

Abstract
The critical roles of TGF-β in the reciprocal differentiation of tolerance-promoting CD4+Foxp3+ regulatory T cells (Treg) and pro-inflammatory Th17 effector cells impact alloimmune reactivity and transplant outcome. We reasoned that a strategy that harnessed TGF-β and blocked pro-inflammatory cytokines would inhibit the differentiation of Th17 cells and strengthen the cadre of Treg to promote tolerance induction and long-term allograft survival. Herein we report the development of a novel, long-lasting auto-active human mutant TGF-β1/Fc fusion protein that acts in conjunction with rapamycin to inhibit T cell proliferation and induce the de novo generation of Foxp3+ Treg in the periphery, while at the same time inhibiting IL-6-mediated Th17 cell differentiation. Short-term combined treatment with TGF-β1/Fc and rapamycin achieved long-term pancreatic islet allograft survival and donor-specific tolerance in a mouse model. This effect was accompanied by expansion of Foxp3+ Treg, enhanced alloantigen-specific Treg function, and modulation of transcript levels of Foxp3, IL-6 and IL-17. Our strategy of combined TGF-β1/Fc and rapamycin to target the IL-6-related Treg and Th17 signaling pathways provides a promising approach for inducing transplant tolerance and its clinical application.
Introduction
Allograft outcome, whether rejection or tolerance, may depend on the balance between the function of effector and regulatory T cells (Treg)3(1-3). Strategies that can promote Treg expansion, while at the same time inhibiting effector T cells (Teff) offer considerable therapeutic potential in transplantation and other immune-mediated disorders. CD4+CD25+ Treg that express the forkhead box p3 (Foxp3) transcription factor, have emerged as critically important for the control of autoimmunity and for the maintenance of allograft tolerance (4, 5). Recent studies demonstrate that naïve peripheral CD4+CD25-Foxp3- T cells can be converted into CD4+CD25+Foxp3+ Treg by TGF-β in the context of TCR signaling and costimulation (6). Indeed there is a consensus that TGF-β may be indispensable for the development and maintenance of Treg in the periphery (7). TGF-β is a multifunctional cytokine that plays a crucial role in fundamental cellular functions, including differentiation proliferation, migration, and survival (8). In the context of an inflammatory cytokine milieu, TGF-β supports the de novo differentiation of naïve CD4+ T cells towards pathogenic IL-17-producing T helper cells (Th17) (9, 10). Thus, control of proinflammatory cytokines plays a key role in the induction of Treg.
Rapamycin is a macrolide antibiotic with potent anti-inflammatory, immunosuppressive, and immunoregulatory properties. It is now evident that the mammalian target of rapamycin (mTOR) has an important role in modulation of innate and adaptive immunity (11). It has been observed that rapamycin can selectively expand murine and human CD4+CD25+Foxp3+ Treg, while at the same time inhibiting Teff, or at least preventing their expansion (12-14). Moreover, binding of rapamycin to the immunophilin FK506 binding protein-12 (FKBP-12) abolishes FKBP-12-induced blockade of TGF-β receptor-mediated signaling events, suggesting a possible signaling link between rapamycin and TGF-β (15). Rapamycin can also program activated lymphocytes to produce TGF-β (16). Thus, the immunosuppressive effects of rapamycin may be mediated in part, by TGF-β. We hypothesized that adjunctive treatment of allograft recipients with rapamycin and TGF-β would (i) block the production of proinflammatory cytokines (such as IL-6), inhibit the differentiation of Th17 cells, and suppress proliferative responses of activated Teff, and (ii) promote the generation of Foxp3+ Treg and thus strengthen the cadre of immunoregulatory T cells, shifting the balance from immunity to tolerance.
Most cell types secrete TGF-β in a biologically inactive form that is composed of a TGF-β dimer in association with the latency-associated protein (17, 18). rTGF-β1 is also secreted in a latent form which can be activated by acidification (19). Active TGF-β1 is a cleaved 24-kDa homodimer. Such small proteins are usually cleared rapidly from the circulation, with t1/2 within minutes to hours (19-22). Several studies have shown that three cysteine (Cys) residues located in the pro region of the TGF-β1 precursor can form interchain disulfide bonds that prevent the release of the mature, active TGF-β1 (19, 23, 24). To produce a long-lasting, biologically active form of TGF-β1, we changed three cysteine codons in the pro region of human TGF-β1 precursor into serine (Ser) codons. We then genetically fused the mutant TGF-β1 cDNA with human IgG4 Fc to produce an auto-active TGF-β1/Fc immunoligand that does not depend on acidification for activation. The Ig component extends the circulating t1/2 to 32 h following systemic administration. Herein, we report on the construction and use of the mutant TGF-β1/Fc fusion protein in combination with rapamycin for the induction of Treg and the promotion of pancreatic islet transplant tolerance.
Materials and Methods
Mice
Male C57BL/6 (B6; H-2b), B6.Foxp3GFP knock-in (H-2b), B6.congenic for CD45.1(H-2b; CD45.1+), B6D2F1 (H-2b,d; CD45.2+), B6AF1 (H-2b/d k), DBA/2 (H-2d) and C3H (H-2K) mice were purchased from The Jackson Laboratory and were maintained under the pathogen-free conditions at the University of Pittsburgh Central Animal Facility.
Genetic construction of TGF-β1/Fc
Human TGF-β1 cDNA was amplified by PCR from the cDNA library of activated human PBMC using TGF-β1-specific synthetic oligonucleotide primers. The primers have a NotI site incorporated into the 5′ end and a BglII site at the 3′ end and their sequences are as follows (5′-3′): GTTAGCGGCCGCCACCATGGCCGCCCTCCGGGCTGC (forward) and TACTAGATCTCTGCACTTGCAGGAGCGCACGAT (reverse). Human Fcγ4 cDNA was generated from an IgG4-secreting hybridoma (HB-8636, ATCC, Manassas, VA) using reverse-transcriptase MMLV-RT and synthetic oligo (dT) oligonucleotides (Life Technologies, Grand Island, NY). The primers are as follows (5′-3′): AATCGGATCCCAAATATGGTCCCCCATGCCCA (forward) and AACTCTAGACTC ATTTACCCAGAGACAGGGAGA (reverse). To generate an auto-active TGF-β1/Fc construct, oligonucleotide site-directed mutagenesis was used to substitute three cysteine residues, Cys-33, Cys-223 and Cys-225 in the pro region of the TGF-β1 precursor with serine residues to prevent the formation of disulfide bonds (23). Ligation of mutant TGF-β1 and Fcγ4 components in the correct translational reading frame at the unique BglII site yielded 1887-bp long open reading frame encoding a 613-amino acid polypeptide. The mature secreted TGF-β1/Fc was predicted to possess the mutated TGF-β1 precursor complex and an m.w. of 190 kDa, exclusive of glycosylation (Fig. 1).
FIGURE 1.

Mutant TGF-β1/Fc fusion protein gene construction. Scheme for the genetic fusion of mutant human TGF-β1 and Fcγ4 cDNAs to create a TGF-β1/Fc immunoligand. Mutations were made in the pro region of TGF-β1 precursor by using site-directed mutagenesis to replace Cys33, Cys223 and Cys225 with Ser residues, which render TGF-β1/Fc immunoligand biologically active.
Expression and purification of TGF-β1/Fc
The appropriate genetic construction of TGF-β1/Fc sequences was confirmed by DNA sequence analysis after cloning of the fusion genes as NotI-XbaI cassettes into the eukaryotic expression plasmid pRc/CMV (Invitrogen, San Diego, CA). The plasmid was transfected into Chinese hamster ovary (CHO) cells by electroporation and selected by G418. High-yield clones were selected and cultured in serum-free medium. TGF-β1/Fc fusion protein was then purified from culture supernatants by protein A-Sepharose affinity chromatography, followed by dialysis against PBS and 0.22-μm filter sterilization. Purified protein was stored at -20 before use (25). Supernatants of the transfected CHO cells yielded ∼0.5μg/ml of TGF-β1/Fc and the endotoxin level was < 0.01 EU/μg of fusion protein.
Confirmation of size, TGF-β1, and Fcγ content specificity
Western blot analysis was performed following SDS-PAGE under reducing (with DTT) and non-reducing (without DTT) conditions using anti-human TGF-β1 mAb as well as anti-human IgG Fc polyclonal Ab (Pierce, Rockford, IL).
Characterization of TGF-β1/Fc biological activity
TGF-β1/Fc biological activity was accessed using the IL-4-dependent HT-2 cell (CRL-1841; ATCC) growth inhibition assay. HT-2 cells (1×105 cells/well) were seeded in 96-well flat-bottom plates in complete medium (RPMI 1640 supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 10% FBS, 2 mM L-glutamine and 50 μM 2-ME) with recombinant human IL-4 (rIL-4, 10 ng/ml, BD PharMingen, San Diego, CA). After 30 min, varying concentrations of TGF-β1/Fc (0.16-2.56 nM) or commercially supplied rTGF-β1 (0.05-0.8 nM, R&D Systems, Minneapolis, MN), were added for a further 24 h incubation. The cells were pulsed with [3H] thymidine (1 μCi/well) for the final 6 h and [3H] thymidine incorporation measured as CPM in a liquid scintillation counter (Perkin Elmer, Waltham, MA). Percent inhibition of proliferation was determined using the following formula: ([1-mean CPM of the treated group/mean CPM of the untreated control] × 100%).
The ability of TGF-β1/Fc to activate the Smads pathway was analyzed using Western blot. Aliquots of IL-4-stimulated HT-2 cells were seeded at 2×106 cells per 100 mm dish and treated with TGF-β1/Fc (2 μg/ml) or rTGF-β (2 ng/ml), or human IgG4 (2 μg/ml) for 24 h. The levels of phosphorylated Smad2 (pSmad2) and total Smad2 were determined in protein extracts from cell lysates with phospho-Smad2 (Ser465/467) antibodies and Smad2 antibodies (Cell Signaling, Boston, MA). 30 μg of proteins were loaded in each lane; protein content was determined by the BCA method (Pierce).
Determination of TGF-β1/Fc circulating t1/2
The serum concentration of TGF-β1/Fc was determined over time after a single bolus i.v. injection of the fusion protein (0.1 mg/mouse) to four 10-wk-old C57BL/6 mice. Serial 100-μl retro-orbital blood samples were obtained at 5 min, 1 h, 5 h, 8 h, 24 h, 48 h, 72 h, and 96 h post injection. A sandwich ELISA was employed using mouse anti-human TGF-β1 mAb as the capture Ab, and a biotin-conjugated mouse anti-human IgG4 mAb as the detection Ab (BD PharMingen), thus ensuring that the assay was specific for the TGF-β1/Fc fusion protein and not for TGF-β1 or IgG4.
T cell proliferation and suppression assays
Single-cell suspensions were prepared from spleens and lymph nodes, and RBC removed using RBC lysis buffer (Qiagen, Valencia, CA). CD4+ or CD8+ T cells were isolated by CD4+ or CD8+ T cell enrichment columns according to the manufacturer's instructions (Miltenyi Biotec, Auburn, CA). CD4+CD25- and CD4+CD25hi populations were sorted using a fluorescence-activated cell sorter (FACS Aria; BD Biosciences, San Diego, CA). To assess T cell proliferation, CD4+ or CD8+ T cells from C57BL/6 mice (2×105/well) were stimulated with plate-bound anti-CD3 (2 μg/ml) and soluble anti-CD28 (2 μg/ml) mAb in 96-well flat-bottom plates for 72h. Different concentrations of rapamycin (5, 10, 25, 50, 100 ng/ml, Sigma) and/or TGF-β1/Fc (1, 5, 10 μg/ml) were added at the start of culture. To quantify suppressive cell function, flow-sorted CD4+CD25- naïve B6AF1 responder T cells (1×105/well) were cultured with various numbers of CD4+CD25hi T cells from naïve or tolerant B6AF1 mice in round-bottomed 96-well plates, using anti-CD3 and anti-CD28 mAb (each 2 μg/ml) or irradiated (3000 rad) splenocytes (5×104/well) from donor (DBA/2) or the third-party (C3H) as stimulators. During 72 h or 6 day MLR, the cells were pulsed with [3H] thymidine (1 μCi/well) for the final 8 h and [3H] thymidine incorporation measured as described above.
In vitro Foxp3 induction and in vivo Treg conversion
For in vitro Foxp3+ induction, flow-sorted CD4+GFP− cells from naive B6.Foxp3GFP knock-in mice were cultured in 96-well plates (2×105/well), and stimulated with anti-CD3 and anti-CD28 mAb (each 2 μg/ml) for 72 h or with LPS-matured DBA/2 bone marrow-derived dendritic cells (DC) for 7 days, in the presence of TGF-β1/Fc (5 μg/ml), rapamycin (5 ng/ml), or TGF-β1/Fc plus rapamycin. The cells were analyzed for GFP(Foxp3) expression by flow cytometry. In some experiments, IL-6 and IL-17 levels in culture supernatant were measured by ELISA using mouse IL-6 and IL-17 ELISA kit (R&D Systems).
For in vivo Treg conversion, flow-sorted CD4+CD25- T cells from naïve congenic B6.CD45.1 mice were injected i.v. into semi-allogeneic non-irradiated B6D2F1 mice (1× 107/mouse). After cell transfer, B6D2F1 hosts were treated on day 0, 1, and 2 with TGF-β1/Fc (0.1 mg, i.p.) or rapamycin (0.6 mg/kg, i.p.) alone or with both agents. On day 4, lymph node and spleen cells harvested from B6D2F1 hosts were stained for surface markers (CD45.1 and CD4) and intracellular Foxp3, then analyzed by flow cytometry, gating on CD45.1+CD4+cells.
Islet allograft transplantation
B6AF1 mice were rendered diabetic by a single i.p. injection of streptozotocin (225 mg/kg), 1 wk prior to transplantation. Approximately 400 DBA/2 islets were transplanted under the renal capsule of each diabetic recipient mouse as previously described (26). Recipients were treated i.p. with TGF-β1/Fc and/or rapamycin as follows: 0.1 mg TGF-β1/Fc per mouse every other day for 10 days, 0.3 mg/kg rapamycin daily for the first 7 days after transplantation, then every other day for 7 days. The recipients with long-term islet allograft survival (150 days) received a second DBA/2 or C3H islet allograft in the contralateral kidney 1 wk after the removal of kidney bearing islet allograft. Allograft function was assessed by monitoring blood glucose levels daily for the first 14 days, then twice weekly, with rejection defined as blood glucose levels of > 300 mg/dl on 2 consecutive measurements.
Flow cytometric analysis
Cells harvested from lymph nodes, spleen, or peripheral blood of host mice, as well as cultured cells were analyzed for the expression of various cell surface or intracellular markers using an LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree star).
Real-Time PCR
Total RNA was extracted from draining lymph nodes or islet allografts using RNeasy mini-Kit (Qiagen) and reverse transcribed to cDNA using SuperScript III RT-kit (Invitrogen). Specific mRNA levels were quantified by real-time PCR using the ABI 7500 Sequence Detection System (Applied Biosystems, Foster City, CA). Gene-specific primers and probes for Foxp3, IL-6 and IL-17 were purchased from Applied Biosystems. Data are expressed using the CT method and normalized to the housekeeping gene GPDH.
Histology and immunohistochemistry
The kidney containing the islet graft was removed at day 8 or 150 after transplantation and embedded in OCT compound, snap frozen in liquid nitrogen, and stored at -80 °C until sectioning. For morphological evaluation, cryostat sections were fixed in methanol and stained with H&E. For immunohistochemical staining, sections were stained by a four-layer peroxidase-antiperoxidase method involving overnight incubation with rat anti-mouse mAb to CD4 (H129.19, BD PharMingen) or α-smooth muscle actin (α-SMA, 1A4, Sigma-Aldrich). Samples were evaluated in a blinded fashion, using two to three different levels of sectioning/sample.
Statistical analysis
Values were expressed as means ± SD. Analyses for statistically significant differences were performed using the Student's ‘t’ test and One-Way ANOVA test. The influence of different treatments on islet allograft survival was analyzed using a Log-Rank test. ‘P’ < 0.05 was considered significant.
Results
Characterization of TGF-β1/Fc fusion protein
To confirm the molecular size and the cytokine/isotype specificity of TGF-β1/Fc, the affinity-purified fusion protein was characterized by Western blot analysis following SDS-PAGE. As shown in Fig. 2, the TGF-β1/Fc migrated under reducing conditions as species of molecular size 95 kDa and 45 kDa (lane 2), indicating that there are two forms TGF-β1 fused to IgG4 Fc components, the mutant pro- TGF-β1 (large form) and the mature chain of TGF-β1 (small form), which corresponds with a previous report that the rTGF-β1 is secreted as a precursor complex (24). Under non-reducing conditions, the TGF-β1/Fc ran as three species with sizes of 190 kDa, 140 kDa and 90 kDa (a, b, c in lane 3) indicating a mixture of three forms of TGF-β1/Fc fusion proteins. The 190 kDa-or 90 kDa- species represents the homodimeric fusion proteins containing mutant pro-TGF-β1 or mature TGF-β1, respectively. The 140 kDa- species represents a homodimer composed of a pro- TGF-β1 and a mature TGF-β1. Moreover, the TGF-β1/Fc fusion proteins were bound by both anti-TGF-β1 mAb and anti-IgG Fc polyclonal Abs, confirming the cytokine and isotype specificity of the TGF-β1 moiety and Fcγ4 domain, respectively.
FIGURE 2.

Western blot analysis of TGF-β1/Fc fusion protein. SDS-PAGE of TGF-β1/Fc was performed under reducing (lane 2) and non-reducing (lane 3) conditions, staining for anti-human TGF-β1. Lane 1 is loaded with a high m.w. protein standard; lanes 2 and 3 are loaded with TGF-β1/Fc. For reference, the letters a, b, and c are noted at the right of the figure to indicate TGF-β1/Fc molecules. Size markers are indicated in kDa.
The biological activity of the TGF-β1/Fc was determined by growth inhibition of HT-2 cells. As shown in Fig. 3A, TGF-β1/Fc inhibited IL-4-stimulated HT-2 cell proliferation in a dose-dependent manner, similar to the function of active rTGF-β1, although the molecules were not compared on a mole-for-mole basis. We further investigated the influence of TGF-β1/Fc on Smad pathway activation using Western blot with specific antibodies. In HT-2 cells, incubation for 24h with either TGF-β1/Fc or rTGF-β1, but not with IgG4 significantly increased the expression of pSmad2 at the protein level (Fig. 3B), supporting an essential role for TGF-β/Smad signaling cascade activation in TGF-β1/Fc-mediated cellular responses.
FIGURE 3.

(A) Concentration-dependent inhibitory effects of TGF-β1/Fc on IL-4-dependent HT-2 cell proliferation. HT-2 cells were stimulated with rIL-4 and cultured in the presence of the indicated concentrations of rTGF-β1, TGF-β1/Fc or IgG4 for 24 h. [3H] thymidine incorporation was measured. Data are expressed as the mean percentage of inhibition from three independent experiments. (B) Western blot analysis for the expression of pSmad2 and Smad2 in HT-2 cells treated with rTGF-β1, TGF-β1/Fc or IgG4 for 24 h. The same lysates were also used to evaluate the expression of β-actin as loading control. One of two similar experiments is shown.
The circulating t1/2 of TGF-β1/Fc after a single i.v. bolus was 32 h, while the t1/2 of active TGF-β1 is 3 min. The TGF-β1/Fc concentration decreased in a biphasic manner, with an initial clearance of 48 h, followed by a slower, terminal component of 96 h (Fig.4). The serum TGF-β1/Fc concentration was 0.9 (± 0.2) μg/ml at 96 h. Thus, the novel fusion protein exhibits a protracted in vivo t1/2.
FIGURE 4.

TGF-β1/Fc circulating t1/2. The time-related serum concentration of TGF-β1/Fc in C57BL/6 mice was determined following a single bolus i.v. dose (0.1mg) of the fusion protein. TGF-β1/Fc was measured in serially obtained serum samples by a sandwich ELISA specific for TGF-β1/Fc. Data are a composite of measurements in four animals.
TGF-β1/Fc acts in conjunction with rapamycin to inhibit T cell proliferation
To examine the influence of TGF-β1/Fc combined with rapamycin on the proliferation of alloreactive T cells, CD4+ or CD8+ T cells were stimulated with anti-CD3 and anti-CD28 mAbs. Rapamycin exerted a dose-dependent inhibitory effect on T cell proliferation (Fig. 5A). A concentration of rapamycin (5 ng/ml) that reduced proliferation approximately 50% was used in subsequent in vitro experiments. TGF-β1/Fc alone at 1, 5, or 10 μg/ml, did not significantly inhibit T cell proliferation. However, combination of TGF-β1/Fc and rapamycin resulted in stronger suppression of CD4+ and CD8+ T cell proliferation than that achieved with rapamycin or TGF-β1/Fc alone (Fig.5B, 5C), suggesting an interactive inhibitory effect of TGF-β1/Fc and rapamycin on T cell proliferation.
FIGURE 5.

Inhibitory effects of TGF-β1/Fc and rapamycin on T cell proliferation. CD4+ or CD8+ T cells from C57BL/6 mice were cultured for 72 h in the presence of anti-CD3 and anti-CD28 mAbs, and treated with the indicated concentrations of rapamycin (5, 10, 25, 50, or 100 ng/ml) alone (A), or treated with the indicated concentrations of TGF-β1/Fc (1, 5, 10 μg/ml) plus rapamycin (5 ng/ml) (B, C). T cell proliferation was measured by [3H] thymidine incorporation. (A) Results are presented as mean percentages of untreated control CPM ± SD. (B, C) Results are presented as mean CPM ± SD. Data are representative of three experiments with similar results. *p < 0.01 versus untreated control.
TGF-β1/Fc and rapamycin promote the de novo induction of CD4+Foxp3+ Treg in vitro and in vivo
Recent studies have shown that both TGF-β and rapamycin can induce CD4+Foxp3+ Treg from naïve T cells (6, 27), but the influence of combined TGF-β1/Fc and rapamycin exposure on Treg conversion remains unclear. To investigate its influence on de novo induction of peripheral Foxp3+ T cells in vitro, flow-sorted naive CD4+GFP- T cells (> 99% pure) were stimulated with anti-CD3 and anti-CD28 mAbs for 72 h. TGF-β1/Fc alone induced 16 (± 4.2)% GFP+(Foxp3+) T cells from naive CD4+GFP− T cells; a similar conversion rate was observed when rTGF-β1 was added to the cultures (15-20%, data not shown). Rapamycin alone also promoted the induction of about 7 (± 2.4) % Foxp3+ T cells. When TGF-β1/Fc and rapamycin were added together to the cultures, the conversion rate increased up to 30 (± 4.5) % (Fig. 6A).
FIGURE 6.

Combined effects of TGF-β1/Fc and rapamycin on the differentiation of Foxp3+ Treg and Th17 cells from naive CD4 T cells. Flow-sorted naïve CD4+GFP- T cells from B6.Foxp3GFP knock-in mice were stimulated with anti-CD3 and anti-CD28 mAbs for 72 h (A), or cultured with LPS-matured allogeneic DC for 7 days (B), alone or in the presence of TGF-β1/Fc (5 μg/ml) and/or rapamycin (5 ng/ml). GFP+(Foxp3+) cells in the responder CD4+ T cell population were analyzed by flow cytometry. (C) IL-6 and IL-17 levels in mature DC-T cell culture supernatants were determined by ELISA. (D) Flow-sorted CD4+CD25- T cells from B6.CD45.1 were transferred to B6D2F1 mice by i.v. injection. The hosts were treated on day 0, 1, 2 with TGF-β1/Fc (0.1 mg) or rapamycin (0.6 mg/kg), alone or together. Data depict flow cytometric analysis of Foxp3+ cells converted from CD45.1+CD4+ T cells harvested from spleens of B6D2F1 mice on day 4. Data are representative of four independent experiments. *p < 0.01 versus untreated control.
There is evidence that TGF-β mediates the reciprocal differentiation of naïve T cells to either Treg or Th17 cells depending on the cytokine milieu and that IL-6 plays an important role in the induction of Th17 cells (10). We investigated whether the combination of TGF-β1/Fc and rapamycin might suppress inflammatory conditions and promote Treg differentiation. Purified naive CD4+GFP- T cells were cultured with LPS-matured DC for 7 days to generate an alloimmune response and an inflammatory cytokine milieu. IL-6 levels in the culture supernatants were reduced by TGF-β1/Fc or rapamycin treatment alone and more markedly by combined treatment (P < 0.01) compared to untreated cultures. In contrast, IL-17 production was increased by TGF-β1/Fc alone, whereas combined treatment reversed this effect and reduced the level of IL-17 significantly (P < 0.01) (Fig. 6C). In these cultures, untreated naïve CD4+ T cells expressed little GFP(Foxp3). Addition of rapamycin slightly increased Foxp3 expression to 7 (± 1.2) %. With TGF-β1/Fc alone, the proportion of CD4+Foxp3+ cells increased to 25 (± 4.5) %, while combined treatment further enhanced the induction of CD4+Foxp3+ cells to 41 (± 3.2) % (Fig. 6B).
We compared the influence of TGF-β1/Fc and rapamycin on in vivo conversion of naive CD4+ T cells into Treg. Flow-sorted CD4+CD25- T cells (> 99% pure) from congeneic B6.CD45.1 mice were adoptively transferred into semi-allogeneic B6D2F1 mice and the recipients were treated with TGF-β1/Fc and/or rapamycin for 3 days. In hosts treated with rapamycin alone, the incidence of CD4+Foxp3+ among the CD45.1+CD4+ T cells in the spleen was 20 (± 3.1) %. TGF-β1/Fc treatment alone resulted in 12 (± 2.7) % Foxp3+ T cells. However, combination of TGF-β1/Fc and rapamycin resulted in approximately 25 (± 2.3) % of CD45.1+CD4+ T cells expressing Foxp3 (Fig. 6D). Taken together, these data indicate that TGF-β1/Fc and rapamycin exert an additive effect on the induction of CD4+Foxp3+ Treg during naïve T cell differentiation in the context of their inhibitory effect on inflammatory cytokine production.
Combined TGF-β1/Fc and rapamycin treatment induces donor-specific transplant tolerance
To determine the influence of TGF-β1/Fc and rapamycin on allograft survival, MHC-mismatched DBA/2 islet allografts were transplanted into diabetic B6AF1 mice. As shown in Fig. 7A, untreated recipients rejected their grafts in a mean survival time (MST) of 19 days (n = 6). Administration of TGF-β1/Fc resulted in delayed islet allograft rejection (MST: 61 ± 21 days, n = 5, P = 0.008). Engraftment was prolonged in recipients treated with rapamycin (MST: 85 ± 15 days, n = 9, P = 0.001). In contrast, combined treatment with TGF-β1/Fc and rapamycin resulted in indefinite allograft survival in 90% of recipients (MST > 120 days, n = 11, p < 0.001) and showed more effective in promoting engraftment than two reagents used separately (p < 0.05). Surgical removal of kidneys bearing islet allografts from three combined treatment recipients 150 days after transplantation led to hyperglycemia, demonstrating that the euglycemic state was maintained by the islet allograft. Two nephrectomized recipients received a second DBA/2 islet allograft under the contralateral renal capsule in the absence of further immunosuppressive treatment and both accepted the second transplant for > 100 days, demonstrating a state of tolerance. One recipient received a third-party C3H islet allograft that was rejected at day 11 post-transplant, thereby confirming that the unresponsiveness was donor-specific and the host immune response was intact (Fig. 7B).
FIGURE 7.

Survival of islet allografts with a combined treatment of TGF-β1/Fc and rapamycin. (A) Diabetic B6AF1 mice were transplanted with DBA/2 islets allograft under the renal capsule and treated with TGF-β1/Fc, or rapamycin, alone or in combination, as indicated in the Materials and Methods. The MST for the untreated recipients was 19 days and for the TGF-β1/Fc-treated recipients 61 days. Recipients treated with rapamycin exhibited prolonged islet graft survival (MST 85 days), whereas combined treatment led to indefinite survival of most (90%) of the islets allografts (MST > 120 days). (B) Second islet transplantation. Three recipients bearing long-surviving islet allografts were nephrectomized to remove their grafts 150 days after transplantation. Two nephrectomized mice received a second DBA/2 islet allograft under the contralateral renal capsule without further immunosuppression and accepted the second transplant for > 100 days. One mouse received a third party (C3H) islet allograft which was rejected at day 11.
CD4+Foxp3+ Treg are expanded in tolerant recipients and express potent ability to suppress Teff responses
To determine if short-term combined TGF-β1/Fc and rapamycin treatment resulted in long-term, stable expansion of CD4+Foxp3+ Treg in graft recipients, we analyzed the expression of Foxp3. The results revealed that a population of CD4+Foxp3+ Treg in PBMC expanded (15.8 ± 2.1% vs 6.4 ± 0.2%; p < 0.05) and that Foxp3mRNA expression in draining lymph nodes and islet allografts was also enhanced significantly (p < 0.05) in tolerant recipients given combined treatment compared with naive mice (Fig. 8A, B). To ascertain whether the expanded Treg retained ability to suppress proliferation of Teff, we tested the ability of flow-sorted CD4+CD25hi cells (> 99% pure) from the lymph nodes/spleens of tolerant B6AF1 mice to suppress the function of naïve B6AF1 CD4+CD25- cells stimulated polyclonally with anti-CD3 and anti-CD28 mAb. CD4+CD25hi cells alone failed to proliferate following stimulation, consistent with the anergic properties ascribed to Treg, while CD4+CD25hi Treg from tolerant mice markedly inhibited the naïve Teff proliferation at a suppressor/responder ratio of 1:4, and exhibited much more potent ability to suppress proliferation than naive CD4+CD25hi Treg (63.4 ± 2.3 % vs 9.8 ± 1.8 %; P < 0.001). We further examined whether the regulatory function of the induced CD4+CD25hi Treg was related to antigen-specific triggering. Naïve CD4+CD25− T cells were stimulated with irradiated splenocytes from either the same donor (DBA/2) or third party (C3H) mice. The CD4+CD25hi Treg from tolerant mice significantly inhibited the proliferation of autologous CD4+CD25- T cells activated by the same donor antigen in MLR, but could not suppress third party antigen-induced alloreactivity (67.7 ± 2.2% vs 35.5 ± 9.7%; P < 0.05) (Fig. 8C).
FIGURE 8.

CD4+Foxp3+ Treg are expanded in TGF-β1/Fc + rapamycin-treated hosts and retain efficient suppressive function after transplantation. Blood, draining lymph nodes and islet allografts were harvested from naïve B6AF1 mice and from the combined-treatment tolerant recipient B6AF1 mice. (A) Foxp3 expression in CD4+ T cells from PBMC was analyzed by flow cytometry. (B) The mRNA expression of Foxp3 (relative to that of GAPDH) in draining lymph nodes and islet allografts was determined by quantitative real-time PCR. (C) Flow-sorted CD4+CD25- T cells from naïve B6AF1 mice were cultured alone, or with flow-sorted CD4+CD25hi Treg from naïve or combined-treatment tolerant B6AF1 mice (suppressor/responder 1 : 4), in the presence of anti-CD3 and anti-CD28 mAbs or irradiated splenocytes from DBA/2 or C3H mice. T cell proliferation was measured by [3H] thymidine incorporation after 72 h or 6 days of culture. Results are mean percentage inhibition (± SD) of proliferation of CD4+CD25- T cells from three independent experiments.
TGF-β1/Fc and rapamycin modulate Treg- and Th17-related gene expression and histological changes in islet allografts
Foxp3, IL-6 and IL-17 mRNA expression was analyzed in islet allografts and draining lymph nodes by quantitative real-time PCR and serum levels of IL-6 and IL-17 measured by ELISA on day 8 post transplantation. As depicted in Fig. 9A-C, combined TGF-β1/Fc and rapamycin markedly upregulated Foxp3 gene expression and profoundly downregulated IL-6 and IL-17 mRNA expression in the islet grafts compared to the untreated or TGF-β1/Fc-treated groups (P < 0.01). Likewise, in the lymph nodes draining the grafts, combined treatment significantly enhanced Foxp3 gene transcripts and decreased IL-6 mRNA expression (P < 0.01). IL-17 mRNA could not be detected in lymph node samples. These changes were accompanied by a reduction in serum levels of IL-6 and IL-17. Rapamycin alone reduced IL-6 expression in both the grafts and draining lymph nodes, and enhanced Foxp3 gene expression in draining lymph nodes, but not in the grafts.
FIGURE 9.

Combined effects of TGF-β1/Fc and rapamycin on the expression of Foxp3, IL-6 and IL-17 in islet allografts and host lymphoid tissue. The mRNA expression of Foxp3, IL-6, and IL-17 (relative to that of GAPDH) in islet allografts (A) and draining lymph nodes (B) of islet allograft recipients was determined by quantitative real-time PCR, 8 days after transplantation with the different treatments shown. IL-6 and IL-17 levels in the serum of islet allograft recipients were measured by ELISA 8 days after transplantation (C). Data are mean ± SD of four independent experiments.
Histopathological changes were assessed in H&E-stained allograft sections at day 8 post transplant. A dense mononuclear cell infiltrate of the islet grafts was observed in untreated recipients (Fig. 10A) with less intense infiltration in TGF-β1/Fc- or rapamycin-treated recipients (Fig. 10B, 10C). In the combined treatment group, cellular infiltration was dramatically reduced and islet architecture was well-preserved (Fig. 10D). As judged by immunohistochemistry, abundant CD4+ cells were present in allografts of untreated hosts (Fig. 10E), while a significant decrease in CD4+ cell infiltration was noted in recipients given TGF-β1/Fc and rapamycin combination therapy (Fig. 10F). This result is in keeping with the hypothesis that TGF-β1/Fc and rapamycin act concertedly to inhibit the alloreactivity of Teff To evaluate whether treatment strategies including short-term use of TGF-β1/Fc promoted fibrosis in allografts of combined treatment long-term graft recipients, we examined immunoreactivity for α-SMA, a valuable marker in determining activated myofibroblasts, which has been shown to be well correlated with progressive fibrosis in various organs (28-30). Only a few, dim α-SMA+ cells were observed in the vessel walls and interstitial areas of the kidney and no α-SMA+ cells were detected in grafted islets at day 150 post transplant (Fig. 10G), similar to that observed in untreated mice (Fig. 10H).
FIGURE 10.
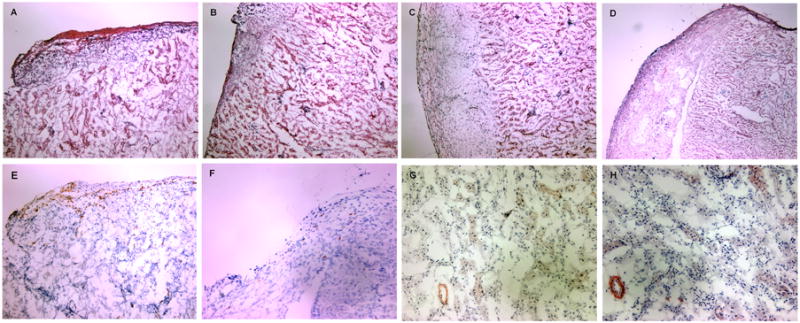
Histologic analysis of islet allografts. (A-D) Routine H&E staining of islet allografts isolated 8 days after islet transplantation. A dense tissue infiltration by mononuclear cells with destruction of islets is observed in untreated recipients (A). Recipients receiving rapamycin (B) or TGF-β1/Fc (C) show a decreased infiltration with some islets preserved. Graft sections from recipients given a combination of TGF-β1/Fc and rapamycin reveal almost normal histology, with minimal mononuclear cell infiltration and intact islets (D). (E-H) Immunohistochemical staining of islet allografts. Abundant CD4+ T cells were present in untreated recipients (E) while rare CD4+ T cell infiltrates were observed in recipients of combined treatment (F) at day 8 post-transplantation. Only a few dim α-SMA+ cells were evident in the vessel walls and the interstitial regions of the allograft in both untreated mice (G) and combined-treated recipients at day 150 post-transplantation (H). Cryostat sections, original magnification, × 100 (A-F), × 200 (G-H). These are representative sections from four allografts of each treatment group.
Discussion
CD4+Foxp3+ Treg have emerged as important factors in promoting tolerance to solid organ transplants (5, 31, 32). Since the elucidation of the immunoregulatory properties of TGF-β1 in Treg-mediated immune tolerance (6, 7), it has been anticipated that TGF-β1 might be a potentially important therapeutic adjunct for the treatment of transplant rejection and autoimmune disease. Nevertheless, the latent form of most cell-secreted precursor TGF-β1 and the short circulating t1/2 of biologically active mature TGF-β1 are major limitations for its in vivo application (18, 22). Previous studies (19, 24) have revealed that CHO cells express wild-type simian TGF-β1 in precursor complex form, consisting of pro- TGF-β1, the pro region of the precursor and mature TGF-β1. Three cysteines located in the pro region of the TGF-β1 precursor influence the maturation and activation of TGF-β1. It has been demonstrated that substitution of Cys-33 with a serine residue results in the generation of a more mature TGF-β1 dimer and that substitution of both Cys-223 and Cys-225 results in the production of only monomeric precursor forms, yet mature TGF-β1 is still able to form a bioactive dimer (23, 24). In this study, we constructed a novel mutant human TGF-β1/Fc immunoligand using genetic engineering. Although the purified mutant TGF-β1/Fc was found to be a mixture of three forms of fusion proteins, both the mutant pro-TGF-β1- and the mature TGF-β1- constituted TGF-β1/Fc fusion proteins could yield biological activity as in previous studies (23). Indeed, our results indicate that the mutant TGF-β1/Fc inhibits IL-4-dependent HT-2 cell proliferation in a dose-dependent manner and induces activation of the receptor-regulated Smad2 pathway (33), confirming the biological function, intracellular signal transduction and specificity of the TGF-β1 moiety in the TGF-β1/Fc fusion protein. Moreover, the Fc fragment ensured TGF-β1/Fc a greatly prolonged plasma t1/2 of 32 h. Hence, TGF-β1/Fc fusion protein can circulate and exert its activity for a greatly extended period following a single injected dose. Although human IgG4 has been demonstrated to be an isotype with relatively ineffective complement-dependent cytotoxicity and Ab-dependent cellular cytotoxicity (34, 35), the neutral effect status of IgG4 may vary due to population Fcγ receptor polymorphisms (36). Thus the specific mutations in Fcγ4 region may further eliminate Fc effector functions to guarantee the lack cytolytic capacity of TGF-β1/Fc (37).
Both TGF-β and rapamycin inhibit T cell proliferation (38, 39). Our results show that TGF-β1/Fc and rapamycin can act together to inhibit the proliferation of both CD4+ and CD8+ T cells more effectively in a dose-dependent manner. This suggests potential interaction between TGF-β1/Fc- and rapamycin-sensitive mTOR signaling pathways in T cell cycle progression. Evidence has accumulated that the control of Treg and Th17 cells may be interlinked (6, 9, 10). Thus, we examined the combined effects of TGF-β1/Fc and rapamycin on Treg and Th17 differentiation. In either mature DC- or anti-CD3/CD28 mAb-stimulated in vitro culture systems, both TGF-β1/Fc and rapamycin independently induced Foxp3+ Treg differentiation from naive CD4+Foxp3- cells, though the ability of rapamycin was lower than that of TGF-β1/Fc. However, combination of the two agents promoted significantly more Foxp3 expression, demonstrating the combined influence of TGF-β1/Fc and rapamycin on de novo induction of Treg. This effect may reflect the ability of rapamycin to stimulate TGF-β production (16, 40) and to suppress cell cycle progression, as Teff undergoing extensive proliferation fail to express Foxp3 (41). Moreover, in the mature DC-T cell culture environment, IL-6, a major cytokine produced by LPS-stimulated DC (42), can inhibit Treg generation and divert T cell differentiation to Th17 cells (10), an effect attributed to the activation by IL-6 of the transcription factor STAT3 (43). Since rapamycin can inhibit IL-6 signal transduction (44), blockade of IL-6 activity may be another important mechanism by which rapamycin favors Treg induction. Indeed, our results show that rapamycin decreases levels of IL-6 and IL-17 under these culture conditions. Although TGF-β1/Fc also inhibits IL-6, it enhances IL-17 production under proinflammatory conditions. We propose that the regulatory influence of TGF-β1/Fc on immune responses is complex and dependent on the extent of inflammatory stimulation. If an excess of IL-6 overrides the immunosuppressive action of TGF-β1/Fc, then the cytokine milieu will support the predominance of TGF-β-mediated Th17 cell differentiation. Thus, our observation that combined treatment with TGF-β1/Fc and rapamycin was more effective in decreasing IL-6 and IL-17 production suggests that their cooperative inhibitory effects on inflammatory responses may ultimately modulate the balance between Treg and Th17 cell generation. In this study, we have also demonstrated that TGF-β1/Fc and rapamycin act concertedly in vivo to promote the conversion of naive CD4+CD25- T cells into CD4+Foxp3+ Treg in a graft-versus-host disease (GVHD)-like model, in which transferred alloreactive CD4+Foxp3- T cells respond to host alloantigen and proliferate (27), though TGF-β1/Fc by itself does not cause significant Treg conversion in vivo, which was different from that shown in vitro. This may reflect the increase in endogenous IL-6 under acute in vivo immune/inflammatory conditions, however, concomitant therapy with rapamycin may abrogate the increase in IL-6 (45) and favor skewing towards TGF-β-driven Treg induction.
On the basis of above observations, we assessed the ability of combined TGF-β1/Fc and rapamycin to promote transplantation tolerance in a murine MHC-mismatched pancreatic islet allograft model. A 14-day course of combined treatment achieved, not only permanent survival of islets allograft in most treated recipients (90%), but also donor/antigen-specific immune tolerance exhibited 150 days post-transplantation. These mice accepted second donor but not third-party islet allografts. Furthermore, combined treatment does yield an improvement upon the effect of TGF-β1/Fc or rapamycin monotherapy, by which only slight to moderate prolongation of islet allograft survival was observed in recipients, indicating that the combined strategy was tolerogenic and that each of the components was required for optimal therapeutic effect. Considering that TGF-β1 is more required for the induction phase of Treg generation than the maintenance phase (46), and that an excess of TGF-β1 could increase the risk of tissue fibrosis (28), we used a regimen comprising only 5 doses of TGF-β1/Fc over the first 10 days after transplantation, in combination with rapamycin, to initiate Foxp3+ T cell differentiation and allow Treg-mediated tolerance to take place (47). Importantly, this protocol did not result in unwanted tissue fibrosis, which we investigated by α-SMA staining in allograft sections 150 days after transplantation and might be attributed to the potential antifibrotic effect of low-dose Rapamycin (48, 49). Peripheral blood, draining lymph nodes and islet allografts of long-term tolerant recipients exhibited an elevated expression of Foxp3 relative to naive animals. Moreover, the CD4+CD25hi Treg purified from tolerant recipients exerted a more potent suppressive effect on naive Teff proliferation following polyclonal stimulation. This suggests that expansion of Foxp3+ Treg and maintenance of their suppressive potency may at least in part underlie allograft tolerance induced by the combined therapy. Furthermore, these CD4+CD25hi Treg potently suppressed the proliferation of naive Teff against donor antigen but not third party alloantigen in MLR, indicating their alloantigen-specificity, their role in the observed donor-specific tolerance.
Active immune suppression by Foxp3+ Treg is a pivotal mechanism underlying peripheral T cell tolerance, while inflammation of local tissue during transplantation not only limits Treg suppression but also promotes proinflammatory Th17 responses (50, 51). Our data obtained on day 8 after islet transplantation revealed that the combined therapy markedly increased Foxp3 and decreased IL-6/IL-17 gene expression in both islet allografts and draining lymph nodes, accompanied by a reduction in serum IL-6 and IL-17 levels and a decrease in islet allograft CD4+ T cell infiltration. This strongly suggests that TGF-β1/Fc acts concertedly with rapamycin to inhibit IL-6-mediated Th17 responses and to provide conditions that favor Foxp3+ Treg generation in the “battlefield” of the alloimmune response. The potential benefit of this combined therapy is also indicated by the recruitment of Foxp3+ Treg to the allografts and draining lymph nodes, in which the continuing presence of Treg may ensure effective control of anti-donor reactivity and promotes allograft tolerance.
This novel immunoligand is as effective biologically as active rTGF-β1, has negligible potential for immunogenicity, and acts in conjunction with rapamycin to inhibit both CD4+ and CD8+ T cell proliferation. We also demonstrate for the first time that TGF-β1/Fc and rapamycin, especially in combination, promote the de novo generation of Foxp3+ Treg in vitro and in vivo, while simultaneously inhibiting IL-6-mediated Th17 cell differentiation. Furthermore, short-term combined treatment with TGF-β1/Fc plus rapamycin achieves donor-specific tolerance in a mouse model of islet transplantation, accompanied by Foxp3+ Treg expansion and their improved alloantigen-specific suppressive function. The concerted influence of these two agents on Foxp3, IL-6 and IL-17 transcripts and the trafficking of Treg after transplantation suggest an important underlying mechanism of tolerance induction.
Footnotes
This work was supported by grants from the Juvenile Diabetes Foundation International (1-2005-1001, XXZ; 3-2008-134, WZ) and the National Institutes of Health (RO1 AI67541, AWT).
Abbreviations used in this paper: Treg, regulatory T cells; Teff, effector T cells; mTOR, mammalian target of rapamycin; CHO, Chinese hamster ovary; DC, dendritic cells; α-SMA, α-smooth muscle actin; MST, mean survival time.
References
- 1.Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, Waldmann H. Infectious transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Fueyo A, Weber M, Domenig C, Strom TB, Zheng XX. Tracking the immunoregulatory mechanisms active during allograft tolerance. J Immunol. 2002;168:2274–2281. doi: 10.4049/jimmunol.168.5.2274. [DOI] [PubMed] [Google Scholar]
- 3.Zheng XX, Sanchez-Fueyo A, Domenig C, Strom TB. The balance of deletion and regulation in allograft tolerance. Immunol Rev. 2003;196:75–84. doi: 10.1046/j.1600-065x.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 4.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 5.Adeegbe D, Bayer AL, Levy RB, Malek TR. Cutting edge: allogeneic CD4+CD25+Foxp3+ T regulatory cells suppress autoimmunity while establishing transplantation tolerance. J Immunol. 2006;176:7149–7153. doi: 10.4049/jimmunol.176.12.7149. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 9.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 11.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–1023. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 13.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 14.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 15.Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, Martin J, Manganaro T, Donahoe PK. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996;86:435–444. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 16.Dodge IL, Demirci G, Strom TB, Li XC. Rapamycin induces transforming growth factor-beta production by lymphocytes. Transplantation. 2000;70:1104–1106. doi: 10.1097/00007890-200010150-00020. [DOI] [PubMed] [Google Scholar]
- 17.Lawrence DA, Pircher R, Kryceve-Martinerie C, Jullien P. Normal embryo fibroblasts release transforming growth factors in a latent form. J Cell Physiol. 1984;121:184–188. doi: 10.1002/jcp.1041210123. [DOI] [PubMed] [Google Scholar]
- 18.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 19.Gentry LE, Webb NR, Lim GJ, Brunner AM, Ranchalis JE, Twardzik DR, Lioubin MN, Marquardt H, Purchio AF. Type 1 transforming growth factor beta: amplified expression and secretion of mature and precursor polypeptides in Chinese hamster ovary cells. Mol Cell Biol. 1987;7:3418–3427. doi: 10.1128/mcb.7.10.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–7160. [PubMed] [Google Scholar]
- 21.Coffey RJ, Jr, Kost LJ, Lyons RM, Moses HL, LaRusso NF. Hepatic processing of transforming growth factor beta in the rat. Uptake, metabolism, and biliary excretion. J Clin Invest. 1987;80:750–757. doi: 10.1172/JCI113130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakefield LM, Winokur TS, Hollands RS, Christopherson K, Levinson AD, Sporn MB. Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J Clin Invest. 1990;86:1976–1984. doi: 10.1172/JCI114932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunner AM, Marquardt H, Malacko AR, Lioubin MN, Purchio AF. Site-directed mutagenesis of cysteine residues in the pro region of the transforming growth factor beta 1 precursor. Expression and characterization of mutant proteins. J Biol Chem. 1989;264:13660–13664. [PubMed] [Google Scholar]
- 24.Gentry LE, Lioubin MN, Purchio AF, Marquardt H. Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol Cell Biol. 1988;8:4162–4168. doi: 10.1128/mcb.8.10.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng XX, Steele AW, Nickerson PW, Steurer W, Steiger J, Strom TB. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccharide-induced septic shock and allogeneic islet transplantation. J Immunol. 1995;154:5590–5600. [PubMed] [Google Scholar]
- 26.Zheng XX, Sayegh MH, Zheng XG, Li Y, Linsley PS, Peach R, Borriello F, Strom TB, Sharpe AH, Turka LA. The role of donor and recipient B7-1 (CD80) in allograft rejection. J Immunol. 1997;159:1169–1173. [PubMed] [Google Scholar]
- 27.Gao W, Lu Y, El Essawy B, Oukka M, Kuchroo VK, Strom TB. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. 2007;7:1722–1732. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 29.Roberts IS, Burrows C, Shanks JH, Venning M, McWilliam LJ. Interstitial myofibroblasts: predictors of progression in membranous nephropathy. J Clin Pathol. 1997;50:123–127. doi: 10.1136/jcp.50.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gauldie J, Sime PJ, Xing Z, Marr B, Tremblay GM. Transforming growth factor-beta gene transfer to the lung induces myofibroblast presence and pulmonary fibrosis. Curr Top Pathol. 1999;93:35–45. doi: 10.1007/978-3-642-58456-5_5. [DOI] [PubMed] [Google Scholar]
- 31.Boros P, Bromberg JS. Human FOXP3+ regulatory T cells in transplantation. Am J Transplant. 2009;9:1719–1724. doi: 10.1111/j.1600-6143.2009.02704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. 2003;3:199–210. doi: 10.1038/nri1027. [DOI] [PubMed] [Google Scholar]
- 33.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 34.Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166:1351–1361. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isaacs JD, Clark MR, Greenwood J, Waldmann H. Therapy with monoclonal antibodies. An in vivo model for the assessment of therapeutic potential. J Immunol. 1992;148:3062–3071. [PubMed] [Google Scholar]
- 36.Greenwood J, Clark M, Waldmann H. Structural motifs involved in human IgG antibody effector functions. Eur J Immunol. 1993;23:1098–1104. doi: 10.1002/eji.1830230518. [DOI] [PubMed] [Google Scholar]
- 37.Reddy MP, Kinney CA, Chaikin MA, Payne A, Fishman-Lobell J, Tsui P, Dal Monte PR, Doyle ML, Brigham-Burke MR, Anderson D, Reff M, Newman R, Hanna N, Sweet RW, Truneh A. Elimination of Fc receptor-dependent effector functions of a modified IgG4 monoclonal antibody to human CD4. J Immunol. 2000;164:1925–1933. doi: 10.4049/jimmunol.164.4.1925. [DOI] [PubMed] [Google Scholar]
- 38.Ahuja SS, Paliogianni F, Yamada H, Balow JE, Boumpas DT. Effect of transforming growth factor-beta on early and late activation events in human T cells. J Immunol. 1993;150:3109–3118. [PubMed] [Google Scholar]
- 39.Nikolaeva N, Bemelman FJ, Yong SL, van Lier RA, ten Berge IJ. Rapamycin does not induce anergy but inhibits expansion and differentiation of alloreactive human T cells. Transplantation. 2006;81:445–454. doi: 10.1097/01.tp.0000194860.21533.b9. [DOI] [PubMed] [Google Scholar]
- 40.Huynh ML, V, Fadok A, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kretschmer KI, Apostolou D, Hawiger K, Khazaie MC, Nussenzweig, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–1227. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 42.Verhasselt V, Buelens C, Willems F, De Groote D, Haeffner-Cavaillon N, Goldman M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: evidence for a soluble CD14-dependent pathway. J Immunol. 1997;158:2919–2925. [PubMed] [Google Scholar]
- 43.Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 44.Kahan BD, Gibbons S, Tejpal N, Stepkowski SM, Chou TC. Synergistic interactions of cyclosporine and rapamycin to inhibit immune performances of normal human peripheral blood lymphocytes in vitro. Transplantation. 1991;51:232–239. doi: 10.1097/00007890-199101000-00038. [DOI] [PubMed] [Google Scholar]
- 45.Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, Wang Z, Thomson AW. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J Immunol. 184:624–636. doi: 10.4049/jimmunol.0900936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daley SR, Ma J, Adams E, Cobbold SP, Waldmann H. A key role for TGF-beta signaling to T cells in the long-term acceptance of allografts. J Immunol. 2007;179:3648–3654. doi: 10.4049/jimmunol.179.6.3648. [DOI] [PubMed] [Google Scholar]
- 47.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, Norton P, Zern MA. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–1204. doi: 10.1016/s0016-5085(99)70406-3. [DOI] [PubMed] [Google Scholar]
- 49.Neef M, Ledermann M, Saegesser H, Schneider V, Reichen J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J Hepatol. 2006;45:786–796. doi: 10.1016/j.jhep.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 50.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 51.Mitchell P, Afzali B, Lombardi G, Lechler RI. The T helper 17-regulatory T cell axis in transplant rejection and tolerance. Curr Opin Organ Transplant. 2009;14:326–331. doi: 10.1097/MOT.0b013e32832ce88e. [DOI] [PubMed] [Google Scholar]