

Abstract
A palladium-catalyzed method for the preparation of sulfonamides is described. The process exhibits significant functional group tolerance and allows for the preparation of a number of arylsulfonyl chlorides and sulfonamides under mild conditions.
The medical significance of aryl sulfonamides can be traced to the 1930’s, with the discovery and development of the first commercially available antibiotics, the so-called sulfa drugs.1 Today, the presence of sulfonamides in medicinal agents is widespread; close to ten percent of the top 100 pharmaceuticals prescribed in 2011 either bear a sulfonamide subunit or are co-administered with a sulfonamide-containing drug.2 In general, aryl sulfonamides can be prepared by the straightforward reaction of a sulfonyl chloride with an amine.3 However, the difficulties associated with sulfonamide synthesis stem not from the amination reaction, but from the preparation of sulfonyl chlorides themselves. Two types of processes represent the current state of the art in arylsulfonyl chloride synthesis: (1) electrophilic aromatic substitution (EAS) with chlorosulfonic acid4 and (2) oxidative chlorination of organosulfur compounds.5–9 The use of both approaches suffers from significant limitations. In particular, the acidic conditions required for EAS processes (and most oxidative chlorination methods) impose severe restrictions on substrate scope. Furthermore, desired substitution patterns may be inaccessible via EAS because the regioselectivity is dictated by the intrinsic properties of the parent arene. Traditionally, oxidative chlorination involves the use of hazardous reagents (e.g., aqueous chlorine)6 or strong chlorinating agents (e.g., SOCl27 and SO2Cl2).8 Although milder conditions have been reported,9 oxidative chlorination of thiophenol derivatives ultimately requires prior formation of a carbon–sulfur bond. 10–11
In principle, a transition metal catalyst could obviate the need for such reagents and permit the convergent synthesis of sulfonamide analogs, allowing variation of both sulfonyl (–SO2–) substituents. Unfortunately, success in this area of catalysis is limited to a single Pd-catalyzed aminosulfonylation process, initially reported by Willis in 2010, to prepare N-amino-sulfonamides (1) from aryl iodides and hydrazines (Eq. 1).12 While this example represents an important achievement in sulfonylation chemistry, amine nucleophiles remain incompatible with these types of couplings.12–13 To address this limitation, we devised an alternative strategy (Eq. 2), which involves oxidative addition of LPd(0) to an electrophile of the type X–SO2–X′ (A), where X and X′ represent leaving groups of different reactivity. Subsequent coupling with an organometallic nucleophile would afford electrophilic species B, which, in turn, would serve as a direct precursor to the sulfonamide product (C).
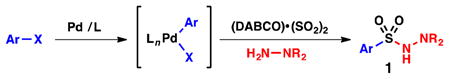 |
Willis and coworkers (Eq. 1) |
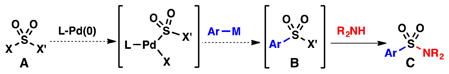 |
Our approach (Eq. 2) |
Early in the investigation, we selected arylboronic acids as coupling partners due to their compatibility with other functional groups, ready availability, and ease of handling. Upon examining the reaction sequence from a retrosynthetic perspective, we envisioned exploiting arylsulfonate esters (3, Scheme 1)14 as the immediate precursors to sulfonamides, and thus potential targets for Suzuki-Miyaura cross-coupling. In turn, we proposed that sulfonate esters (3) could be prepared from an aryl chlorosulfate derivative (2)15 via Pd-catalysis. However, contrary to our expectations, we discovered serendipitously that this coupling process generates arylsulfonyl chlorides (4) in preference to 3, and we describe herein the development and scope of a Pd-catalyzed chlorosulfonylation reaction.
Scheme 1.
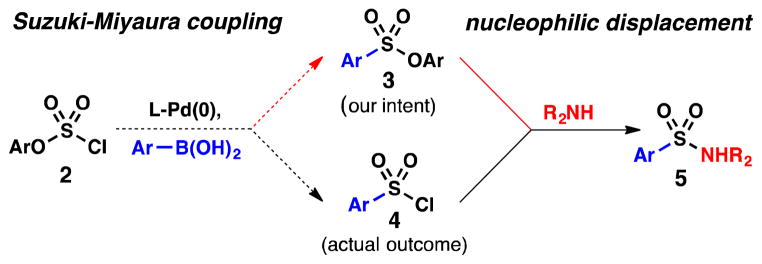
Sulfonylation of Arylboronic Acids
Initial experiments focused on identifying conditions to prepare 3a from the simplest aryl chlorosulfate, phenyl chlorosulfate (2a, Table 1), which is an easy to prepare, easily handled liquid.16 Results from a preliminary survey of bases are outlined in Table 1, utilizing 4-methoxyphenylboronic acid in combination with a catalyst derived from Pd(OAc)2 and tert-BuBrettPhos (L8) or DavePhos (L1). Unexpectedly, we observed mixtures of 3a and the corresponding sulfonyl chloride (4a) when excess oxygen base was used (entries 1–3). More importantly, we discovered that 4a could be formed exclusively in the absence of base (entries 4 & 5), and that neither product is formed in the absence of Pd or phosphine ligand.17 Additional control experiments verified that aryl sulfonate esters (3) are not converted to the corresponding sulfonyl chlorides (4) under the reaction conditions;18 conversely, ester 3a is generated when crude reaction mixtures of 4a are treated with excess K3PO4 or K2CO3 (following complete consumption of 2a in the absence of base).
Table 1.
Pd-Catalyzed Chlorosulfonylationa

| ||||
|---|---|---|---|---|
| Entry | Ligand | Base | 3a (%)b | 4a (%)b |
| 1 | L8 | K3PO4 | 31 | 4 |
| 2 | L8 | K2CO3 | 15 | 22 |
| 3 | L8 | Na2CO3 | 4 | 36 |
| 4 | L8 | none | 0 | 59 |
| 5 | L1 | none | 0 | 87 |
Performed on a 0.5 mmol scale with respect to 2a.
GC yields.
The above results suggest a catalytic cycle in which Pd(0) inserts into the SO2–OPh bond of 2a in preference to the SO2–Cl bond (Figure 1).19 The phenoxy substituent of the resulting Pd-sulfinate complex (6) would be expected to facilitate transmetallation without the aid of an oxygen base;20 subsequent reductive elimination from 7 would yield 4 and regenerate the active Pd(0) catalyst. Oxygen bases likely promote the release of phenoxide, which in turn would react with the sulfonyl chloride to provide 3. Examination of the chemical literature revealed that Buncel and coworkers have also observed the displacement of aryloxide from aryl chlorosulfate derivatives (2), but in the context of direct nucleophilic additions.15,21 Nonetheless, this precedent demonstrates the lability of the SO2–OPh bond, as we have observed for the transfer of –SO2Cl in the analogous Pd-catalyzed process.
Figure 1.

Proposed Catalytic Cycle
In light of these results, we focused our efforts on the preparation of sulfonyl chlorides as they represent ideal precursors to sulfonamides. However, the system represented in Table 1, entry 5 proved ineffective for both electron-deficient and ortho-substituted substrates. For example, subjecting 2-methoxyphenylboronic acid to these conditions resulted in near complete recovery of phenyl chlorosulfate (2a). Further optimization revealed that yields of aryl-sulfonyl chlorides could be increased by employing anhydrous acetone as solvent in combination with a catalytic amount of Na2CO3 (5 mol %). Although Pd(OAc)2 proved to be a competent palladium source, the use of palladacyclic precatalysts22 (P1–P7) allowed for these experiments to be conducted at lower temperatures. As exemplified in the coupling of 2a with 2-methoxyphenylboronic acid (Figure 2), precatalysts based on diphenyl-(L3 and L5) and di-tert-butylbiaryl phosphine ligands (L2 and L7) proved to be the most effective. In particular, the PhCPhos precatalyst (P5) afforded 4b in the highest yield (82%), albeit with only a slight improvement over that derived from tert-BuDavePhos, L2 (80%). In contrast, the use of XPhos (L6),23 previously reported to be an excellent ligand for Suzuki-Miyaura reactions, provided little product.24
Figure 2.

Ligand Effects
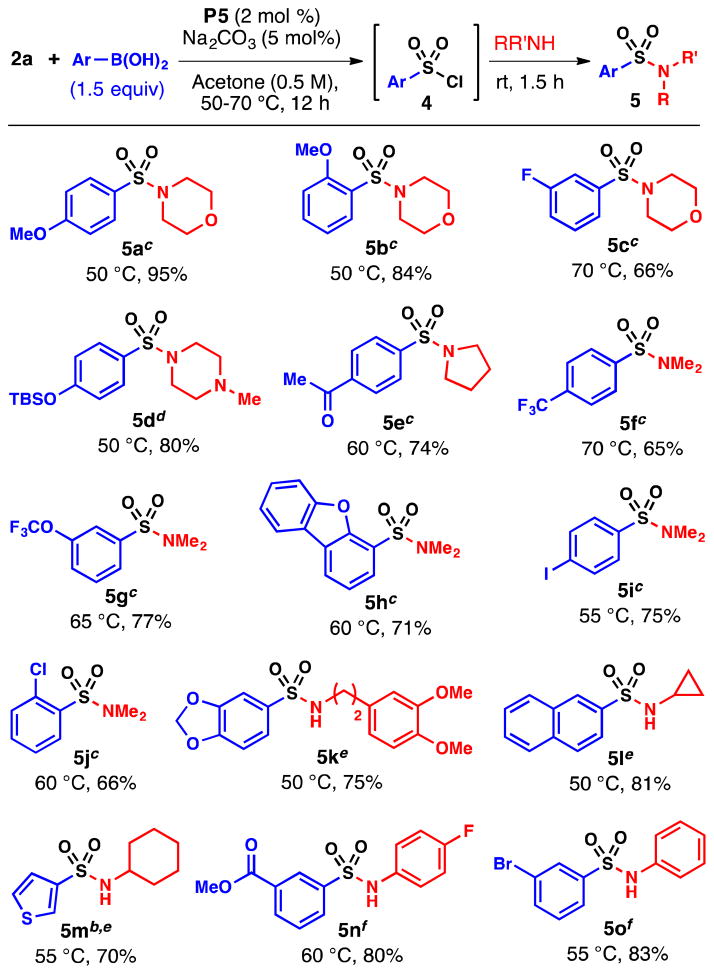
With this improved protocol, utilizing 2 mol % P5 and 5 mol % of Na2CO3 in acetone, we prepared and isolated a number of sulfonyl chlorides in good yields (Table 2). In general, electron-rich, -neutral and -deficient arylboronic acid reagents represent compatible substrates. Couplings with electron-rich substrates can be conducted at 50 °C, while reactions with electron-deficient substrates are typically slower and require higher temperatures.25–26 While most of the sulfonyl chlorides are stable to chromatography, electron-deficient compounds, such as 4h and 4j, were found to decompose to varying degrees upon attempted purification.27 Thus to circumvent this problem, the sulfonyl chloride intermediates were converted to sulfonamides (5a–m) directly by simply adding a primary or secondary amine to the crude reaction mixtures of 4 (Table 3).28 Weakly nucleophilic aniline derivatives may also be incorporated into the sulfonamide moiety, but pyridine is required to facilitate amination in these cases. As illustrated in Tables 2 and 3, the chlorosulfonylation reaction tolerates chloro-, bromo-, and iodo-substituted arylboronic acids as well as substrates containing TBS-ethers, ester and acetyl functional groups. Heteroaryl substrates, such as 3-thiophene and 2-dibenzofuran boronic acid, also represent suitable coupling partners.29
Table 2.
Chlorosulfonylation of Boronic Acidsa

|
Yields represent isolated yields (average of two runs): 2a (1 mmol), ArB(OH)2 (1.5 mmol), Na2CO3 (5 mol %), P5 (2 mol %), degassed, anhydrous acetone (2 mL), 50–70 °C, 12 h.
Table 3.
Preparation of Sulfonamides
|
Isolated yields (average of two runs). Step 1: See conditions in Table 2.
P4 (2 mol %) was used. Step 2:
R2NH (2.2 mmol), rt, 1.5 h.
1-methylpiperazine (1.2 equiv.) and DIPEA (2.0 equiv.) were used.
RNH2 (3.0 equiv.), rt, 1.5 h.
ArNH2 (1.2 equiv.), pyridine (3.0 equiv.), rt, 5 h.
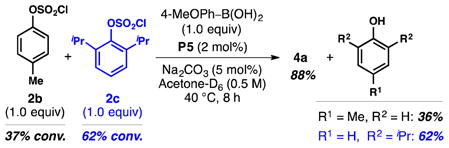
Several additional features of this chemistry are noteworthy. First, Pd(0) reacts with phenyl chlorosulfate (2a) in preference to aryl iodide groups bearing electron-withdrawing sulfonyl groups para to the iodo-substituents (e.g., 4e and 5i). Second, while others have demonstrated that arylsulfonyl chlorides themselves are efficient cross-coupling partners for various Pd-catalyzed processes,30–32 these intermediates are essentially unreactive under the chlorosulfonylation conditions. Third, palladium typically catalyzes the desulfonylation of arylsulfonyl chlorides; as a result, these substrates (4) are often used as aryl halide equivalents for carbon–carbon bond-forming processes.32 In contrast, the Pd-catalyst derived from L5 promotes carbon– sulfur bond formation, thus enabling the installation of a sulfonyl chloride (–SO2Cl) functional group.33 Finally, regarding the oxidative addition step, we surmised that the sp3 oxygen atom of 2a might direct the insertion of Pd(0) into the proximal PhO–SO2 bond; however, results from a competition experiment between 2b and the bulkier 2c (Eq. 3) revealed that 2c is slightly more reactive than 2b (ratio of recovered 2b/2c = 1.7:1).34 Moreover, this counterintuitive trend in reactivity is not specific to Pd-catalysis. For example, reacting a 1:1 mixture of 2b and 2c with piperidine (Eq. 4) resulted in a comparable ratio of recovered 2b/2c (1.6:1) along with formation of the sulfamoyl chloride (8) derived from piperidine. To explain the displacement of phenoxide from non-catalyzed nucleophilic additions to 2, Buncel has invoked an SN2 mechanism in which elongation of the S–O bond relieves considerable strain in the bipyramidal transition state; according to the authors, such relief of strain would not accompany partial rupture of the S–Cl bond.21 In this regard, increasing the size of the aryloxy substituent of 2 may intensify this stereoelectronic effect and further weaken the S–O bond.
 |
(Eq. 3) |
 |
(Eq. 4) |
In summary, we demonstrated that phenyl chlorosulfate (2a) represents an excellent [SO2Cl]+ synthon in the context of Pd-catalyzed Suzuki-Miyaura cross-coupling. The chlorosulfonylation reaction exhibits considerable functional group tolerance and the transformation is inherently regioselective; the substitution patterns of many of the products shown (such as 4h–4j) cannot be accessed by EAS processes. Furthermore, the sulfonyl chloride intermediates can be derivatized in situ and isolated as the corresponding sulfonamides. Therefore, both the aryl and amine components of arylsulfonamides can be installed in a single synthetic operation from readily available reagents. Investigations aimed at broadening the scope of this transformation and further delineating the mechanism of sulfur-oxygen bond scission are currently in progress.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for financial support of this project (GM58160) and a fellowship to J.R.D. (1F32GM099202). The Varian 300 and 500 MHz NMR spectrometers used for portions of this work were purchased with funds from NSF (Grants CHE-9808061 and DBI-9729592).
Footnotes
Experimental procedures and characterization data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest( s): MIT has patents on ligands that are described in the paper from which S.L.B. receives royalty payments.
References
- 1.Drews J. Science. 2000;287:1960. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 2.Bartholow M. Top 200 Drugs of 2011. Pharmacy Times. http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011.For a list of top drugs by year see: (b)http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster
- 3.Anderson KK. In: Sulfonic Acids and Their Derivatives in Comprehensive Organic Chemistry. Barton DHR, Ollis WD, Jones DN, editors. Vol. 3. Pergamon Press; Oxford: 1979. pp. 331–350. [Google Scholar]
- 4.Cremlyn RJ. Chlorosulfonic Acid: A Versatile Reagent. Royal Society of Chemistry; Cambridge: 2002. pp. 7–21. [Google Scholar]
- 5.(a) Baharami K, Khodaei MM, Khaledian D. Tetrahedron Lett. 2012;53:354. [Google Scholar]; (b) Prakash GKS, Mathew T, Olah GA. J Org Chem. 2007;72:5847. doi: 10.1021/jo070907g. [DOI] [PubMed] [Google Scholar]; (c) Wright SW, Hallstrom KN. J Org Chem. 2006;71:1080. doi: 10.1021/jo052164+. [DOI] [PubMed] [Google Scholar]
- 6.(a) Percec V, Bera TK, De BB, Sanai Y, Smith J, Holerca MN, Barboiu B, Grubbs BBB, Fréchet JMJ. J Org Chem. 2001;66:2104. doi: 10.1021/jo001694x. [DOI] [PubMed] [Google Scholar]; (b) Watson RJ, Batty D, Baxter AD, Hannah DR, Owen DA, Montana JG. Tetrahedron Lett. 2002;43:683. [Google Scholar]
- 7.(a) Alapafuja SO, Nikas SP, Shukla VG, Papanastasiou I, Makriyannis A. Tetrahedron Lett. 2009;50:7028. doi: 10.1016/j.tetlet.2009.09.167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bahrami K, Khodaei MM, Soheilizad M. J Org Chem. 2009;74:9287. doi: 10.1021/jo901924m. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pandya R, Murashima T, Tedeschi L, Barret AGM. J Org Chem. 2003;68:8274. doi: 10.1021/jo034643j. [DOI] [PubMed] [Google Scholar]; (b) Woolven H, González-Rodrígues C, Marco I, Thompson AL, Willis MC. Org Lett. 2011;13:4876. doi: 10.1021/ol201957n. [DOI] [PubMed] [Google Scholar]; (c) Monovich LG, Tommasi RA, Fujimoto RA, Blancuzzi V, Clark K, Cornell WD, Doti R, Doughty J, Fang J, Farley D, Fitt J, Ganu V, Goldberg R, Goldstein R, Lavoie S, Kulathila R, Macchia W, Parker DT, Melton R, O’Byrne E, Pastor G, Pellas T, Quadros E, Reel N, Roland DM, Sakane Y, Singh H, Skiles J, Somers J, Toscano K, Wigg A, Zhou S, Zhu L, Shieh W, Xue S, McQuire LW. J Med Chem. 2009;52:3523. doi: 10.1021/jm801394m. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ho DKH, Chan L, Brennan PE. Tetrahedron Lett. 2011;52:820. [Google Scholar]; (b) Bonk JD, Amos DT, Olson SJ. Synth Commun. 2007;37:2039. [Google Scholar]
- 10.Diazonium salts have been used as direct precursors to sulfonyl chlorides. For a recent example, see: Malet-Sanz L, Madrzak J, Ley SV, Baxendale IR. Org Biomol Chem. 2010;8:5324. doi: 10.1039/c0ob00450b.
- 11.Alternatively, sulfonyl compounds can be prepared in one-pot from Grignard additions to SO2 (see refs 8a-b), but subsequent chlorination requires SO2Cl2, which readily chlorinates aromatic rings.
- 12.(a) Nguyen B, Emmet EJ, Willis MC. J Am Chem Soc. 2010;132:16372. doi: 10.1021/ja1081124. [DOI] [PubMed] [Google Scholar]; (b) Emmet AJ, Richards-Taylor CS, Nguyen B, Garcia-Rubia A, Hayter BR, Willis MC. Org Biomol Chem. 2012;10:4007. doi: 10.1039/c2ob07034k. [DOI] [PubMed] [Google Scholar]
- 13.(a) Shengqing Y, Wu J. Chem Comm. 2012;48:7753. doi: 10.1039/c2cc33725h. [DOI] [PubMed] [Google Scholar]; (b) Shengqing Y, Wu J. Chem Comm. 2012;48:10037. doi: 10.1039/c2cc34957d. [DOI] [PubMed] [Google Scholar]
- 14.(a) Caddick S, Wilden JD, Judd DB. Chem Commun. 2005:2727. doi: 10.1039/b501212k. [DOI] [PubMed] [Google Scholar]; (b) Wilden JD, Geldeard L, Lee CC, Judd DB, Caddick S. Chem Commun. 2007:1074. doi: 10.1039/b614604j. [DOI] [PubMed] [Google Scholar]; (c) Bornholdt J, Fjaere KW, Felding J, Kristensen JL. Tetrahedron. 2009;65:9280. [Google Scholar]
- 15.Buncel E. Chem Rev. 1970;70:323. [Google Scholar]
- 16.Phenyl chlorosulfate is prepared by reacting phenol SO2Cl2 and pyridine at −78 °C. Compound 2a is stable in neat form for at least one year when stored at 4 °C.
- 17.No reaction was observed when solutions of 4-methoxyphenylboronic acid (1.5 equiv) and 2a (1.0 equiv) in toluene or 1,4-dioxane were stirred at 110 °C for 24 h.
- 18.Subjecting a phenyl sulfonate ester (3) to the reaction conditions in the presence of 2a and ArB(OH)2 resulted in complete recovery of 3. See Supporting Information for details.
- 19.An alternative mechanism might involve reversible oxidative addition to 2a, with transmetallation occurring at the –OPh group of 7 in preference to the –Cl group of the related Pd-Cl species. Oxidative addition is typically considered an irreversible process, although examples of reductive elimination of aryl halides have been reported. See: Roy AH, Hartwig JF. Organometallics. 2004;23:1533.Newman SG, Lautens M. J Am Chem Soc. 2010;132:11416. doi: 10.1021/ja1052335.
- 20.Alkoxo- and (phenoxo)Pd(II) complexes are known to undergo efficient transmetallation without the aid of base. For reviews and discussions, see: Miyaura N. J Organomet Chem. 2002;653:54.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.
- 21.(a) Buncel E, Raoult A. Can J Chem. 1972;50:1907. [Google Scholar]; (b) Buncel E, Choong LI, Raoult AJ. Chem Soc, Perkin Trans II. 1972:691. [Google Scholar]
- 22.Bruno NC, Tudge MT, Buchwald SL. Chem Sci. 2012;4:916. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For examples of cross-couplings using XPhos precatalysts, see: Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799.Oberli MA, Buchwald SL. Org Lett. 2012;14:4606. doi: 10.1021/ol302063g.
- 24.Similarly, little to no product was observed using SPhos or RuPhos ligands. For a review of Suzuki–Miyaura reactions employing dialkylbiaryl phosphine ligands, see: Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461. doi: 10.1021/ar800036s.
- 25.Both Pd(0) and phosphine ligands can potentially react with 4; thus to limit product decomposition and promote complete conversion of 2a, we chose to increase the reaction temperatures accordingly rather than adjust the respective catalyst loadings.
- 26.Electron-poor arylboronic acids typically undergo transmetallation at a slower rate than electron-neutral and electron-rich arylboronic acids.
- 27.The volatility of 4d–4i may also contribute to lower yields.
- 28.The stoichiometry of the amine can be reduced to 1.2 equivalents if DIPEA is used as a sacrificial base. See conditions for product 5d (Table 3).
- 29.Currently, boronic acids derived from nitrogen heterocycles do not couple in useful yields. This inefficiency is due to deboronation of the starting material and/or the instability of the corresponding sulfonyl chlorides. See: Caldwell WT, Kornfeld EC. J Am Chem Soc. 1950;72:4890.
- 30.The order of reactivity of electrophiles for Suzuki-Miyaura cross-coupling is: ArI > ArSO2Cl > ArBr ≫ ArCl (see ref 32c).
- 31.Aryl sulfonyl chlorides have been used as precursors to diaryl sulfones. See: Bandgar BP, Bettigeri SV, Phopase J. Org Lett. 2004;6:2105. doi: 10.1021/ol049692c.
- 32.For examples of desulfonylative cross-couplings of sulfonyl chlorides to form C—C bonds, see: Miura M, Itoh K. Chem Lett. 1989;18:77.Dubbaka SR, Vogel P. J Am Chem Soc. 2003;125:15292. doi: 10.1021/ja038328q.Dubbaka SR, Vogel P. Org Lett. 2004;6:95. doi: 10.1021/ol036131x.Dubbaka SR, Vogel P. Tetrahedron Lett. 2006;47:3345.
- 33.We observe homocoupling (< 15% biaryl) of certain electron-deficient and ortho-substituted arylboronic acids (e.g. corresponding to 5f and 5j). This side reaction may occur at the expense of 2a via an oxidative addition/desulfonylation pathway.
- 34.A control experiment in which 2c is introduced after complete conversion of 2b verified that 2c is not converted to 2b. See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.