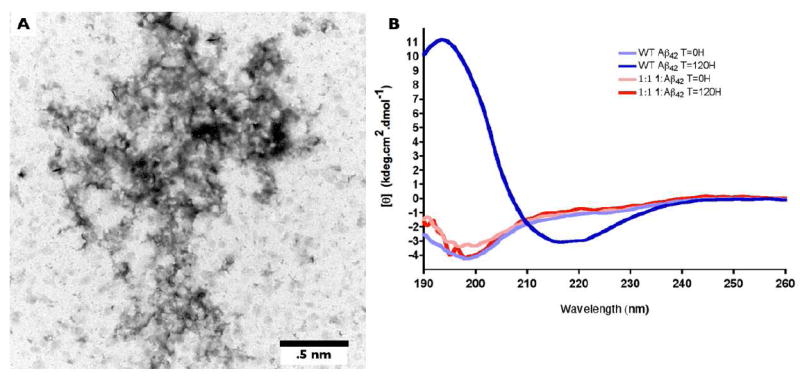
Analysis of the effect of 1 on the fibril and secondary structure formation of Aβ42. (A) TEM image of a 1:1 mixture of 25 μM 1 and 25 μM Aβ42. Samples were incubated for four days at 37 °C. After incubation, samples were transferred to carbon-coated Formvar grids and stained with uranyl acetate. (B) CD spectra of 25 μM Aβ42 and a 1:1 mixture of 25 μM 1 and 25 μM Aβ42 incubated for five days at 37 °C. Spectra for Aβ42 alone and the 1:1 mixture were obtained at t=0d (light blue and pink, respectively), and after a 5 day incubation (dark blue and red, respectively).
To supplement our data on the effect of 1 on Aβ42 fibril formation, we investigated the formation of fibrils by TEM in solutions of the compound at a 1:1 ratio to Aβ42. As reported above, WT Aβ42 forms mature fibrils over the course of a four-day aggregation experiment (Figure 4A,B). However, in the 1:1 mixture no fibrils were observed and only amorphous aggregates were seen (Figure 6B). In comparison with [Nle35,D-Pro37]Aβ42 which did not completely block fibril formation at this concentration ratio, 1 was more effective than [Nle35,D-Pro37]Aβ42 at inhibiting fibril growth.
Neurotoxicity of [Nle35,D-Pro37]Aβ42 and 1
We then examined the effect of [Nle35,D-Pro37]Aβ42 and 1 on cell viability using the (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay in differentiated PC-12 cells. Cells exposed to 5, 10, or 20 μM [Nle35,D-Pro37]Aβ42 demonstrated a dose-dependent decrease in cell viability (Figure 7). The toxicity was weaker than that of Aβ42, which led to a 45% decrease in viability at 10 μM, while [Nle35,D-Pro37]Aβ42 showed only 14% decrease in viability at this concentration. 1 also showed a dose dependent increase in toxicity, reducing cell viability by 40% at 50 μM. (Figure 6)
Figure 7.
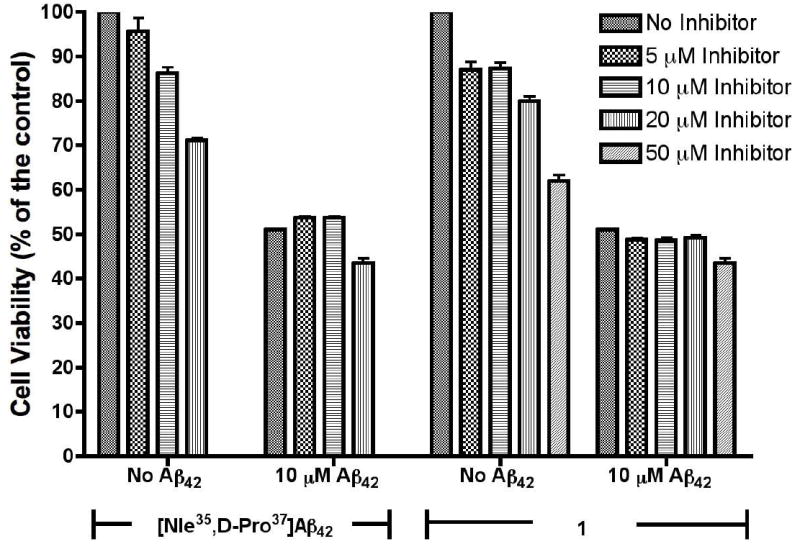
Results of MTT reduction assay to assess the effect of [Nle35,D-Pro37]Aβ42 or 1 on the viability of PC-12 cells, in the presence and absence of 10 μM Aβ42. Assay was performed after maintaining cells with Aβ42 mixtures for 48 h.
Stability assessment of 1 and identification of active fragment
Given the intriguing effects of 1 on Aβ42, we sought to verify its stability under experimental conditions by LC-MS. We found that upon entering an aqueous environment 1 reached an equilibrium with several degredation products (Figure 8). Dechlorination of 1 at the para chlroine, followed by epoxidation, yeilds a product with a mass 20 g/mol lighter than 1, identified as 1-20 (Mass spectra and NMR available in the Supplementary Material). Either 1 or 1-20 undergoes hydrolysis of the imine to yeild the degredation products 2 or 2-20, respectively, and 3. Unexpectedly, mass chromatograms and UV spectra of samples incubated up to 24 hours were identical within error, indicating that the equlibrium does not trend towards any product on timescales relevant to these experiments. When compound 2 was analyzed alone with the same protocol, the product 2-20 was not identified, suggesting that the dechlorination/epoxidation occurs only in the parent compound, 1.
Figure 8.
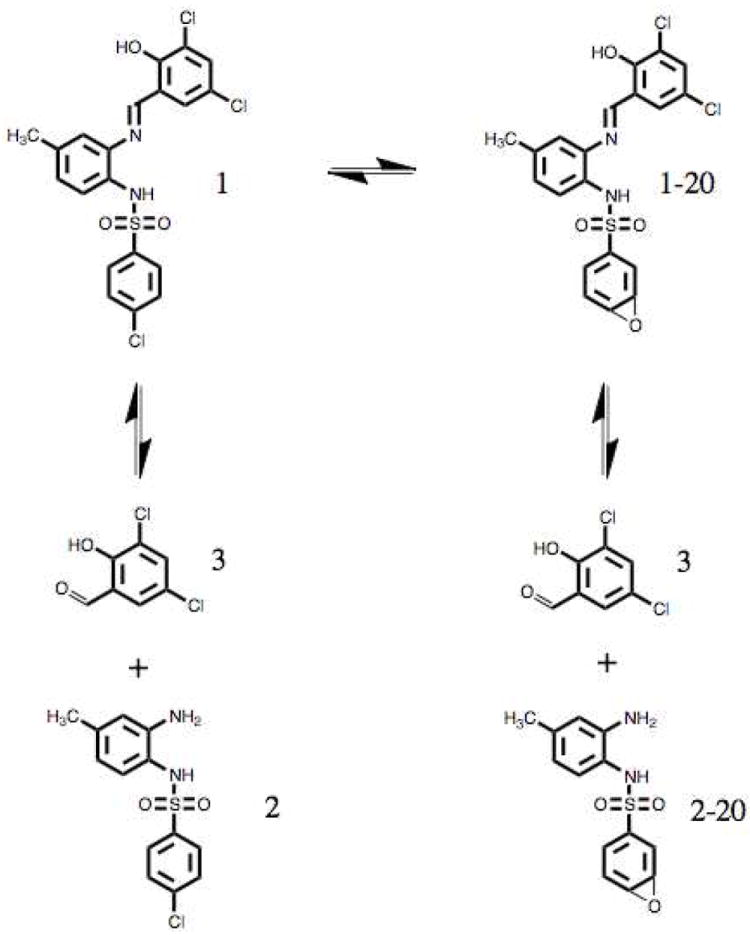
Equilibrium reaction diagram of the products created upon dissolution of 1 in aqueous buffer, pH = 7.4. Dechlorination of 1 at the para position yields product 1-20. Hydrolysis of the imine bond of 1 yields 2, and analogous hydrolysis of 1-20 yields 2-20, in addition to the aldehyde 3.
To assess the activity of the fragments relative to compound 1, we obtained 2 and 3 to evaluate in ThT assays. While compound 3 was found to be inactive (data not shown), compound 2 exhibited a dose dependent inhibition of Aβ42 aggregation (Figure 9). Interestingly, the inhibition of Aβ42 by 2 exhibited notably different kinetics in comparison to 1. At t=0d (Figure 9A), the IC50 of 1 as measured by percent of flourescence of an untreated control was 62 μM and the IC50 of 2 was 30 μM. Over the course of the four day aggregation, 1 increased in efficacy in comparison to control with an IC50 of 13 μM at t=4d (Figure 9B). Conversely, compound 2 showed similar efficacy after the aggregation period, with an IC50 of 23 μM at t=4d.
Figure 9.
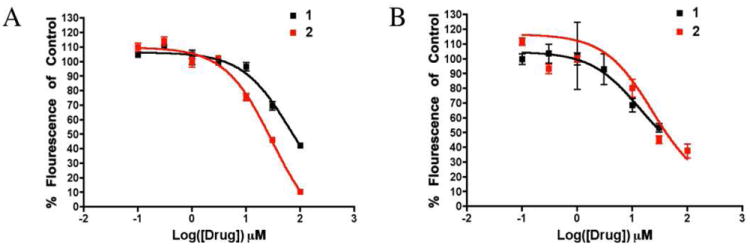
Results from ThT assays measuring the dose dependence of ThT flouresence in solutions of Aβ42 aggregated with the indicated concentrations of 1 and 2. A) Flouresence measured immediately after resuspending Aβ42 and combining with eith 1 or 2. B) Flouresence of identical samples, measured after a 4 day aggregation period.
Discussion
In our effort to design a stable oligomer of Aβ42, we have predicted21 that the substitution Gly37→D-Pro would result in enhanced β-hairpin formation and increased stability of low order oligomers, particularly trimers. Here, our PICUP results confirmed these computational predictions demonstrating a 70.7±5.5% increase in trimer abundance of [Nle35,D-Pro37]Aβ42 compared to Aβ42 (Figure 4). The CD, Thioflavin T, and TEM experiments demonstrated that the Gly37→D-Pro substitution prevented formation of β-sheet-rich fibrils and promoted formation of amorphous assemblies devoid of regular secondary structure elements (Figures 1-3). The data suggest that the β-turn stabilized by the Gly37→D-Pro mutation either a) stabilizes the small, soluble oligomers to such a degree as to prohibit further aggregation into fibrils and/or, b) the conformation assumed by [Nle35,D-Pro37]Aβ42 represents a conformation that is off pathway of fibril formation all together. Interestingly, although other parts of Aβ are unaltered, most notably, the central hydrophobic cluster (residues 17–21), which is known to be important for β-sheet formation, the Gly37→D-Pro substitution is sufficient for preventing formation of β-sheets.
Importantly for the development of our research on [Nle35,D-Pro37]Aβ42 towards a lead compound for Alzheimer’s Disease, the change in aggregation induced by the Gly37→D-Pro substitution is not limited to solutions containing only [Nle35,D-Pro37]Aβ42, but is observed in mixtures of [Nle35,D-Pro37]Aβ42 and WT Aβ42 as well. The results from ThT and TEM analysis of mixtures of [Nle35,D-Pro37]Aβ42 and WT Aβ42 clearly indicate that [Nle35,D-Pro37]Aβ42 interacts with WT Aβ42 to reduce the extent of aggregation and fibril formation when compared to Aβ42 alone (Figures 1,3). At a 1:1 ratio, aggregation as read by ThT was reduced by 60% when compared to WT peptide alone. In TEM images, fibrils were reduced in the solution containing a mixture of [Nle35,D-Pro37]Aβ42 and Aβ42.
We further attempted to recapitulate the effects of [Nle35,D-Pro37]Aβ42 on Aβ42 aggregation with a small molecule. Structural and chemical motifs from the NMR structure of [Nle35,D-Pro37]Aβ42 were used in a ligand-based drug discovery effort. Using this approach, we discovered 1 and found that it inhibited Aβ42 β-sheet formation with an IC50 of 13 μM over a 4 day aggregation (Figure 5). Results from CD and TEM experiments were consistent with the ThT data and indicated that 1 completely inhibited Aβ42 fibril and β-sheet formation at a 1:1 molar ratio (Figure 6). Considering the results of the LC-MS stability assessment in combination with the ThT studies with compound 2, the substructure represented by 2 clearly is the primary fragment of 1 responsible for inhibition of Aβ42. Although 3 does not inhibit aggregation on its own, the addition of fragment 3 to fragment 2 (yielding 1) significantly alters the kinetics of the aggregation in comparison to 2 alone. The data suggest that 2 has a faster rate of association with Aβ42 in comparison to 1.
Both [Nle35,D-Pro37]Aβ42 and 1 were found to cause a dose-dependent decrease in cell viability. Notably, [Nle35,D-Pro37]Aβ42 is significantly less toxic than WT Aβ42 (Figure 7). Recently, O’Nuallain et al. found that freshly prepared dimers of [Cys26]Aβ42 did not block long term potentiation (LTP), but that protofibrils built from these dimers did reduce LTP.27 Similarly, our results suggest that the trimer stabilized by [Nle35,D-Pro37]Aβ42 is a minimally toxic species, in comparison to WT Aβ42. Given the results, it is hypothesized that [Nle35,D-Pro37]Aβ42 and 1 reduce Aβ42 aggregation by either stabilizing soluble oligomers to such an extent that further aggregation is prohibited, and/or by stabilizing a conformation of Aβ42 that is off pathway altogether of protofibril and fibril formation.
Conclusions
We designed [Nle35,D-Pro37]Aβ42 based on a structural motif observed in the structure of the Aβ42 trimer predicted from molecular dynamics simulations. Recently, we have reported solution NMR structures confirming the existence of the predicted β-hairpin, and the data herein demonstrate a 70% increase in the relative population of the trimer in freshly prepared solutions. The use of computation to predict the structure of a stable species in a heterogeneous aggregation pathway, and guide the development of an experimental investigation is notable, and suggests a methodology for developing aggregation inhibitors for other amyloid-related diseases, which are complicated due to a lack of structural information. Both [Nle35,D-Pro37]Aβ42 and the compounds identified based on its structure, 1 and 2, are able to interact in solution with Aβ42 to stabilize soluble oligomers and reduce fibril formation. The shift in oligomer distribution induced by [Nle35,D-Pro37]Aβ42 correlated to a reduction in toxicity compared to Aβ42. Given the evidence herein that [Nle35,D-Pro37]Aβ42, 1, and 2 inhibit Aβ42 aggregation and β–sheet formation, we hope to further exploit their structural motifs to develop more efficacious and less toxic lead compounds.
Materials and Methods
Sample Preparation
WT Aβ42 was purchased from a single production lot from Bachem (Bachem H-6466 lot 1013710). Synthetic peptide [Nle35,D-Pro37]Aβ42 (DAEFRHDSGY10EVHHQKLVFF20AEDVGSNKGA30IIGLNLVDPGVV40IA) was synthesized by Anaspec (San Jose, CA). Because the work was carried out in two different laboratories, with slightly different protocols, two sample preparations exist. Peptides were prepared for all experiments in Eppendorf Protein Lo-Bind tubes (Eppendorf # 022431081). For Thioflavin T studies, the entire contents of the 1 mg Aβ42 peptide vial was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (Sigma # 325244) to a concentration of 1 mg/mL, and incubated for 2 hours at room temperature. After aliquoting the solution, samples were covered with Parafilm in which holes were poked. The solvent was evaporated using a SpeedVac for 1 h and the dry peptide films stored at −80 °C. Immediately before experiments, samples were dissolved in DMSO (Sigma # D2438), Biotechnology Performance Grade) at 2 mM, and sonicated for 5 min. The peptide solutions were diluted with PBS, pH 7.4 (GIBCO # 10010), to the desired concentration. Final DMSO concentration was kept below 3%. For all other experiments, peptides were prepared as described previously.28 Dried peptide films were dissolved by adding sequentially 10% of the final volume 60 mM NaOH, 45% water, and 45% 20 mM NaH2PO4 buffer. After dissolution in 60 mM NaOH, peptides were sonicated for 1 minute in a bath sonicator. Peptide concentration was measured by Bradford protein assay (Bio-Rad) or quantitative amino acid analysis, and was generally 70-80% of the nominal concentration. Peptide mixtures were prepared immediately prior to the beginning of experiments.
Compounds were purchased from the Maybridge Screening Collection (Maybridge Chemical Company, Cornwall, UK) and Sigma. Compounds were dissolved in DMSO to 10 mM, and then diluted with PBS to the desired concentration. Purity of the compounds was assessed by NMR, and the degredation of the parent compound was investigated by LC-MS. For MTT and CD experiments, which are sensitive to the presence of DMSO, 1 was dissolved to 10 mM in NaOH and then diluted further with PBS.
PICUP/SDS-PAGE
Photo-Induced Cross-linking of Unmodified Proteins (PICUP) was performed as described previously.25 Briefly, 1 μL of 40 mM ammonium persulfate (APS, Aldrich) and 1 μL of 2 mM Tris(2,2’-bipyridyl)dichlororuthenium(II) (Ru(Bpy)) (Aldrich) were added to 18 μL of 25 μM WT Aβ42 sample in a clear PCR tube. Irradiation was carried out for 1 second. The cross-linking reaction is quenched immediately by adding 1 μL 1M DTT and 10 μL of Tricine sample buffer (Invitrogen). Samples are then analyzed using 1-mm-thick, 10-20% Tris-Tricine gradient gels (Invitrogen), and silver stained (SilverXpress, Invitrogen). Following air drying, gels were analyzed using ImageJ. Reported values are from averaging of three separate gels.
Circular Dichroism
Samples of 25 μM WT peptide, in the absence or presence of [Nle35,D-Pro37]Aβ42 or 1, were incubated at 37°C with continuous agitation using an orbital shaker at 200 rpm. Spectra were recorded every 24 h during 5 days using a J-810 spectropolarimeter (Jasco, Easton, MD) equipped with a thermostable sample cell at 37°C using 1-mm path-length cuvettes. Spectra were collected from 190 to 260 nm with 1-s response time, 100-nm/min scan speed, 0.2-nm resolution and 1-nm bandwidth. Spectra were averaged after background subtraction.
Thioflavin T Fluoresence
Samples were incubated at 37 °C for four days in triplicates with shaking. Ten μL samples were added in black, flat- and clear-bottom 96-well plates (Corning # 3631) to 190 μL 25 μM Thioflavin T (Sigma # T3516) in 50 mM glycine, pH 8 (Fisher # G48-212). After a five-minute incubation with the dye, the fluorescence of ThT was measured using a Spectromax M5 fluorometer (Molecular Devices) at an excitation wavelength of 446 nm and emission at 490 nm.
Transmission Electron Microscopy
Samples of Aβ42 were incubated in the absence or presence of [Nle35,D-Pro37]Aβ42 or 1 for four days at 37°C. Aliquots of 7 μL were spotted onto 400-mesh, glow-discharged, carbon-coated Formvar grids (Electron Microscopy Science, Hatfield, PA) for 2 min and stained with 7 μL 1% uranyl acetate. The samples were analyzed using a transmission electron microscope JEM1200-EX (JOEL).
MTT assay
MTT assays were conducted as previously reported.26 Rat pheochromocytoma (PC-12) cells were maintained in F-12 nutrient mixture with Kaighn’s modification (F-12K) with 15% heat-inactivated horse serum and 2.5% FBS at 37°C in an atmosphere of 5% CO2. For cell viability assays, cells were plated in 96-well plates at a density of 30,000 cells per well in differentiation media (F-12K, 0.5% FBS, 100 μM nerve growth factor) and maintained for 48 h.
To assess the biological activity of Aβ42, [Nle35,D-Pro37]Aβ42, and 1, solutions were prepared as indicated above and diluted into RPMI media to yield concentrations 10-times higher than experimental concentrations. Aliquots of 10 μL were added to PC-12 cells to yield final concentrations of 10 μM WT Aβ42 and incubated for 15 h. Cell viability was assessed qualitatively by visual observation and quantitatively by the CellTiter 96 Non-Radioactive Cell Proliferation Assay (Promega). Briefly, 15 μL of dye solution was incubated with the cells for 3 h. Then 100 μL of solubilization/stop solution was added and the plates were incubated overnight in the dark to ensure complete solubilization. Plates were read by using a Synergy HT microplate reader (BioTek), and the absorbance at 570 nm (formazan product) minus the absorbance at 630 nm (background) was recorded. At least three independent experiments with six replicates (n = 18) were carried out, and the results were averaged.
Virtual Screening
A representative structure from the previously reported NMR studies into the [Nle35,D-Pro37]Aβ42 peptide was used as a basis for the ligand-based screen. Residues 35-40 were extracted from the full structure, and side chain atoms were deleted except for the proline atoms. The Maybdrige Screening Collection was used as a screening database. Molecules were stripped of salts, and conformers of the remaining molecules were created using Omega (version 2.3, OpenEye Scientific Software). The screening database was then compared to the modified peptide structure using the program ROCS (version 2.3, OpenEye Scientific Software). The Explicit Mills Dean force field was used, with the exception that ring interactions were turned off. The top 100 compounds ranked by combo tanimoto after optimization by shape and color, and were selected for evaluation by ThT. Compound 1 was ranked 48th out of 56,897 compunds screened (Details available as supplementary information).
Supplementary Material
Acknowledgments
We would like to acknowledge the UCLA Jim Easton Consortium Alzheimer’s Drug Discovery and Biomarker Development, the NIH Nanomedicine Center (PN1 EY016525-07), and the Stanford Center for Molecular Analysis and Design (CMAD) for funding. We would also like to NSF award CNS-0619926 and #0960306 (MRI-R2: Acquisition of a Hybrid CPU/GPU and Visualization Cluster for Multidisciplinary Studies in Transport Physics with Uncertainty Quantification) for computing resources.
Footnotes
Abbreviations: Aβ, amyloid β-protein; AD, Alzheimer’s Disease, Nle, norleucine; D-Pro, D-proline; ThT, Thioflavin T; PICUP; Photo-Induced Cross-linking of Unmodified Peptides
Supporting Information Available: Results from experiments conducted with Aβscramble and [Sem35,D-Pro37]Aβ42 are provided in the supplementary material. The LC-MS and 1HNMR of 1, 2, and 3, are also available in the supporting information. Additionally, the rankings and structures of top scoring compounds from the virtual screen are provided.
References
- 1.World Alzheimer Report 2010. Alzheimer’s Disease International. 2010 [Google Scholar]
- 2.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 3.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 4.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid β variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 5.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 6.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of beta-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 7.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 9.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 10.Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci USA. 2005;102:2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, Allsop D, Nakagawa M. High-molecular-weight {beta}-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010;24:2716–2726. doi: 10.1096/fj.09-150359. [DOI] [PubMed] [Google Scholar]
- 12.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 13.Neve RL, Robakis NK. Alzheimer’s disease: a re-examination of the amyloid hypothesis. Trends Neurosci. 1998;21:15–19. doi: 10.1016/s0166-2236(97)01168-5. [DOI] [PubMed] [Google Scholar]
- 14.Giannakopoulos P, Herrmann FR, Bussière T, Bouras C, Kövari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 15.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 16.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watson D, Castaño E, Kokjohn TA, Kuo YM, Lyubchenko Y, Pinsky D, Connolly ES, Esh C, Luehrs DC, Stine WB, Rowse LM, Emmerling MR, Roher AE. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer’s disease. Neurol Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 18.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bitan G, Lomakin A, Teplow DB. Amyloid beta-protein oligomerization: prenucleation interactions revealed by photo-induced cross-linking of unmodified proteins. J Biol Chem. 2001;276:35176–35184. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]
- 20.Ono K, Condron MM, Teplow D. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA. 2009;106:14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelley N, Vishal V, Krafft G, Pande V. Simulating oligomerization at experimental concentrations and long timescales: A Markov state model approach. J Chem Phys. 2008;129:214707. doi: 10.1063/1.3010881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid beta-protein folding and oligomerization. Proc Natl Acad Sci USA. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajadas J, Liu CW, Kelley NW, Novick PA, Inayathullah M, LeMieux MC, Pande VS. Rationally designed turn promoting mutation in the amyloid-β peptide sequence stabilizes oligomers in solution. PLOS One. 2010 doi: 10.1371/journal.pone.0021776. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maiti P, Piacentini R, Ripoli C, Grassi C, Bitan G. Surprising toxicity and assembly behaviour of amyloid β-protein oxidized to sulfone. Biochem J. 2011;433:323–332. doi: 10.1042/BJ20101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bitan G, Teplow D. Rapid photochemical cross-linking--a new tool for studies of metastable, amyloidogenic protein assemblies. Acc Chem Res. 2004;37:357–364. doi: 10.1021/ar000214l. [DOI] [PubMed] [Google Scholar]
- 26.Maiti P, Lomakin A, Benedek GB, Bitan G. Despite its role in assembly, methionine 35 is not necessary for amyloid β-protein toxicity. J Neurochem. 2010:11. doi: 10.1111/j.1471-4159.2010.06692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’nuallain B, Freir D, Nicoll A, Risse E, Ferguson N, Herron C, Collinge J, Walsh D. Amyloid β-Protein Dimers Rapidly Form Stable Synaptotoxic Protofibrils. J Neurosci. 2010;30:14411–14419. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahimi F, Maiti P, Bitan G. Photo-induced cross-linking of unmodified proteins (PICUP) applied to amyloidogenic peptides. J Vis Exp. 2009 doi: 10.3791/1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.