

Abstract
The genetic defect in Friedreich's ataxia (FRDA) is the expansion of a GAA·TCC triplet in the first intron of the FXN ene, which encodes the mitochondrial protein frataxin. Previous studies have established that the repeats reduce transcription of this essential gene, with a concomitant decrease in frataxin protein in affected individuals. Since the repeats do not alter the FXN protein coding sequence, one therapeutic approach would be to increase transcription of pathogenic FXN genes. Histone posttranslational modifications near the expanded repeats are consistent with heterochromatin formation and FXN gene silencing. In an effort to find small molecules that would reactivate this silent gene, histone deacetylase inhibitors were screened for their ability to up-regulate FXN gene expression in patient cells and members of the pimelic 2-aminobenzamide family of class I histone deacetylase inhibitors were identified as potent inducers of FXN gene expression and frataxin protein. Importantly, these molecules up-regulate FXN expression in human neuronal cells derived from patient induced pluripotent stem cells and in two mouse models for the disease. Preclinical studies of safety and toxicity have been completed for one such compound and a phase I clinical trial in FRDA patients has been initiated. Further medicinal chemistry efforts have identified improved compounds with superior pharmacological properties.
Keywords: Friedreich's ataxia, histone deacetylase inhibitor, heterochromatin, neurodegenerative disorder
Introduction
Most individuals affected with Friedreich's ataxia (FRDA) carry a homozygous mutation consisting of the expansion of GAA•TTC trinucleotide repeats within the first intron of the frataxin (FXN) gene (Campuzano et al., 1996), which results in inhibited gene expression. A few FRDA subjects (about 4%) are compound heterozygotes for the expanded repeat mutation in one allele and a different loss-of-function mutation (missense, nonsense, indel) in the other one (Cossée et al., 1999). Frataxin amounts in affected individuals range between 5 and 35% of the levels in healthy individuals, while heterozygous subjects with no sign of disease have approximately 50% (Campuzano et al., 1997). The degree of frataxin expression reduction correlates with the length of the expanded GAA•TTC sequences and with the severity of clinical symptoms.
Although the exact function of frataxin is still under debate, available evidence supports a role in the biogenesis of iron-sulfur (Fe-S) clusters in mitochondria (Tsai & Barondeau, 2010). Fe-S clusters are essential cofactors for a variety of enzymes localized in all cellular compartments (Rouault & Tong, 2008). Frataxin deficiency results in impaired activities of Fe-S enzymes, altered cellular iron metabolism with iron accumulation in mitochondria, decreased mitochondrial energy production, and increased oxidative stress (these aspects of FRDA pathogenesis are treated in detail in other review articles in this issue, e.g. González-Cabo and Palau, 2012). To counteract these abnormalities, antioxidants, iron chelators and stimulants of mitochondrial biogenesis have been proposed as therapeutics (Pandolfo, 2013). However, no clear results supporting a benefit of any of these drugs have so far been obtained in randomized controlled trials. While continuing investigation of these therapeutics is warranted, the alternative of restoring frataxin expression in affected cells appears an appealing approach to slow down or stop disease progression, and stabilize or reduce the severity of disabilities. Gene replacement and protein replacement can in principle be used for this purpose. However, while preliminary studies in cell cultures and animal models do support their validity (Hebert and Whittom, 2007; Fleming et al., 2005; Vyas et al., 2012), further development depends on the resolution of many general problems in the field, in particular targeted delivery, controlled expression, and, for gene therapy, potential genotoxicity. An alternative is offered by the fact that, unlike the situation in diseases caused by coding repeat expansions (e.g. Huntington's disease or the autosomal dominant spinocerebellar ataxias), or by mutations resulting in a truncated or dysfunctional protein, in FRDA the protein encoded by the disease gene is structurally normal. Boosting the expression of the endogenous frataxin genes is therefore an appealing therapeutic approach. Here, we review the progress in the development of drugs targeting the frataxin gene silencing caused by the GAA•TTC repeat expansion. But first, we summarize the evidence that Friedreich's ataxia is a gene silencing disease with an epigenetic basis.
Frataxin deficiency is due to transcriptional silencing
Two early reports documented that expanded GAA•TTC repeats cause transcriptional silencing of the FXN gene (Bidichandani et al., 1998; Ohshima et al., 1998). In the first study, plasmids containing various numbers of repeats were shown to cause a length- and orientation-dependent inhibition of reporter gene expression (Ohshima et al., 1998). In the second study, the activity of the endogenous FXN gene was analyzed, with the result that patients who are homozygous for this expansion have a marked deficiency in FXN mRNA (Bidichandani et al., 1998). This study also documented that the repeats interfere with in vitro transcription, again in a length- and orientation-dependent manner, with both prokaryotic and eukaryotic RNA polymerases. No evidence has been obtained for aberrant splicing of the primary transcript; that is, generation of the mature messenger RNA from the primary transcript is not affected by the repeats (Bidichandani et al., 1998; Ohshima et al., 1998). Although an effect of the repeats on RNA splicing from a reporter construct has been reported, this study did not extend the results to the endogenous FXN transcript in patient cells (Baralle et al., 2008). There is also no evidence that long GAA-repeat intron 1 RNA is stable and could lead to an RNA-toxicity disease, such as found in myotonic dystrophy or fragile X-associated tremor/ataxia (reviewed in (Orr & Zoghbi, 2007)). Together, both early papers (Bidichandani et al., 1998; Ohshima et al., 1998) presented strong evidence that the loss of frataxin protein in FRDA is due to repression of FXN transcription by the GAA•TTC repeats, and the data presented were fully consistent with the negative correlation between repeat length and age of onset and severity of the disease in patients.
Having established that RNA transcription is impaired by the GAA•TTC repeats, the question remained as to how the repeats interfere with RNA polymerase at the FXN gene. An impressive series of papers from Wells and colleagues documented that expanded GAA•TTC repeats adopt unusual DNA structures in vitro, such as triplexes and “sticky” DNA (Sakamoto et al., 1999; Wells, 2008). These structures could very well impede the progress of RNA polymerase II through the repeats and lead to stalled or aborted transcription. Another model is that a DNA-RNA triplex formed at the repeats is responsible for blocking transcription elongation (Grabczyk & Usdin, 2000a; 2000b). While the biochemical results on DNA structure are fully consistent with the observed correlation between repeat length, triplex/“sticky” DNA formation and the age at onset and severity of disease, confirmation of the role of such structures in FXN gene silencing must await experimental evidence that expanded GAA•TTC repeats exist in a non-B DNA structure (DNA or DNA-RNA triplexes, or “sticky” DNA) at the chromosomal FXN in patient cells. Chemical probing and triplex-specific antibody-based approaches are needed to resolve this issue.
Repeat induced heterochromatin formation at pathogenic FXN alleles
An alternative, but not mutually exclusive, mechanism for silencing pathogenic FXN alleles is epigenetic gene silencing through heterochromatin. Heterochromatin is characterized by histone hypoacetylation, histone H3 lysine 9 and lysine 27 methylation, and the association of histone deacetylase enzymes, specific histone methyltransferases and heterochromatin proteins, such as members of the HP1 family and polycomb group proteins. The first report in support of an epigenetic silencing mechanism in FRDA came from Festenstein and colleagues (Saveliev et al., 2003), who showed that a transgene containing GAA•TTC repeats was silenced in vivo, in a manner reminiscent of position effect variegated gene silencing. In this study, repeat-induced silencing was augmented by over-expression of the heterochromatin protein HP1, and the silenced transgene was packaged into condensed chromatin, as evidenced by resistance to nuclease digestion (Saveliev et al., 2003). Interestingly, the repeats could be located outside of the transcribed region of the transgene, suggesting that models for a block in transcription elongation due to non-B DNA structures need to be reconsidered. While this study pointed to heterochromatin formation by the GAA•TTC repeats as the mechanism for FXN silencing in FRDA (Saveliev et al., 2003), these authors did not examine the chromatin structure of endogenous FXN alleles in FRDA patient cells.
The first such report came from Herman et al. (Herman et al., 2006), and subsequently other laboratories have used similar chromatin immunoprecipitation methods to monitor the histone modifications on the endogenous FXN alleles in cell lines derived from Friedreich's ataxia patients and in patient primary cells (peripheral lymphocytes) (Herman et al., 2006; Greene et al., 2007; Rai et al., 2010; Kim et al., 2011; Kumari et al., 2011). The general consensus from these studies is that the first intron of active FXN alleles in cells from unaffected individuals is enriched in acetylated histones H3 and H4, compared with the inactive alleles in Friedreich's ataxia cells. Additionally, lysine 9 of histone H3 (H3K9) is highly methylated in Friedreich's ataxia cells compared with the normal cells. Along with hypoacetylation, trimethylation of H3K9 is a hallmark of heterochromatin, and provides the binding site for heterochromatin protein HP1 (Saveliev et al., 2003) (Figure 1). Although these studies were conducted in Friedreich's ataxia lymphoid cells, the same epigenetic differences between active and inactive FXN alleles have also been found in the affected tissues (brain and heart) from mouse models for the disease (Al-Mahdawi et al., 2008; Rai et al., 2008) and in Friedreich's ataxia autopsy brain, cerebellum, and heart (Al-Mahdawi et al., 2008). Ongoing studies have also documented similar heterochromatin marks on pathogenic FXN alleles in neurons derived from patient induced pluripotent stem cells (Soragni et al., unpublished). Recent reports have also suggested that the chromatin changes associated with pathogenic FXN alleles prevent transcript elongation by RNA polymerase II through expanded GAA•TTC repeats (Punga & Bühler, 2010; Kim et al., 2011; Kumari et al., 2011), although one report suggested that both transcription initiation and elongation may be affected (Kumari et al., 2011).
Figure 1. Putative silencing pathway in Friedreich's ataxia.
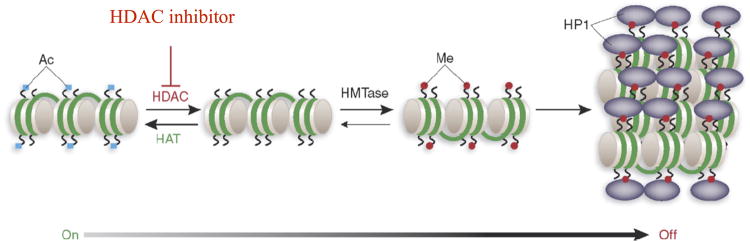
Figure first published in and modified from Festenstein (2006).
Taken together, there is now a strong body of evidence supporting the notion that the repeats induce silencing through changes in the chromatin landscape of the FXN gene. Just how the repeats signal heterochromatin formation is still a subject of debate. Perhaps a non-B DNA structure is the key signal for recruitment of the cellular machinery for heterochromatin formation. Alternatively, one report has implicated the chromatin insulator protein CTCF in repeat-induced silencing (De Biase et al., 2009). CTCF is depleted from pathogenic FXN alleles in FRDA cells, but just how the repeats cause this depletion remains a mystery.
Histone Deacetylase Inhibitors to Correct Frataxin Deficiency in Friedreich's Ataxia
Numerous studies have shown that small molecule inhibitors of the histone deacetylase (HDAC) enzymes are able to revert silent heterochromatin to an active chromatin conformation, and restore the normal function of genes that are silenced in various human diseases, including neurodegenerative and neuromotor diseases (Di Prospero & Fischbeck, 2005; Kazantsev & Thompson, 2008). Eighteen histone deacetylase enzymes have been identified in the human genome, including the zinc-dependent (class I, class II, and class IV) and the NAD+-dependent enzymes (class III or sirtuins). Histone deacetylase enzymes 1, 2, 3, and 8 belong to class I, showing homology to the yeast enzyme RPD3. Class II is further divided into class IIa (histone deacetylase enzymes 4, 5, 7, and 9) and IIb (histone deacetylase enzymes 6 and 10), according to their sequence homology and domain organization. Histone deacetylase enzyme 11 is the lone member of class IV. The sirtuins (class III) are related to the yeast silent information regulator 2 protein and are involved in regulation of metabolism and aging. Several small molecule chemical families have been shown to act as histone deacetylase inhibitors (Cole, 2008). These include small carboxylates (such as sodium butyrate, valproic acid (VPA), and sodium phenylbutyrate), hydroxamic acids (such as trichostatin A, TSA, suberoylanilide hydroxamic acid, SAHA, and suberoyl bishydroxamic acid, SBHA), benzamides (such as MS-275), epoxyketones (trapoxins), and cyclic peptides (including apicidin and depsipeptide). While the development of HDAC inhibitors as anti-cancer therapeutics is quite advanced, and two such compounds have been approved by the FDA (SAHA, under the generic name of vorinostat, and the cyclic peptide romidepsin) (Marks, 2010), interest in the development of HDAC inhibitors for neurodegenerative and neuromotor diseases has increased in the past few years, based on an increasingly recognized role of epigenetic modifications in neurodegeneration (Kazantsev & Thompson, 2008).
Based on our finding of epigenetic silencing marks associated with pathogenic FXN alleles, we asked whether HDAC inhibitors could reverse silencing in FRDA lymphoblasts and primary lymphocytes (Herman et al., 2006). We screened a small collection of the HDAC inhibitors discussed above and found that only 2-aminobenzamides (Fig. 1) were active in restoring FXN gene expression in FRDA cells. While each of the HDAC inhibitors tested (at their reported IC50 value for HDAC inhibition), including TSA, SAHA, SBHA and VPA increased the fraction of total acetylated histones in FRDA cells,,only 2-aminobenzamides increased acetylation at FXN intron 1 and FXN gene expression (Herman et al., 2006). Chromatin immunoprecipitation experiments also showed that the 2-aminobenzamide HDAC inhibitor 4b increased acetylation at particular lysine residues of histones H3 and H4 within intron 1 of FXN (H3-K14, H4-K5, and H4-K12) (Herman et al., 2006), while SAHA and TSA did not. Taken together, our results suggest that a specific histone deacetylase, or histone deacetylase-protein complex, is involved in FXN silencing and 2-aminobenzamide histone deacetylase inhibitors selectively target this protein in cells. Alternatively, a different mechanism of action of these inhibitors could account for our results (see below).
These results were initially obtained with lymphoblast cell lines (Herman et al., 2006), but have been extended to primary lymphocytes from FRDA patients (Rai et al., 2010), and to mouse models (Rai et al., 2008; 2010; Sandi et al., 2011). We have recently shown increases in FXN gene expression in human FRDA neuronal cells derived from patient induced pluripotent stem cells (Soragni et al., unpublished). Importantly, the HDAC inhibitors increase the levels of frataxin protein in both cellular and animal models, paralleling the observed changes in FXN messenger RNA. These findings demonstrate that expanded GAA repeats in the primary FXN RNA transcript do not interfere with RNA processing. Additionally, we find that the 2-aminobenzamide HDAC inhibitors only increase FXN expression in patient cells, with minor effects on the normal alleles (Soragni et al., 2012), indicating that these compounds are likely acting on the mechanism of silencing induced by the repeats. Members of this compound class have increased levels of FXN messenger RNA in lymphocytes from >100 FRDA patients, and the level of FXN messenger RNA in their lymphocytes is generally increased to at least that of lymphocytes from carrier siblings or parents. Since heterozygous individuals do not exhibit symptoms of Friedreich's ataxia, we believe our compounds have elicited a therapeutically useful increase in FXN messenger RNA in patient cells.
Which HDAC isoform is the target of our 2-aminobenzamide inhibitors?
We felt that it was essential to know the cellular HDAC enzyme target of our inhibitors for future drug development. Previous studies with other benzamide-type HDAC inhibitors, such as MS-275, indicated that these compounds target the class I HDAC family (Beckers et al., 2007; Bradner et al., 2010). We find that the pimelic 2-amimobenzamides, exemplified by compound 106, are also class I histone deacetylase inhibitors, with a moderate preference for HDAC3 over HDACs 1 and 2, with little activity against the other class I HDAC, HDAC8, and little or no activity against class II HDACs (Chou et al., 2008). To try to understand why the 2-aminobenzamides but not other potent HDAC inhibitors, such as the hydoxamates SAHA and TSA, fail to activate FXN gene expression, we determined the kinetic parameters for these compounds with recombinant class I HDACs (Chou et al., 2008). While the hydroxamates are rapid-on/rapid-off, classical competitive inhibitors, compound 106 inhibits HDACs 1 and 3 through a slow-on/slow-off mechanism (Chou et al., 2008). Ki measurements show that HDACi 106 has a ∼10-fold preference for HDAC3 over HDAC1, and this difference is reflected in proteomic profiling of the HDAC enzymes with a chemical probe version of HDAC inhibitor 106 in cell extracts (Xu et al., 2009). Other class I HDAC inhibitors, such as MS-275 and related benzamides, exhibit a ∼4 to 10-fold preference for HDAC1 over HDAC3 (Hu et al., 2003; Beckers et al., 2007; Siliphaivanh et al., 2007); thus, 106 is one of the first examples of a HDACi that shows selectivity for HDAC3 over all other class I HDAC enzymes. Small interfering RNA approaches also point to a role for HDAC3 in FXN gene silencing in FRDA cell models (Soragni et al., unpublished).
We also examined the cellular histone deacetylase inhibition activities of SAHA and 106 in a Friedreich's ataxia lymphoblast cell line (Chou et al., 2008). While both compounds show increased global acetylation of histone H3, after washing cells free of the compounds, acetylated histones are rapidly lost in the SAHA-treated cells. In contrast, acetylation persists for several hours in the 106-treated cells, paralleling the in vitro slow dissociation described above. Thus, the active 2-aminobenzamides differ from hydroxamates in their mechanism of inhibition of class I HDACs, and this difference could well account for the efficacy of this class of compounds in reactivation of silent FXN alleles in Friedreich's ataxia cells and mouse models.
Do compounds targeting other HDACs activate FXN gene expression?
While the pimelic 2-aminobenzamides are potent activators of FXN gene expression in FRDA cells, the question remained as to whether compounds with other HDAC isoform selectivities would also activate FXN gene expression. To address this issue, we tested compounds with different selectivity profiles in the FRDA lymphoblast cell line (Xu et al., 2009). Appending a phenyl group at the 5 position of the 2-aminobenzamide ring results in a compound that is ∼300-fold selective for HDACs 1 and 2 over HDAC3 (Methot et al., 2008; Xu et al., 2009). While this compound is a potent HDAC inhibitor in these cells, it is without effect on FXN gene expression. Similarly, we find that potent inhibitors of class II and class III (sirtuins) histone deacetylase also fail to activate FXN expression in the FRDA cells, although each of these compounds is active against known substrates (Xu et al., 2009). Recent work has suggested that compounds with moderate selectivity for HDAC3 over HDACs 1 and 2 are the best activators of FXN gene expression (Soragni et al., unpublished).
Efficacy of Histone Deacetylase Inhibitors in Mouse Models for Friedreich's Ataxia
The efficacy of 2-aminobenzamide HDAC inhibitors has been tested in two mouse models for FRDA (Rai et al., 2008; 2010; Sandi et al., 2011). Homozygous knock-in mice carrying 230 GAA•TTC triplets inserted in the endogenous frataxin gene (KIKI, FXN230GAA/230GAA) express 66 to 75% of wild-type Fxn mRNA and protein in the brain and other organs (Miranda et al., 2002). Similar to FRDA patient cells and tissue samples, characteristic histone modifications occur in the first intron of the Fxn gene in the KIKI mouse (Herman et al., 2006), including increased H3-K9 trimethylation and hypoacetylation of histones H3 and H4. Treatment of KIKI mice with the HDAC inhibitor 106 (Figure 1) restored frataxin levels in the nervous system and heart to those of wild-type mice and increased histone H3 and H4 acetylation near the GAA•TTC repeat. Microarray analysis indicated that most of the differentially expressed genes in KIKI mice, compared with wild-type mice of the same genetic background, reverted toward wild-type levels, indicating correction of changes induced by frataxin downregulation. The HDAC inhibitor only increased Fxn mRNA levels from the knock-in allele harboring GAA repeats; as it had no effect in wild-type mice (Rai et al., 2008). A second study in KIKI mice confirmed the effect of 106 and of a similar molecule, 109 on frataxin mRNA and protein. This study also showed that the elevation of frataxin mRNA and protein was sustained, persisting 48 hours after drug exposure in the case of the protein (Rai et al., 2010).
Pook and colleagues investigated the long-term effects of three 2-aminobenzamide HDAC inhibitors, 106, 109, and 136, in a second GAA repeat expansion mouse model (Sandi et al., 2011). This mouse model harbors a yeast artificial chromosome containing the human FXN locus with expanded repeats. Frataxin expression from the YAC transgene is sufficient to rescue embryonic lethality when crossed into a Fxn-/- background, but the resulting line, called YG8R, shows reduced levels of frataxin mRNA, protein, heterochromatin formation at the repeats, motor deficits, and neuronal pathology (Al-Mahdawi et al., 2008). During 5 months of treatment with the compounds 109, 106 and 136 (Figure 2), no overt toxicity was observed. Both 109 and 106 improved motor coordination, whereas 109 and 136 increased locomotor activity. All three compounds increased global histone H3 and H4 acetylation of brain tissue, but only 109 significantly increased acetylation of specific histone residues at the FXN locus. Furthermore, compound 109 significantly increased frataxin protein expression in brain tissue, improved brain aconitase enzyme activity, which is reduced in frataxin-deficient cells as a consequence of impaired iron-sulfur cluster biogenesis (Rötig et al., 1997; Tsai and Barondeau, 2010), and reduced neuronal pathology in the dorsal root ganglia (Sandi et al., 2011). Taken together, the results in mouse models support the use of 2-aminobenzamides as therapeutics for Friedreich's ataxia.
Figure 2.
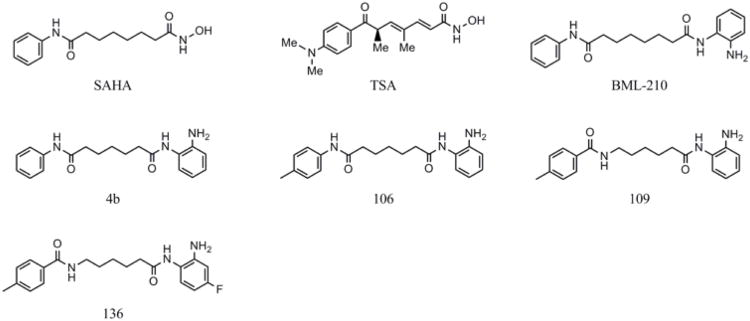
Structures of the histone deacetylase inhibitors.
Generation of Compounds with Improved Pharmacological Properties
While the 2-aminobenzamides are potent inducers of FXN gene expression in both cellular and animal models, this compound class suffers from two liabilities that need to be addressed for their use as human therapeutics in Friedreich's ataxia. These are less than optimal brain penetration and conversion of the active molecule into an inactive metabolic product under the acidic conditions of the stomach and in serum. For example, on subcutaneous injection of histone deacetylase inhibitor 109 in rodents, this molecule has only a 10% to 15% brain concentration compared with serum. In addition, the 2-aminobenzamides are converted to an inactive benzimidazole in vivo. Through a medicinal chemistry effort at Repligen, two structural features that individually improve brain distribution and metabolic stability were identified. These are replacement of the “left” amide with an ether, olefin, or ketone to improve brain penetration and introduction of an unsaturated linkage adjacent to the “right” amide to prevent formation of a benzimidazole metabolic byproduct (Figure 3a). On the basis of these results, we devised and synthesized a new lead compound click-1 using Cu(I)-catalyzed click chemistry (Figure 3b). This synthetic route allows for the generation of compounds containing both modifications mentioned above, but introduces a triazole into the aliphatic linker region of the standard pimelic 2-aminobenzamide scaffold. Initially, we did not know whether a heterocycle in the linker would retain activity compared to the active parent molecules, such as 4b or 106, etc. (Figure 2).
Figure 3.

Compounds with improved pharmacological properties. (A) Brain penetration can be improved by elimination of the left amide, and replacement with an ether, olefin, or ketone. Metabolic stability can be improved by introducing a non-saturated α/β linkage adjacent to the right amide, which prevents formation of a benzimidazole. (B) Synthetic route to compounds with these replacements using Cu(I)-catalyzed click chemistry, where azide A is reacted with alkyne B, and after Boc deprotection, a triazole is generated in the linker region of the histone deacetylase inhibitor.
We therefore monitored click-1 for its activity against recombinant HDACs 1, 2, and 3 in vitro, in-cell histone deacetylase activity, and activity in restoring FXN transcription in patient cells. We find that click-1 has comparable activity to histone deacetylase inhibitor 106 or 109 in these assays (Xu et al., 2009). Blood-brain barrier penetration was measured in the rat for compound 109 and click-1. Brain penetration (as measured by brain/plasma ratio at tmax) was determined to be 0.15 for 109 and 0.33 for click-1 (Cmax: 800 ng/g in the brain at Tmax = 5 min post-dose [5 mg/kg IV rat]), representing a significant increase in brain penetration for click-1 compared with the standard 2-aminobenzamide 109. The half-life of the compounds in acidic conditions was used to quantify their relative stability. At pH = 2 and 50 °C, t1/2 = 6 hours for 109 versus t1/2 = 33 hours for click-1, representing a 5.5-fold improvement relative to histone deacetylase inhibitor 109. These results demonstrate that click-1 has improved acid stability and better brain penetration, through placing a double bond next to the right amide bond and removing the left amide. Based on these findings, new generations of molecules have been synthesized and are currently in preclinical testing.
Future Directions and Clinical Studies
Full preclinical assessment of a lead clinical compound (109/RG2833) has been accomplished and an Investigational New Drug application has been filed with the US Food and Drug Administration for initiation of a phase I clinical trial of RG2833 in man. Additionally, Repligen has received Orphan Drug status for its clinical candidate. In Europe, the European Medicine Agency (EMA) as granted RG2833 orphan drug status as well and authorized a phase I study to determine whether this drug can safely increase frataxin levels in peripheral blood mononuclear cells (PBMCs) from FRDA patients. After further authorization from national health authorities and ethics approval, a trial has started in Italy to assess the safety of single ascending doses of RG2833, its pharmacokinetics and its effect on frataxin in PBMCs. This study is ongoing at the time of submission of this review article.
Such information will provide a proof of principle for initiating the clinical efficacy stage of clinical studies. If oral delivery of a histone deacetylase inhibitor can be effective at drug exposures that are well-tolerated, it will provide impetus to further advance the 2-aminobenzamide class of compounds to target histone deacetylase inhibition as a viable therapeutic strategy for this devastating disease.
Acknowledgments
Studies in our laboratories have been supported by grants from the National Institute of Neurological Disorders and Stroke, National Institutes of Health (to Dr Gottesfeld), the Friedreich's Ataxia Research Alliance (Dr Xu and Repligen), GoFAR (Dr Soragni and Repligen), Ataxia UK (Dr Soragni), Friedreich's Ataxia Society Ireland (Dr Soragni), and the Muscular Dystrophy Association (Repligen Corporation).
Footnotes
The authors declare no conflict of interest.
References
- Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- Baralle M, Pastor T, Bussani E, Pagani F. Influence of Friedreich ataxia GAA noncoding repeat expansions on pre-mRNA processing. Am J Hum Genet. 2008;83:77–88. doi: 10.1016/j.ajhg.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, Sanders K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer. 2007;121:1138–1148. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62:111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J Biol Chem. 2008;283:35402–35409. doi: 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossée M, Dürr A, Schmitt M, Dahl N, Trouillas P, Allinson P, Kostrzewa M, Nivelon-Chevallier A, Gustavson KH, Kohlschütter A, Müller U, Mandel JL, Brice A, Koenig M, Cavalcanti F, Tammaro A, De Michele G, Filla A, Cocozza S, Labuda M, Montermini L, Poirier J, Pandolfo M. Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes. Annals of Neurology. 1999;45:200–206. doi: 10.1002/1531-8249(199902)45:2<200::aid-ana10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- De Biase I, Chutake YK, Rindler PM, Bidichandani SI. Epigenetic silencing in Friedreich ataxia is associated with depletion of CTCF (CCCTC-binding factor) and antisense transcription. PLoS ONE. 2009;4:e7914. doi: 10.1371/journal.pone.0007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet. 2005;6:756–765. doi: 10.1038/nrg1690. [DOI] [PubMed] [Google Scholar]
- Festenstein Breaking the silence in Friedreich's ataxia. Nat Chem Biol. 2006;2:512–513. doi: 10.1038/nchembio1006-512. [DOI] [PubMed] [Google Scholar]
- Fleming J, Spinoulas A, Zheng M, Cunningham SC, Ginn SL, McQuilty RC, Rowe PB, Alexander IE. Partial correction of sensitivity to oxidant stress in Friedreich ataxia patient fibroblasts by frataxin-encoding adeno-associated virus and lentivirus vectors. Hum Gene Ther. 2005;16:947–956. doi: 10.1089/hum.2005.16.947. [DOI] [PubMed] [Google Scholar]
- González-Cabo P, Palau F. Mitochondrial Pathophysiology in Friedreich's Ataxia. J Neurochem S2 Special Issue. 2012 doi: 10.1111/jnc.12303. [DOI] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. Alleviating transcript insufficiency caused by Friedreich's ataxia triplet repeats. Nucleic Acids Res. 2000a;28:4930–4937. doi: 10.1093/nar/28.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich's ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000b;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat-induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res. 2007;35:3383–3390. doi: 10.1093/nar/gkm271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert MD, Whittom AA. Gene-based approaches toward Friedreich ataxia therapeutics. Cell Mol Life Sci. 2007;64:3034–3043. doi: 10.1007/s00018-007-7293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat Chem Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kim E, Napierala M, Dent SYR. Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res. 2011;39:8366–8377. doi: 10.1093/nar/gkr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari D, Biacsi RE, Usdin K. Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J Biol Chem. 2011;286:4209–4215. doi: 10.1074/jbc.M110.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19:1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific CM, Kral AM, Meinke PT, Middleton RE, Ozerova N, Sloman DL, Stanton MG, Szewczak AA, Tyagarajan S, Witter DJ, Secrist JP, Miller TA. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, Cossée M, Koenig M, Sequeiros J, Kaplan J, Pandolfo M. Frataxin knockin mouse. FEBS Lett. 2002;512:291–297. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- Ohshima K, Montermini L, Wells RD, Pandolfo M. Inhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. J Biol Chem. 1998;273:14588–14595. doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Treatment of Friedreich's ataxia. Expert Opinion on Orphan Drugs. 2013;1:221–234. [Google Scholar]
- Punga T, Bühler M. Long intronic GAA repeats causing Friedreich ataxia impede transcription elongation. EMBO Mol Med. 2010;2:120–129. doi: 10.1002/emmm.201000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pandolfo M. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich's ataxia patients and in a mouse model. PLoS ONE. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, Geschwind DH, Gottesfeld JM, Pandolfo M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich's ataxia. Mol Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- Sandi C, Pinto RM, Al-Mahdawi S, Ezzatizadeh V, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pook MA. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol Dis. 2011;42:496–505. doi: 10.1016/j.nbd.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveliev A, Everett C, Sharpe T, Webster Z, Festenstein R. DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature. 2003;422:909–913. doi: 10.1038/nature01596. [DOI] [PubMed] [Google Scholar]
- Siliphaivanh P, Harrington P, Witter DJ, Otte K, Tempest P, Kattar S, Kral AM, Fleming JC, Deshmukh SV, Harsch A, Secrist PJ, Miller TA. Design of novel histone deacetylase inhibitors. Bioorg Med Chem Lett. 2007;17:4619–4624. doi: 10.1016/j.bmcl.2007.05.080. [DOI] [PubMed] [Google Scholar]
- Soragni E, Xu C, Plasterer HL, Jacques V, Rusche JR, Gottesfeld JM. Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for friedreich ataxia. J Child Neurol. 2012;27:1164–1173. doi: 10.1177/0883073812448533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- Vyas PM, Tomamichel WJ, Pride PM, Babbey CM, Wang Q, Mercier J, Martin EM, Payne RM. A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich's ataxia mouse model. Hum Mol Genet. 2012;21:1230–1247. doi: 10.1093/hmg/ddr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RD. DNA triplexes and Friedreich ataxia. FASEB J. 2008;22:1625–1634. doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- Xu C, Soragni E, Chou CJ, Herman D, Plasterer HL, Rusche JR, Gottesfeld JM. Chemical probes identify a role for histone deacetylase 3 in Friedreich's ataxia gene silencing. Chem Biol. 2009;16:980–989. doi: 10.1016/j.chembiol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]