

Abstract
Objective
Prenatal cocaine exposure (PCE) can cause persistent neuropsychological and motor abnormalities in affected children, but the physiological consequences of PCE remain unclear. Conclusions drawn from clinical studies can sometimes be confounded by poly-substance abuse and nutritional deprivation. However, existing observations suggest that cocaine exposure in utero, as in adults, increases synaptic dopamine and promotes enduring dopamine-dependent plasticity at striatal synapses, altering behaviors and basal ganglia function.
Methods
We used a combination of behavioral measures, electrophysiology, optical imaging, and biochemical and electrochemical recordings to examine corticostriatal activity in adolescent mice exposed to cocaine in utero.
Results
We show that PCE caused abnormal dopamine-dependent behaviors, including heightened excitation following stress and blunted locomotor augmentation to repeated treatment with amphetamine. These abnormal behaviors were consistent with abnormal GABA interneuron function, which promoted a reversible depression in corticostriatal activity. PCE hyperpolarized and reduced tonic GABA currents in both fast-spiking and PLTS-type GABA interneurons to increase tonic inhibition at GABAB receptors on presynaptic corticostriatal terminals. While D2 receptors paradoxically increased glutamate release following PCE, normal corticostriatal modulation by dopamine was reestablished with a GABAAR antagonist.
Interpretation
The dynamic alterations at corticostriatal synapses that occur in response to PCE parallel the reported effects of repeated psychostimulants in mature animals, but differ in being specifically generated through GABA. Our results indicate that approaches which normalize GABA and D2 receptor-dependent synaptic plasticity may be useful for treating the behavioral effects of PCE and other developmental disorders that are generated through abnormal GABAergic signaling.
Introduction
PCE can cause debilitating neuropsychological and motor abnormalities in humans and is an important public health concern.1 In spite of prevention-related measures, over 4% of pregnant women in the United States use illicit drugs,2 with up to 1% using cocaine.1 Cocaine readily crosses the placenta3 and can cause withdrawal symptoms in neonates and cognitive and behavioral abnormalities in adolescence.4,5 Unfortunately, the prospect for treating affected children remains poor, as the drug’s effect on the developing nervous system remains unclear.6
Observations in the clinic1,4,5 and laboratory7–10 suggest that abnormal symptoms and signs following PCE reflect alterations in corticostriatal function. The striatum is the primary nucleus for cortical information entering the basal ganglia and regulates motor control, cognition, and habit learning.11,12 Attention to salient behavioral cues, as well as the selection and execution of movements and decisions, requires the striatum to collect cortical signals, modulate information, and activate appropriate corticofugal pathways.13 Signal integration is thought to occur at a ‘striatal microcircuit’, where glutamate from cortical inputs, dopamine from nigrostriatal terminals, and gamma-aminobutyric acid (GABA) from striatal interneurons interact at the dendritic spines of striatal medium spiny projection neurons (MSNs) to select behaviorally-relevant synapses (Fig 1A).12,14–16 Here we show that PCE causes abnormal dopamine-dependent motor behaviors and striatal synaptic plasticity in adolescent mice through abnormal GABA interneuron function, providing insights into novel therapeutic approaches that may improve outcome in children with PCE.
FIGURE 1.

The striatal ‘microcircuit’ and the proposed mechanism for over-inhibition of corticostriatal activity following PCE. (A) This simplified striatal circuit is composed of medium spiny neurons (MSNs) that are excited by glutamate and inhibited by dopamine and GABA. Glutamate (green) released from cortical afferents stimulates MSNs through post-synaptic AMPA receptors (R).30 Dopamine (purple) modulates corticostriatal activity through D1- or D2-class dopamine receptors on MSNs and ‘filters’ corticostriatal activity though D2Rs acting on corticostriatal terminals.16 Synaptic ‘filtering’ occurs when dopamine inhibits a subset of cortical terminals with a low probability of release.12,16 GABA (blue) is capable of providing strong inhibition of corticostriatal activity through ionotropic GABAARs on MSNs, and also through metabotropic GABABRs located on corticostriatal terminals. GABABRs are relatively inactive in the resting state and provide little tonic inhibition.41 GABAergic fast-spiking and PLTS-type interneurons are excited by glutamate and are inhibited by dopamine D2Rs,32 as well as by GABAA autoreceptors33 that are tonically-activated by GABAergic inputs from the pallidum and other sources.41 (A1) In saline and SPF mice, D2R stimulation with an agonist inhibits glutamate release from a subset of cortical terminals (red box). D2R stimulation slightly depolarizes (activates) PLTS interneurons (red oval), but provides no downstream modulation of corticostriatal activity via GABABRs on cortical terminals. (B) PCE reduces GABA interneuron migration38 and hyperpolarizes (inhibits) both FS and PLTS-type interneurons (double blue arrow). The putative reduction in GABA availability promotes overexpression of GABABR1a-receptor subunits which sensitizes GABABRs on corticostriatal terminals (red arrow) to produce tonic over-inhibition of glutamate release (blue arrow). Phasic dopamine release is also suppressed. (B1) Stimulation of D2Rs (red oval) suppresses GABA interneurons, by reducing glutamate release from cortical afferents (blue arrow) and by hyperpolarizing PLTS-type GABA interneurons. The reduction in GABA availability (double blue arrow) relieves tonic inhibition at GABABRs on corticostriatal terminals to produce a paradoxical increase in glutamate release (red arrow). D2Rs located on corticostriatal terminals (red box) remain inhibitory, but since GABABRs are more potent modulators of presynaptic release,30 corticostriatal activity is dominated by the change in GABA. (B2) Blockade of inhibitory GABAA autoreceptors (blue oval) has little effect on interneuron function following PCE, but competes with the D2R-dependent reduction in GABA inhibition (red oval) and prevents dopamine-dependent corticostriatal excitation following PCE. Therefore, when D2R are stimulated in the presence of a GABAAR antagonist, the synapse may remain suppressed by tonic inhibition at GABABRs, but dopamine filtering (red box) is restored.
Materials and Methods
Animals
Procedures were approved by the University of Washington Institutional Animal Care and Use Committee and are detailed in the Supplementary Methods. Timed-pregnant Swiss Webster-strain (n=78), C57Bl6-strain (n=9), and Lhx6-GFP BAC transgenic (n=10; MMRRC-GENSAT project, www.gensat.org) dams received either cocaine (20mg/kg, s.q.) or saline twice daily from embryonic day 8 through 18.7 As cocaine causes anorexia,7 controls included a saline-treated, pair-fed group (SPF) whose nutritional intake was matched to that of cocaine-treated dams, and a saline-treated group that were allowed to feed ad libitum (saline). Litters were fostered to an untreated surrogate dam at birth (P0), weaned by P22, and utilized as indicated below.
Behavior
Tail-flick, rotarod, open-field, locomotor, and tail suspension tests were performed in 181 mice, aged 60–80 days, as described17–21 in the Supplementary Methods.
Electrophysiology
Whole-cell electrophysiological recordings were made in 318 neurons from 125 mice aged 30–32 and 55–94 days, as described22 in the Supplementary Methods.
Multiphoton Optical Imaging with FM1-43
Optical recordings of presynaptic corticostriatal release were made in slices from 46 mice, aged 30–33 days and 60–80 days, as described16 in the Supplementary Methods.
Western Blotting
Tissue was extracted from the dorsal striatum of 14 mice, aged 63–70 days and biotinylated for both total and surface receptor (R) expression of GABABR subunit proteins, as described23 in the Supplementary Methods.
Fast Scan Cyclic Voltammetry
Dopamine efflux was measured at sub-second resolution in striatal slices from 14 mice, aged 63–79 days using cyclic voltammetry, as described15,20,24 in the Supplementary Methods.
Statistics
Values given in the text and in the figures are Mean ± SEM. Differences, considered significant if p<0.05, were assessed with appropriate t-tests, ANOVAs or the non-parametric Mann-Whitney test (see Supplementary Methods).
Results
Abnormal growth and dopamine-dependent behaviors
Timed-pregnant cocaine-exposed (n=19) and SPF dams (n=17) consumed similar quantities of food throughout gravidity (p=0.95), but 14% less than saline dams (n=20; p<0.001, t-test; Fig 2A). Maternal weights reflected these differences in food intake, as saline dams gained more weight than either cocaine or SPF dams (p<0.001, repeated-measures ANOVA; Fig 2B). Following birth, cocaine-exposed pups (n=59) weighed less than either saline (n=96) or SPF pups (n=51) until week 6 (p<0.01, t-test; Fig 2C), while the cranial diameter of saline pups (n=100) was greater than that of SPF (n=24) or cocaine pups (n=35) after week 1 (p<0.01, t-test; Fig 2D). The reduction in postnatal weight was dependent on PCE, while the smaller head size was a consequence of nutrition.
FIGURE 2.
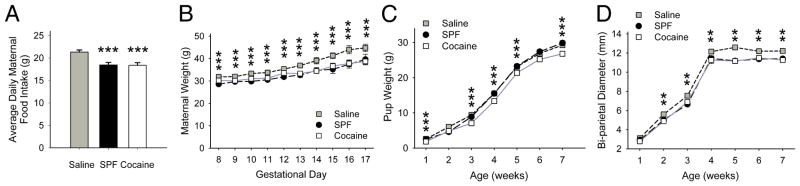
PCE reduces growth. (A) Cocaine (18.38±0.6 g/d) and SPF (18.44±0.6 g/d) dams consumed less food than saline dams (21.33±0.5 g/d). For panels A, B and D, **p<0.01, ***p<0.001 for saline compared to SPF or cocaine, t-test. (B) The weight of cocaine-treated and SPF dams were similar throughout pregnancy and both weighed less than saline-exposed dams. (C) Cocaine pups weighed less than saline pups through week 6 and weighed less than SPF pups through week 7. SPF pups weighed less than saline pups on weeks 1 and 2 (p<0.05, t-test), but weighed more than saline pups after week 3 (p<0.01, t-test). (D) Saline pups had a larger cranial (bi-parietal) diameter than either cocaine or SPF pups after week 1 and there was no difference in the cranial diameter of cocaine and SPF mice (p>0.1 t-test).
Dopamine-dependent reflexes and behaviors were assessed in adolescent mice. Cocaine-exposed mice (n=19) showed increased nociceptive latencies on tail-flick testing compared to either saline (n=27) or SPF mice (n=20; p<0.001, Mann-Whitney; Fig 3A). Rotarod testing for motor coordination revealed increasing falling latencies for saline (n=26), SPF (n=20), and cocaine-exposed mice (n=8; p<0.001, repeated-measures ANOVA; Fig 3B), consistent with motor learning.19 However, falling latencies in cocaine-exposed mice decreased in later trials (p<0.05 compared to either saline or SPF mice, ANOVA) and cocaine mice gripped the rod more frequently than either saline or SPF mice (p<0.001, Mann-Whitney; Fig 3C).
FIGURE 3.

PCE causes abnormal behaviors. (A) The latency for removing the tail from warm water was higher in cocaine-exposed mice over three trials, while the flick-latencies of saline and SPF mice were similar (p=0.48, Mann-Whitney). For panels A and C, **p<0.01, ***p<0.001 for cocaine compared with saline or SPF, Mann-Whitney test. (B) Saline, SPF and cocaine-exposed mice spent a similar amount of time on the rotarod over the first 15 trials (p=0.1, t-test), but the falling latency decreased in cocaine mice after trial 15, while latencies of saline and SPF mice increased. For panels B, D, and E, *p<0.05, **p<0.01, ***p<0.001 for cocaine compared with either saline or SPF mice, t-test. (C) Instead of falling, some mice would grip onto, and rotate with the rod. Cocaine mice gripped the rotarod more than saline or SPF mice, while there was no difference between the SPF and saline control groups (p=0.1, Mann-Whitney). (D) Open-field ambulations of saline, SPF and cocaine-exposed mice were similar on test day 1. A saline injection, administered immediately following the first test increased ambulations in cocaine-exposed mice on test day 2. (E) Saline, SPF, and cocaine mice were treated with saline for 2 days and then received amphetamine (2 mg/kg, i.p.) for 5 consecutive days. Mice were later challenged with amphetamine on days 10 and 28. Locomotor activity measured for 90 min following each amphetamine treatment revealed no stereotypic behaviors (data not shown). Locomotor activity increased in response to repeated amphetamine in all treatment groups. Ambulations in saline and SPF mice remained similar throughout testing (p=0.07, repeated-measures ANOVA). Cocaine-exposed mice demonstrated reduced ambulations on experiment days 4–7, but not following a drug challenge in withdrawal (days 10 and 28), as locomotor activity was similar in all groups (p=0.1, ANOVA). (F) When suspended by the tail, saline, SPF, and cocaine-exposed mice showed an equivalent increase in immobility over time, as there was no significant Treatment*Time effect (p=0.16, two-way ANOVA). These groups of mice also spent a similar amount of time immobile at most time points during the test (min 1, 2, 4, and 6). However, compared to saline, both cocaine and SPF mice spent a longer time immobile during min 3 and min 5. *p=0.03 and **p=0.01 for saline compared to either SPF or cocaine mice, t-test.
Locomotion in the open-field was similar in saline (n=35), SPF (n=38), and cocaine-exposed mice (n=24; Fig 3D). Following a saline injection, the ambulations of saline and SPF mice remained similar while cocaine-exposed mice became more active (p<0.001, t-test). In the absence of injection, similar responses were found in all groups (not shown).
To test for dopamine-dependent plasticity,20 locomotor responses were measured in response to repeated amphetamine (2mg/kg/d i.p.). Saline (n=14), SPF (n=20), and cocaine-exposed mice (n=12) all demonstrated locomotor sensitization with an increase in locomotor activity following repeated amphetamine (p<0.01, repeated-measures ANOVA). Similar to prior studies,25 cocaine-exposed mice had blunted locomotor augmentation to repeated amphetamine (p<0.001, repeated-measures ANOVA; Fig 3E), but comparable sensitized responses to amphetamine challenges in withdrawal.
As a control experiment to test for hypodopaminergia21 and depression,26 mice were suspended by the tail and time spent immobile was measured. While there was no overall difference between saline (n=49), SPF (n=29), and cocaine mice (n=29; Fig 3F), both SPF and cocaine mice displayed small increases in immobility compared to saline mice, suggesting an effect of nutritional deficiency as opposed to PCE.
Presynaptic inhibition of corticostriatal activity
These behaviors suggested that PCE might alter dopamine-dependent excitatory neurotransmission within the dorsal (motor) striatum.27 Whole-cell electrophysiological recordings in MSNs from SPF and cocaine-exposed mice revealed similar passive and active membrane properties (Fig 4A and Supplementary Table S1). However, compared to SPF mice, neurons from cocaine-exposed mice were 36% less responsive to current injection (p<0.001, ANOVA; Fig 4B) and cortical stimulation >0.6 mA evoked lower amplitude excitatory postsynaptic currents (eEPSC; p<0.05, two-way ANOVA; Fig 4C and D). This inhibition of corticostriatal activity following PCE was likely of presynaptic origin,28 since the eEPSC paired-pulse ratio (PPR) was higher in cocaine (1.2±0.05; n=49 cells) than in SPF (1.07±0.03; n=60; p=0.01) and the frequency of miniature (m) EPSCs was 18% less in cocaine-exposed cells (n=19) compared to SPF (n=14; p=0.03, t-test), while the cumulative mEPSC amplitude distributions were unchanged (Fig 4E).
FIGURE 4.

PCE causes presynaptic depression through GABABRs. (A) Current clamp recordings in MSNs from SPF (n=15) and cocaine (n=11) mice displayed similar current-voltage curves with inward rectification, typical for MSNs (responses were measured at arrows in panel B). (B) Representative traces (above) demonstrate that fewer action potentials were generated in cells from cocaine mice in response to depolarizing current pulses (below). (C) The corticostriatal slice stained with FM1-43 and diaminobenzidine shows the areas of stimulation and recording. Corticostriatal activity was provoked using a bipolar stimulating electrode placed over cortical layers V–VI. Electrophysiological, optical, and biochemical recordings were obtained from the corresponding motor striatum (recording region), located 1.5–2.0 mm from the site of stimulation. (D) Representative traces (above) of voltage-clamp recordings show that similar cortical stimulation intensities evoked lower amplitude currents in MSNs from cocaine-exposed mice, compared to SPF. The AMPAR antagonist NBQX prevented evoked currents. Graph (below) shows the mean peak current evoked by the series of increasing cortical stimulation intensities in MSNs from SPF (n=10) and cocaine (n=12) mice. The GABABR antagonist CGP52432 had no effect on SPF cells, but blocked the reduction in eEPSC amplitudes in cocaine MSNs. *p<0.05, **p<0.01, t-test. Cells were voltage clamped at −70 mV to minimize post-synaptic GABAAR-mediated conductances (calculated ECl− = −74.2 mV).47 (E) Representative traces of mEPSCs (above; recorded in the presence of tetrodotoxin (1μM) show a reduction in low-release probability (5–20 pA) inward currents in cells from cocaine mice. The average frequency of mEPSCs (inset, left) was lower in cocaine MSNs (3.4±0.4 Hz), compared to SPF (4.2±0.6 Hz), while the cumulative mEPSC amplitude distributions (inset, right) were similar. *p<0.05, t-test. (F) Stimulation of axons or cell bodies of projection neurons in layers V–VI of the cortex overlying the motor striatum resulted in endocytosis of FM1-43 dye by recycling synaptic vesicles, characteristic of corticostriatal afferents.16 Following dye loading, cortical stimulation at 20 Hz (beginning at t=0) resulted in exocytosis of FM1-43 dye from the terminals, which decreased in a manner approximated by a single exponent, characteristic of synaptic vesicle fusion.22 Feedback from MSNs was prevented using glutamatergic receptor antagonists (see Supplemental Methods). FM1-43 destaining was activity and calcium-dependent since no stimulation (n=30) or bath-applied cadmium (200μM; n=25) prevented stimulated release of the dye from presynaptic terminals. As FM1-43 destaining generally followed first-order kinetics, corticostriatal release was characterized by the halftime (t1/2) of release, defined as the time required for terminal fluorescence to decay to half its initial value.22 (G) Mean ± SEM halftimes of FM1-43 release for destaining curves shown in panel F. FM1-43 destining was similar in slices from saline (t1/2=205 sec) and SPF (t1/2=200 sec) mice (p=0.6, Mann-Whitney), but was reduced in slices from cocaine mice (t1/2=233 sec; *p<0.05, Mann-Whitney). (H) An advantage of this optical technique is that we are able to examine vesicular release kinetics from individual cortical terminals. When the halftimes of individual terminals are presented relative to their standard deviation from the median value, a straight line indicates a normally-distributed (or single) population.15 Normal probability plots of individual terminal halftimes of release for experiments in panel F show that PCE decreased exocytosis from the slowest-releasing terminals (those with the highest t1/2). This depression in corticostriatal release following PCE was not due to an inadequate innervation since the number of active corticostriatal terminals was higher in cocaine mice (61.4±10 puncta vs. 41.9±5 for saline; p=0.02, ANOVA). Bars: B, 40 mV, 25 ms; C, 1 mm; D, 50 pA, 5 ms; E, 10 pA, 250 ms. Curves were fit with a Hill equation.
Exocytosis from corticostriatal terminals was directly examined by multiphoton microscopy using the endocytic tracer FM1-43.15 The halftime of FM1-43 release was 14% lower in cocaine-treated mice (n=125 puncta; p=0.02, Mann-Whitney) and compared to saline (n=102) or SPF (n=100), PCE selectively inhibited the subset (~50%) of cortical terminals with a low probability of release, while the faster-releasing terminals remained unperturbed (Fig 4F–H). This synaptic depression was long-lasting, since exocytosis in younger 30-day-old mice was 18% lower in cocaine (t1/2=245 sec; n=114) compared to saline (t1/2=201 sec; n=68; p=0.01, Mann-Whitney not shown).
GABABR-dependent over-inhibition at corticostriatal synapses
We tested whether this presynaptic depression might occur through GABAergic neurotransmission, which regulates presynaptic corticostriatal activity14 via metabotropic GABABRs located on corticostriatal terminals.29 Tonic inhibition by GABABRs was absent in SPF mice, since the GABABR antagonist CGP52432 (10μM) did not change the eEPSC amplitude (Fig 4D) or the frequency of mEPSCs (10±14%; n=7 cells; Fig 5A). However, CGP52432 prevented the tonic inhibition of MSNs from cocaine-exposed mice (Fig 4D) and increased the frequency of mEPSCs (19±3%; n=6; p=0.02, paired t-test; Fig 5B).
FIGURE 5.

(A) Representative traces show that CGP52432 had no effect on the frequency of mEPSC in cells from SPF mice (3.2±0.8 Hz in vehicle vs. 3.3±0.9 Hz following CGP52432; p=0.1, paired t-test) and the cumulative distribution of mEPSC amplitudes was unchanged. (B) CGP52432 increased the frequency of mEPSCs in cells from cocaine mice (3.1±0.8 Hz in vehicle vs. 3.9±0.8 Hz following CGP52432) and the cumulative mEPSC amplitude distribution was unchanged. #p<0.05, ##p<0.01, paired t-test. (C) Representative traces show the average responses to paired-pulses delivered at 50 ms every 30 sec before (above, left) and 5 to 7.5 min following bath application of baclofen (above, right). Graph shows the normalized amplitude of the first eEPSC (of the pair) and the normalized PPR. Baclofen reduced the amplitude of the first eEPSC (−92±23 pA for vehicle vs. −21±5 pA following baclofen) and increased the PPR (1.1±0.1 in vehicle to 1.6±0.2 following baclofen) in MSNs from SPF mice. (D) In cells from cocaine-exposed mice, baclofen reduced the amplitude of the first eEPSC (−59±6 pA for vehicle vs. −13±4 pA in baclofen) and increased the PPR (1.2±0.1 in vehicle vs. 1.8±0.4 in baclofen). (E) Concentration-curves demonstrate that baclofen (100 nM – 50 μM) reduced the amplitude of the first eEPSC and (F) increased the PPR to a greater degree in MSNs from cocaine-exposed mice than in SPF controls (IC50= 4.2 μM;48 n=6, 5, 12, 5, 6 cells for SPF and n=4, 5, 5, 4 for cocaine. *p<0.05, **p<0.01, ANOVA). Bars: A and B, 10 pA, 250 ms; C and D, 100 pA, 12.5 ms.
To determine if PCE might change the sensitivity of presynaptic GABABRs, eEPSCs from MSNs were measured in response to cortical stimulation with 50 ms paired-pulses, applied every 30 sec.30 In MSNs from SPF mice, the GABABR agonist baclofen (5μM) decreased the amplitude of the first current of the pair (−67±3%; p=0.002) and the PPR increased (44±8%; n=14; p<0.001, paired t-test; Fig 5C), consistent with strong presynaptic inhibition by GABABRs.30 In cocaine-exposed mice, baclofen also decreased the amplitude of the first eEPSC (−79±4%; n=8; p=0.03) and the PPR increased (61±6%; p=0.01, paired t-test; Fig 5D). Over a range of concentrations, baclofen depressed corticostriatal activity to a greater degree in cocaine-exposed MSNs (p<0.05, 2-way ANOVA; Fig 5E and F), suggesting that PCE increases GABABR sensitivity.
Overexpression of GABABR1a-receptor subunits
Western blots determined striatal GABABR subunit expression in the dorsal striatum and the membrane surface protein pool was detected with biotinylation. Results showed a 25±5% increase in the GABABR1a-receptor subunit surface protein membrane pool from cocaine-exposed striatum (1.41±0.3) compared to SPF (1±0.1; n=7 mice each, p=0.02, t-test; Fig 6). The total protein level of GABABR1a and levels of both GABABR1b and GABABR2 were unchanged, indicating that PCE selectively increases the expression of GABABR1a-receptor subunits, which are preferentially expressed on corticostriatal terminals.31
FIGURE 6.
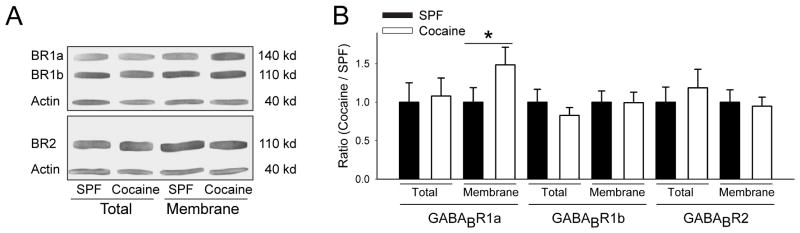
PCE increases the surface expression of GABABR1a receptor subunits in the dorsal striatum. (A) Representative immunoblots of lysates from the dorsal striatum of 60-day-old SPF and cocaine-exposed mice compare levels of total and surface membrane-bound fractions of GABABR1, GABABR2, and actin. Antibody against GABABR1 labeled 2 bands, GABABR1a and GABABR1b. (B) Densitometry measurements of receptor protein bands presented as the ratio of densities for SPF and cocaine-exposed mice measured in the same gel (*p<0.05, t-test).
Paradoxical Excitation by D2Rs is dependent on GABABRs
Dopamine modulates corticostriatal activity through D2Rs16 and in MSNs from SPF mice, the D2R agonist quinpirole decreased the amplitude of the first eEPSC (−15±7%; n=9 cells; p=0.004) and increased the PPR (30±8%; p=0.01, paired t-test; Fig 7A). In cocaine mice, quinpirole paradoxically increased the eEPSC amplitude (43±19%; n=8; p=0.02) and decreased the PPR (−25±6%; p=0.05, paired t-test; Fig 7B). This aberrant excitation of corticostriatal activity by D2Rs following PCE was likely presynaptic since quinpirole reduced the frequency of mEPSCs in SPF cells (−18±6%; n=7; p=0.003; Fig 7C), but enhanced their frequency in cocaine-exposed neurons (38±14%; n=8; p=0.04, paired t-test; Fig 7D). Similar plasticity was found in MSNs from mice exposed to half the dose of cocaine and also in younger 30-day-old C57Bl6-strain mice (Supplementary Data and Fig S1).
FIGURE 7.

D2Rs provoke GABAAR-dependent paradoxical responses following PCE. (A) In MSNs from SPF mice, representative traces (above) and graph demonstrate that the D2R agonist quinpirole reduces the amplitude of the first eEPSC (in each pair; −160±27 pA for vehicle vs. −140±29 pA following quinpirole) and increases the PPR (1.2±0.1 in vehicle to 1.6±0.2 in quinpirole). (B) In MSNs from cocaine mice, quinpirole increased the amplitude of the first eEPSC (−81±18 pA for vehicle vs. −106±20 pA for quinpirole) and the PPR decreased (1.4±0.2 in vehicle vs. 1.0±0.1 in quinpirole). (C) In MSNs from SPF mice, quinpirole diminished the frequency (inset, left; 5.1±0.9 Hz in Veh vs. 4.1±0.8 Hz in quinpirole), but not amplitude (inset, right) of low-release probability (5–10 pA) inward currents. For panels C and D, #p<0.05, ##p<0.01, paired t-test. (D) In MSNs from cocaine-exposed mice, quinpirole increased the frequency (3.4±0.7 Hz in Veh vs. 4.7±1 Hz in quinpirole) of low-release probability mEPSCs, while having no effect on the cumulative amplitude distribution. (E) Amphetamine (left) and quinpirole (right) decreased FM1-43 destaining in slices from saline and SPF mice, but increased release in slices from cocaine mice. *p<0.05, **p<0.01, Mann-Whitney. (F) In cells from SPF mice, the GABABR antagonist CGP52432 did not change the eEPSC amplitude (−132±20 pA for Veh vs. −122±16 pA for CGP52432) or the PPR (1.3±0.1 in Veh vs. 1.4±0.1 in CGP52432). When quinpirole was added to CGP52432, the eEPSC amplitude decreased (−115±15 pA) and the PPR increased (1.6±0.1). (G) In MSNs from cocaine-exposed mice, the GABABR antagonist increased the eEPSC amplitude (−94±23 pA for Veh vs. −121±26 pA for CGP52432) and decreased the PPR (1.5±0.1 in Veh vs. 1.0±0.1 in CGP52432). When quinpirole was added to CGP52432, there was a slight reduction in eEPSC amplitude (−112±12 pA) and an increase in the PPR (1.3±0.1). (H) In MSNs from SPF mice, bicuculline did not change the amplitude of the first evoked current (−143±31 pA for Veh vs. −148±33 pA for bicuculline; p=0.1) or the PPR (1.1±0.1 in Veh vs. 1.2±0.2 in bicuculline; p=0.06, paired t-test). When quinpirole was added to bicuculline, the eEPSC amplitude decreased (−132±22 pA) and the PPR increased (1.5±0.1). (I) In MSNs from cocaine mice, bicuculline did not change the amplitude of the eEPSC (−103±24 pA for Veh vs. −101±21 pA for bicuculline) or the PPR (0.9±0.1 in Veh vs. 0.9±0.1 in bicuculline). When quinpirole was added to bicuculline, the eEPSC amplitude decreased (−82±21 pA) and the PPR increased (1.3±0.1). Bars: A, B, F, and G–I, 100 pA, 12.5 ms; C and D, 10 pA, 250 ms.
These changes in corticostriatal activity were accompanied by long-term adaptations in dopamine transmission, as measurements of electrically-evoked dopamine release and reuptake using cyclic voltammetry showed that PCE produced region-specific alterations in phasic dopamine release without affecting clearance (Supplementary Data and Fig S2). However, optical experiments in slices from saline and SPF mice, confirmed that both the dopamine releaser amphetamine15 (10μM) and quinpirole reduced exocytosis by specifically modulating glutamatergic inputs with a low probability of release (Fig 7E, Supplementary Data and Fig S3), but boosted release from those same synapses in slices from cocaine-exposed mice. Consistent with the lack of D1Rs on cortical terminals within the dorsal striatum,15 D1R ligands did not change exocytosis or the modulatory response to amphetamine (Supplementary Data and Fig S4).
While D2Rs inhibit corticostriatal release in untreated mice,15 they also suppress GABA interneuron function,32 and might increase corticostriatal activity by reducing tonic inhibition at sensitized GABABRs. In MSN from SPF mice, inhibition by D2Rs was not dependent on GABABRs, as the GABABR antagonist CGP52432 did not change the eEPSC amplitude (−5±3%; n=6) or the PPR (13±5%), whereas CGP52432 with quinpirole decreased the eEPSC amplitude (−23±4%; p<0.03) and increased the PPR (33±11%; p<0.02, compared with vehicle or CGP52432, paired t-test; Fig 7F). In MSNs from cocaine-exposed mice however, CGP52432 was excitatory, as the eEPSC amplitude increased (38±15%; n=8; p=0.03) and the PPR decreased (−26±6%; p=0.02, paired t-test; Fig 7G). CGP52432 with quinpirole remained excitatory, but the eEPSC amplitude decreased (−18±8%; p<0.04) and the PPR increased (19±12%; p<0.05, compared with vehicle or CGP52432, paired t-test), possibly due to concurrent corticostriatal inhibition by D2Rs.30
GABAAR blockade prevents D2R-dependent excitation
GABAARs promote tonic inhibition of GABA interneurons,33 but are absent from presynaptic corticostriatal terminals.29 In MSNs from SPF mice, the GABAAR antagonist bicuculline (10μM) did not change the eEPSC amplitude (5±3%; n=9) or the PPR (12±7%; Fig 7H) and D2Rs remained inhibitory, as quinpirole in the presence of bicuculline decreased the eEPSC amplitude (−15±3%; p=0.02) and increased the PPR (50±9%; p<0.01, compared with vehicle or bicuculline, paired t-test). In cocaine cells, bicuculline did not change the eEPSC amplitude (2±7%; n=9; p=0.3) or the PPR (3±4%; p=0.6), but bicuculline blocked excitation by D2Rs since quinpirole with bicuculline reduced the eEPSC amplitude (−25±8%; p<0.02) and increased the PPR (38±7%; p<0.04, compared with vehicle or bicuculline, paired t-test; Fig 7I).
PCE alters GABA interneuron function
As results indicated that cocaine-induced corticostriatal plasticity is generated through GABA interneurons, we used Lhx6-GFP transgenic mice to target striatal fast-spiking (FS) and persistent-low-threshold-spiking (PLTS) interneurons. Whole-cell voltage- and current-clamp recordings identified FS and PLTS interneurons by their distinctive physiological properties14,34 and showed that PCE lowered their resting membrane potentials (FS, −11.6%, p=0.04; PLTS, −18%, p=0.02) and action potential thresholds (FS, −18%, p=0.02; PLTS, −27%, p=0.001, compared to saline, t-test; Supplementary Table S2–S4, Fig 8A and B).
FIGURE 8.

Abnormal GABA interneuron function. (A) Current clamp recordings show characteristic responses of FS and (B) PLTS interneurons from Lhx6-GFP transgenic mice to hyperpolarizing and depolarizing current injections (below). Both FS and PLTS interneurons from cocaine cells had lower resting membrane potentials (RMP) than saline cells. Current-voltage plots (right) show similar responses in saline and cocaine-exposed cells after subtraction of their resting membrane potentials (responses were measured at arrows). FS cells were silent at rest, and displayed a high firing rate with little adaptation following depolarizing current injection. Spikes were short and followed by a large after-hyperpolarization. PLTS interneurons exhibited a marked time-dependent sag in response to hyperpolarizing current injections and a rebound persistent low-threshold spike and/ or a plateau potential persisted after termination of hyperpolarizing current. During current injections, both FS and PLTS interneurons displayed a variable pattern of spike bursts (1–48 action potentials) interspersed by membrane oscillations. Compared to FS interneurons, PLTS interneurons exhibited a much higher input resistance, a lower resting membrane potential (RMP), and a much lower input current was required to produce action potentials (Supplementary Tables S3–S5), with values similar to those reported previously.14,49 (C) Representative current-clamp recordings in FS and (D) PLTS interneurons from saline-(above) and cocaine-exposed mice (below) demonstrate typical responses to the GABAAR antagonist bicuculline before and after bath application of the sodium channel blocker tetrodotoxin. In FS and PLTS interneurons, bicuculline depolarized saline-exposed cells (the RMP became more positive) to a much greater degree than cocaine-exposed cells. GABA likely produced tonic inhibition at GABAA autoreceptors14 since the change in membrane potential by bicuculline in saline-exposed mice (FS, 37±2%, p<0.001; PLTS, 23±7%, p=0.03) and cocaine-exposed mice (FS, 3±1%, p=0.09; PLTS, 4±1%, p=0.003, paired t-test) persisted when synaptic transmission was blocked by tetrodotoxin. Note that the cellular input resistance was monitored by (250 ms, 100 pA) current pulses applied every 10 sec. Changes in input resistance during depolarization were measured after transiently repolarizing the cell to resting membrane potential levels. Interestingly, bicuculline reduced the input resistance (Supplementary Fig S5), suggesting recruitment of additional ion channels with depolarization that are critical for sustained high-frequency firing.39 (E) Representative current-clamp recordings in FS and (F) PLTS interneurons from saline-(above) and cocaine-exposed mice (below) demonstrate typical responses to the D2 receptor agonist quinpirole before and after bicuculline. Quinpirole had no effect in FS interneurons, but slightly depolarized saline-exposed PLTS cells, while hyperpolarizing cocaine-exposed PLTS cells. For all interneurons, the membrane potential became more positive and the cell depolarized when bicuculline was added to quinpirole. A summary of membrane potentials and input resistance for FS and PLTS interneurons under all conditions tested can be found in Supplementary Fig S5. (G) Excitatory inputs onto GFP+ fluorescent interneurons from Lhx6-GFP transgenic mice were activated with paired-pulses using cortical bipolar stimulating electrodes. The PPR was similar in cells from saline (1.35±0.11; n=11) and cocaine-exposed mice (1.13±0.09; n=8; p=0.2, t-test). In saline-exposed mice, representative traces (above) and graph show that quinpirole did not change the amplitude of the first eEPSC (−32±3 pA in vehicle vs. −35±6 pA following quinpirole) or the PPR (1.3±0.1 in vehicle vs. 1.3±0.1 in quinpirole). When quinpirole was combined with bicuculline, the eEPSC amplitude increased (−45±6 pA), but the PPR remained unchanged (1.3±0.3). The AMPA-receptor antagonist NBQX (10 μM) abolished the eEPSC. (H) In GFP+ interneurons from cocaine-exposed mice, quinpirole reduced the amplitude of the first eEPSC (−62±5 pA in vehicle vs. −49±9 pA following quinpirole) and the PPR increased (1.1±0.1 in vehicle vs. 1.4±0.2 in quinpirole). Paired-pulse depression was observed in the presence of bicuculline, which prevented the change in PPR (1.1±0.2) and the eEPSC amplitude increased (−95±13 pA). Bars: A and B, 30 mV, 20 ms; C–F, 30 mV, 2 min; G and H, 50 pA, 12.5 ms.
In saline-exposed mice, ambient GABA inhibited interneurons as the GABAAR antagonist bicuculline depolarized both FS (32±2%, n=4, p<0.001) and PLTS cells (26±8%, n=5, p=0.04, paired t-test; Fig 8C and D). However, cocaine-exposed FS and PLTS cells demonstrated little depolarization following bicuculline (FS, 2±3%, p=0.4; PLTS, 4±2%, p=0.1), consistent with the reported reduction in GABAAR subunit expression following PCE.23,27
D2Rs can modify tonic GABA inhibition through G-protein-coupled phosphorylation of GABAAR subunits by PKA.35 The D2R agonist quinpirole did not change the membrane potential in FS interneurons from saline- (1±1%, n=7) and cocaine-exposed mice (1±1%, n=7), but it depolarized PLTS cells from saline-exposed mice (5±1%, n=8, p=0.003) and hyperpolarized PLTS cells from cocaine-exposed mice (4±1%, n=6; p=0.02, paired t-test; Fig 8E and F). While quinpirole reduced the extent of depolarization by bicuculline in saline-exposed FS (29%, n=7, p=0.003, t-test) and had little effect on quinpirole in PLTS interneurons (4%, n=8), quinpirole promoted substantial depolarization by bicuculline in both cocaine-exposed FS (29±8%, p<0.001) and PLTS interneurons (24±8%; p=0.02, paired t-test). Thus, the paradoxical reduction in GABA interneuron function following D2R activation was alleviated by the GABAAR antagonist.
PCE also provoked abnormal excitatory, AMPA receptor-mediated responses in GABA interneurons. In saline-exposed mice, quinpirole had no effect on the eEPSC amplitude (7±10%; n=11; p=0.4) or the PPR (0.5±9%; p=0.9). The addition of bicuculline to quinpirole did not change the PPR (−0.2±10%), but enhanced cell excitability as the eEPSC amplitude increased (55±11%; p<0.02, compared with vehicle or quinpirole, paired t-test; Fig 8G). Following PCE, quinpirole reduced the eEPSC amplitude (−18±5%; n=8; p=0.02) and increased the PPR (27±7%; p=0.01). The addition of bicuculline to quinpirole increased the eEPSC amplitude (29±2%; p=0.01), while the PPR approached baseline (−2±5%; Fig 8H; p=0.9, compared with vehicle; p=0.02, compared to quinpirole, paired t-test).
Discussion
PCE can produce long-lasting behavioral and motor disturbances in affected children,4,5 and it reduced dopamine-dependent reflex and motor task performance in mice. These abnormal behaviors were paralleled by a long-lasting and reversible depression of corticostriatal activity that was generated though abnormally-functioning GABA interneurons (Fig 1B). PCE promoted inappropriate dopamine filtering of corticostriatal activity; rather than boosting stronger cortical connections by inhibiting the weak,15 D2Rs strengthened weaker glutamatergic synapses while stronger signals remained unperturbed (Fig 1).
GABA interneurons are created in the medial ganglionic eminence36 and become potent regulators of corticostriatal signalling,37 but their tangential migration is reduced following PCE.38 PCE reduced FS and PLTS interneuron excitability and the downstream increase in GABABR1a-receptor subunit sensitivity would inhibit corticostriatal activity when extracellular GABA surges during synaptic activity.39
While normally promoting inhibition at corticostriatal synapses,15,30 D2Rs boosted corticostriatal activity in cocaine-exposed mice. D2Rs likely reduced tonic inhibition of corticostriatal GABABRs by inhibiting excitatory inputs on GABA interneurons and by hyperpolarizing PLTS interneurons. Because only small currents are required to activate PLTS interneurons, minor decreases in the membrane potential become physiologically significant, requiring recruitment of additional excitatory inputs39, which are also aberrantly controlled by D2Rs.
We also found that a GABAAR antagonist can prevent this paradoxical excitation of corticostriatal activity by D2Rs. While GABAAR blockade had no direct effect on corticostriatal activity,29 it prevented the reduction in tonic GABA currents that occurred in response to D2R activation. Inhibition of GABA interneuron activity by GABAA autoreceptors33 is dependent on ambient GABA supplied by striatal neurons and inputs from the external pallidum.33,40,41 In controls, GABAAR blockade suppressed inhibitory tonic GABA currents, the interneuron depolarized, but any increase in synaptic GABA remained undetected at corticostriatal GABABRs. PCE hyperpolarized GABA interneurons and also suppressed bicuculline-sensitive tonic GABA currents, which were reactivated by the D2R agonist. Thus, the combination of GABAAR antagonist and D2R agonist would strengthen corticostriatal inhibition at GABABRs and oppose any reduction in tonic GABA caused by the D2R agonist alone.
D2Rs located on GABA interneurons33 modulate tonic GABA currents through PKA35 which is essential to the physiological states of both dopamine30 and GABAA receptors.14,34 In striatal MSNs, tonic GABAAR currents are dependent on β3-subunit phosphorylation which is inhibited by D2R inactivation of PKA through intracellular Gi/o protein coupling.35 PCE down-regulates cortical β3-GABAAR subunits,23 suggesting that these aberrant responses to D2R and GABAAR activation may be due to alterations in receptor subunit expression, G-protein coupling, phosphorylation, or GABA transport.35,40
Synaptic depression and dopamine-dependent excitation at corticostriatal synapses are also elicited by repeated psychostimulant exposure in adult mice.20 In contrast to PCE, these phenomena in mature mice are produced through D1 and cholinergic receptors. PCE failed to elicit excitatory D1R responses, perhaps due to D1R uncoupling42 or because the dopamine-releasing effects of cocaine in rodents may not be possible until the first week of life.43 Differences in plasticity were also reflected in the observed behaviors, as PCE reduced the magnitude of augmented locomotor responses to repeated amphetamine, but achieved comparable locomotion with controls in withdrawal. Thus, PCE did not spare the adaptive behavior that is linked to incentive saliency and drug dependence.44
Other observations following PCE might also arise from paradoxical D2R responses generated by over-inhibited synapses. The reduction in body weight is consistent with dysregulated D2R-mediated hypophysiotrophic function45 (as well as through reduced uteroplacental blood flow6) and decreased thermal reactivity suggests an abnormal D2R-dependent reflex arc.17,18 Cocaine-exposed mice also performed poorly on late rotarod trials and demonstrated a heightened response to aversive stimulation, suggesting that stress or unanticipated environmental changes cause inconsistent behaviors, similar to those reported in adolescent boys with a history of PCE.1,9
These findings support and extend reports of abnormal GABA function in the cortex and hippocampus of mouse models for PCE.23 Since GABA receptor ligands can reverse synaptic depression and allow appropriate D2R filtering at corticostriatal synapses, these findings provide insight into novel therapeutic approaches that may correct behaviors in cocaine-exposed mice and improve outcome in children with PCE, as well as a broad range of disorders that involve abnormal GABAergic signaling.46
Supplementary Material
Acknowledgments
Supported by the University of Washington Alcohol & Drug Abuse Institute (GPS and NSB), NS052536, NS060803, NS060803-2S1, HD02274 (NSB), Mary Gates Endowment for Students research scholarship (IN and JSL), F31MH08626 (JCL), 5T32DA007278 (GPS), DA016782 (PEMP), the University of Washington Vision Research Center and Seattle Children’s Hospital, Seattle, WA. We thank Drs. Mu-ming Poo, Laura Jansen and Claire Walker for advice on the Western blot and Drs. Richard Palmiter and Kyle Steinman for their critical review.
References
- 1.Church MW, Crossland WJ, Holmes PA, Overbeck GW, Tilak JP. Effects of prenatal cocaine on hearing, vision, growth, and behavior. Ann N Y Acad Sci. 1998 Jun 21;846:12–28. doi: 10.1111/j.1749-6632.1998.tb09723.x. [DOI] [PubMed] [Google Scholar]
- 2.SAMHSA. National Survey on Drug Use & Health. 2010 [database online], updated on December 30, 2008. Available at http://www.oas.samhsa.gov/nhsda.htm.
- 3.Downs T, Padbury J, Blount L, Kashiwai K, Chan K. Ovine fetal-placental cocaine pharmacokinetics during continuous cocaine infusion. J Soc Gynecol Investig. 1996 Jul-Aug;3(4):185–90. [PubMed] [Google Scholar]
- 4.Chiriboga CA, Bateman DA, Brust JC, Hauser WA. Neurologic findings in neonates with intrauterine cocaine exposure. Pediatr Neurol. 1993 Mar-Apr;9(2):115–9. doi: 10.1016/0887-8994(93)90045-e. [DOI] [PubMed] [Google Scholar]
- 5.Bada HS, Das A, Bauer CR, et al. Impact of prenatal cocaine exposure on child behavior problems through school age. Pediatrics. 2007 Feb;119(2):e348–59. doi: 10.1542/peds.2006-1404. [DOI] [PubMed] [Google Scholar]
- 6.Bandstra ES, Morrow CE, Mansoor E, Accornero VH. Prenatal drug exposure: infant and toddler outcomes. J Addict Dis. 2010 Apr;29(2):245–58. doi: 10.1080/10550881003684871. [DOI] [PubMed] [Google Scholar]
- 7.Ren JQ, Malanga CJ, Tabit E, Kosofsky BE. Neuropathological consequences of prenatal cocaine exposure in the mouse. Int J Dev Neurosci. 2004 Aug-Oct;22(5–6):309–20. doi: 10.1016/j.ijdevneu.2004.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrnes JJ, Pritchard GA, Koff JM, Miller LG. Prenatal cocaine exposure: decreased sensitization to cocaine and decreased striatal dopamine transporter binding in offspring. Neuropharmacology. 1993 Jul;32(7):721–3. doi: 10.1016/0028-3908(93)90087-j. [DOI] [PubMed] [Google Scholar]
- 9.Church MW, Tilak JP. Differential effects of prenatal cocaine and retinoic acid on activity level throughout day and night. Pharmacol Biochem Behav. 1996 Dec;55(4):595–605. doi: 10.1016/s0091-3057(96)00285-7. [DOI] [PubMed] [Google Scholar]
- 10.Keller RW, Jr, Maisonneuve IM, Nuccio DM, Carlson JN, Glick SD. Effects of prenatal cocaine exposure on the nigrostriatal dopamine system: an in vivo microdialysis study in the rat. Brain Res. 1994 Jan 21;634(2):266–74. doi: 10.1016/0006-8993(94)91929-1. [DOI] [PubMed] [Google Scholar]
- 11.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989 Oct;12(10):366–75. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 12.Bamford NS, Cepeda C. The Corticostriatal Pathway in Parkinson’s Disease. In: Tseng KY, editor. Cortico-Subcortical Dynamics in Parkinson’s Disease. New York: Humana Press; 2009. pp. 87–104. [Google Scholar]
- 13.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1992;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- 14.Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999 May;2(5):467–72. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- 15.Bamford NS, Zhang H, Schmitz Y, et al. Heterosynaptic dopamine neurotransmission selects sets of corticostriatal terminals. Neuron. 2004 May 27;42(4):653–63. doi: 10.1016/s0896-6273(04)00265-x. [DOI] [PubMed] [Google Scholar]
- 16.Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004 Oct 27;24(43):9541–52. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saade NE, Atweh SF, Bahuth NB, Jabbur SJ. Augmentation of nociceptive reflexes and chronic deafferentation pain by chemical lesions of either dopaminergic terminals or midbrain dopaminergic neurons. Brain Res. 1997 Mar 14;751(1):1–12. doi: 10.1016/s0006-8993(96)01164-x. [DOI] [PubMed] [Google Scholar]
- 18.Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005 Dec 8;438(7069):854–7. doi: 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- 19.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005 Feb 8;102(6):2174–9. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bamford NS, Zhang H, Joyce JA, et al. Repeated exposure to methamphetamine causes long-lasting presynaptic corticostriatal depression that is renormalized with drug readministration. Neuron. 2008 Apr 10;58(1):89–103. doi: 10.1016/j.neuron.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori A, Ohashi S, Nakai M, Moriizumi T, Mitsumoto Y. Neural mechanisms underlying motor dysfunction as detected by the tail suspension test in MPTP-treated C57BL/6 mice. Neurosci Res. 2005 Mar;51(3):265–74. doi: 10.1016/j.neures.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Joshi PR, Wu NP, Andre VM, et al. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009 Feb 25;29(8):2414–27. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu H, Lim B, Poo MM. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci. 2009 Oct 7;29(40):12664–74. doi: 10.1523/JNEUROSCI.1984-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips PE, Johns JM, Lubin DA, et al. Presynaptic dopaminergic function is largely unaltered in mesolimbic and mesostriatal terminals of adult rats that were prenatally exposed to cocaine. Brain Res. 2003 Jan 24;961(1):63–72. doi: 10.1016/s0006-8993(02)03840-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simansky KJ, Kachelries WJ. Prenatal exposure to cocaine selectively disrupts motor responding to D-amphetamine in young and mature rabbits. Neuropharmacology. 1996 Jan;35(1):71–8. doi: 10.1016/0028-3908(95)00151-4. [DOI] [PubMed] [Google Scholar]
- 26.Cryan JF, O’Leary OF, Jin SH, et al. Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc Natl Acad Sci U S A. 2004 May 25;101(21):8186–91. doi: 10.1073/pnas.0401080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beutler LR, Wanat MJ, Quintana A, et al. Balanced NMDA receptor activity in dopamine D1 receptor (D1R)- and D2R-expressing medium spiny neurons is required for amphetamine sensitization. Proc Natl Acad Sci U S A. 2011 Feb 22; doi: 10.1073/pnas.1101424108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995 Oct 1;488 (Pt 1):85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nisenbaum ES, Berger TW, Grace AA. Depression of glutamatergic and GABAergic synaptic responses in striatal spiny neurons by stimulation of presynaptic GABAB receptors. Synapse. 1993 Jul;14(3):221–42. doi: 10.1002/syn.890140306. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Dever D, Lowe J, et al. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol. 2012 Aug 15;590(Pt 16):3743–69. doi: 10.1113/jphysiol.2012.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charara A, Heilman TC, Levey AI, Smith Y. Pre- and postsynaptic localization of GABA(B) receptors in the basal ganglia in monkeys. Neuroscience. 2000;95(1):127–40. doi: 10.1016/s0306-4522(99)00409-1. [DOI] [PubMed] [Google Scholar]
- 32.Centonze D, Grande C, Usiello A, et al. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. J Neurosci. 2003 Jul 16;23(15):6245–54. doi: 10.1523/JNEUROSCI.23-15-06245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ibanez-Sandoval O, Tecuapetla F, Unal B, Shah F, Koos T, Tepper JM. Electrophysiological and morphological characteristics and synaptic connectivity of tyrosine hydroxylase-expressing neurons in adult mouse striatum. J Neurosci. 2010 May 19;30(20):6999–7016. doi: 10.1523/JNEUROSCI.5996-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bracci E, Centonze D, Bernardi G, Calabresi P. Dopamine excites fast-spiking interneurons in the striatum. J Neurophysiol. 2002 Apr;87(4):2190–4. doi: 10.1152/jn.00754.2001. [DOI] [PubMed] [Google Scholar]
- 35.Janssen MJ, Ade KK, Fu Z, Vicini S. Dopamine modulation of GABA tonic conductance in striatal output neurons. J Neurosci. 2009 Apr 22;29(16):5116–26. doi: 10.1523/JNEUROSCI.4737-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000 Aug 15;20(16):6063–76. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tepper JM, Wilson CJ, Koos T. Feedforward and feedback inhibition in neostriatal GABAergic spiny neurons. Brain Res Rev. 2008 Aug;58(2):272–81. doi: 10.1016/j.brainresrev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crandall JE, Hackett HE, Tobet SA, Kosofsky BE, Bhide PG. Cocaine exposure decreases GABA neuron migration from the ganglionic eminence to the cerebral cortex in embryonic mice. Cereb Cortex. 2004 Jun;14(6):665–75. doi: 10.1093/cercor/bhh027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Semyanov A, Walker MC, Kullmann DM, Silver RA. Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci. 2004 May;27(5):262–9. doi: 10.1016/j.tins.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Wang W, Richerson GB. The transmembrane sodium gradient influences ambient GABA concentration by altering the equilibrium of GABA transporters. J Neurophysiol. 2006 Nov;96(5):2425–36. doi: 10.1152/jn.00545.2006. [DOI] [PubMed] [Google Scholar]
- 41.Tepper JM. GABAergic Interneurons of the Striatum. In: Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function. San Diego: Elsevier; 2010. pp. 151–66. [Google Scholar]
- 42.Friedman E, Wang HY. Prenatal cocaine exposure alters signal transduction in the brain D1 dopamine receptor system. Ann N Y Acad Sci. 1998 Jun 21;846:238–47. [PubMed] [Google Scholar]
- 43.Kim DS, Froelick GJ, Palmiter RD. Dopamine-dependent desensitization of dopaminergic signaling in the developing mouse striatum. J Neurosci. 2002 Nov 15;22(22):9841–9. doi: 10.1523/JNEUROSCI.22-22-09841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005 Aug;162(8):1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 45.Bosse R, Fumagalli F, Jaber M, et al. Anterior pituitary hypoplasia and dwarfism in mice lacking the dopamine transporter. Neuron. 1997 Jul;19(1):127–38. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- 46.Fernandez F, Garner CC. Over-inhibition: a model for developmental intellectual disability. Trends Neurosci. 2007 Oct;30(10):497–503. doi: 10.1016/j.tins.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Purves D, Augustine GJ, Fitzpatrick D, et al. In: Neuroscience. 4. Purves D, editor. Sunderland, MA: Sinauer Associates; 2008. [Google Scholar]
- 48.Arias Montano JA, Martinez-Fong D, Aceves J. GABAB receptor activation partially inhibits N-methyl-D-aspartate-mediated tyrosine hydroxylase stimulation in rat striatal slices. Eur J Pharmacol. 1992 Aug 6;218(2–3):335–8. doi: 10.1016/0014-2999(92)90187-9. [DOI] [PubMed] [Google Scholar]
- 49.Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993 Nov;13(11):4908–23. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.