

Abstract
Objectives
Recent genetic association studies have provided convincing evidence that several novel loci and single nucleotide polymorphisms (SNPs) are associated with the risk of developing type 2 diabetes mellitus (T2DM). The aims of this study were: 1) to develop a predictive model of T2DM using genetic and clinical data; and 2) to compare misclassification rates of different models.
Methods
We selected 212 individuals with newly diagnosed T2DM and 472 controls aged in their 60s from the Korean Genome and Epidemiology Study. A total of 499 known SNPs from 87 T2DM-related genes were genotyped using germline DNA. SNPs were analyzed for significant association with T2DM using various classification algorithms including Quest (Quick, Unbiased, Efficient, Statistical tree), Support Vector Machine, C4.5, logistic regression, and K-nearest neighbor.
Results
We tested these models using the complete Korean Genome and Epidemiology Study cohort (n = 10,038) and computed the T2DM misclassification rates for each model. Average misclassification rates ranged at 28.2–52.7%. The misclassification rates for the logistic and machine-learning algorithms were lower than the statistical tree algorithms. Using 1-to-1 matched data, the misclassification rate of the statistical tree QUEST algorithm using body mass index and SNP variables was the lowest, but overall the logistic regression performed best.
Conclusions
The K-nearest neighbor method exhibited more robust results than other algorithms. For clinical and genetic data, our “multistage adjustment” model outperformed other models in yielding lower rates of misclassification. To improve the performance of these models, further studies using warranted, strategies to estimate better classifiers for the quantification of SNPs need to be developed.
Keywords: classification, early predictive model, single nucleotide polymorphism (SNP), type 2 diabetes mellitus (T2DM)
1. Introduction
The prevalence of type 2 diabetes mellitus (T2DM) is increasing and the disease is becoming a worldwide epidemic [1]. T2DM is a complex disease affected by both genetic and environmental factors. Recent genetic association studies have provided convincing evidence that several novel loci and specific single nucleotide polymorphisms (SNPs) are associated with an increased risk of T2DM [2–6]. However, whether these SNPs have predictive value for future development of T2DM by an individual is controversial [7,8]. Hence development of an optimal and useful predictive model for T2DM using both genetic and clinical data is needed, because early prediction of T2DM can facilitate early control of blood glucose and lead to better clinical outcomes for patients with T2DM, or who may develop T2DM. However, there are few reports regarding prediction models that incorporate both genetic and clinical data.
We developed a new risk-prediction model for T2DM based on various statistical algorithms, and compared our model with preexisting models in terms of the error rate, also referred to as misclassification. We used SNPs and clinical data derived from a large community-based prospective cohort. The aims of this study were: (1) to develop a predictive model of T2DM using both genetic and clinical data; and (2) to compare the misclassification rates of our model versus other models.
2. Methods
2.1. Study population
The study samples were drawn from the Korean Genome and Epidemiology Study (KoGES), an ongoing prospective community-based epidemiological study in the communities of Ansung (rural) and Ansan (urban). From this study cohort (n = 10,038), we selected subjects who were aged in their 60s. From blood, it had measured fasting glucose, insulin, 1 hour and 2 hour glucose levels after the ingestion of 75 grams of glucose. Using these measures, we diagnosed new T2DM. We excluded 264 subjects who were already diagnosed with T2DM using questionnaire and selected newly diagnosed T2DM cases and age-matched controls from the KoGES cohort.
The diagnosis of T2DM was based on WHO criteria [9]. Control individuals had no past history of T2DM and had HbA1C values <5.8%.
2.2. Selection of polymorphisms in candidate genes
Using the NCBI (National Center for Biotechnology Information) database and published reports, we selected 87 known candidate genes associated with T2DM. The selected genes included 15 genes involved in insulin metabolism, 8 genes involved in fatty acid binding/translocation, 13 genes involved in GLUT4 translocation, and 51 others. Subjects (control and T2DM) were genotyped by Taqman/Goldengate method, and 499 SNPs in 87 genes analyzed.
The association of a given SNP with T2DM were determined by χ2 tests. We then selected significantly associated SNPs to optimize the predictive model and summarized the associations with allele types (dominant, recessive, heterozygous). Subsequently, we generated predictive models for T2DM using selected SNPs and clinical data. We then tested these models using data on the original study cohort (n = 10,038) and computed the misclassification rates for each model.
2.3. Statistical and classification analysis methods
We used a statistical decision tree classification algorithm to select SNPs because of the difficulty in optimizing high-risk SNPs.
We merged epidemiological, clinical, and genetic data to optimize classification. We then randomly divided the dataset into two parts, one for training or constructing the model and the other for testing the performance of the model (70% of the dataset was used for training and 30% for validation). The simulations resampled the data randomly and each simulation was repeated 100 times. We used various classification algorithms in this analysis based on the work of Ripley [10], Breiman [11], Hastie [12], and Quinlan [13] as follows. After first analyzing and screening for significantly associated SNPs using the χ2 test, we used all the selected SNPs in various multidimensional models. The misclassification rate of the test data was then estimated using selected variables (SNPs).
The QUEST binary tree-growing algorithm [14] was used for SNP selection and estimation of misclassification rates (significance level = 0.05; split methods = exhaustive search using Pearson’s χ2; 10-fold cross-validation sample pruning).
We also analyzed another tree classification algorithm first developed by Quinlan [13], C4.5, which splits by Information Gain or Entropy Reduction (70% of the dataset was used for training and variance reduction).
After χ2 analysis of the significantly correlated SNPs, we also used logistic regression (additive model) and K-nearest neighbor (KNN) in Euclidean distance algorithms.
Support Vector Machines (SVMs) are machine-learning algorithms originally developed by Hastie [12] that provide an optimization method for forming a separating hyperplane between cases and controls. We also analyzed the misclassification rate using SVM-light algorithms (regularization parameter c = 1.0; Kernal function = linear).
3. Results
3.1. SNP selection and predictive models using classification
A total of 684 subjects (472 controls and 212 subjects with T2DM) were analyzed to construct predictive models for T2DM. The average age of the test group subjects was 64.1 ± 2.9 years (mean ± standard deviation) for controls and 64.5 ± 2.8 years for subjects with T2DM; the average ages of the test and control groups were not significantly different. Supplementary Table 1 shows the baseline clinical data for the T2DM and control subjects.
The 684 subjects were genotyped for 499 SNPs that have previously been associated with T2DM. Among these known 499 SNPs, 18 SNPs were significantly associated with T2DM in our study subjects (Supplementary Table 2).
We generated predictive models for T2DM and calculated various misclassification rates (Supplementary Tables 3–7). In Supplementary Tables 3–5, listing of >1 variable indicates that the variables were treated as covariates. For example, “BMI + SNPs” means we have estimated the misclassification rates when both SNPs and body mass index (BMI) were used as variables. Additionally, SNPs stands for the SNPs that were significantly correlated with T2DM by χ2 test. The data in Supplementary Tables 3 and 6 were derived from all 684 subjects whereas those in Supplementary Tables 4 and 7 were derived from 1-to-1 matched data (212 controls, 212 subjects with T2DM).
In Supplementary Table 3 the misclassification rate of the logistic algorithm using triacylglycerides (TG), BMI, and SNPs was the lowest among the five classification algorithms. The misclassification rate of the logistic algorithm using TG, BMI, and SNPs generated the best results because the estimated misclassification rate was lowest (27.98 ± 2.76) for any of the variables and algorithms tested. However, the sensitivity was low (19.97 ± 8.00) in spite of the high specificity (91.29 ± 10.82).
Using 1-to-1 matched data, the misclassification rate of the QUEST algorithm using both BMI and SNPs as variables was lowest (Supplementary Table 4). The estimated misclassification rate was 34.85 ± 3.00; sensitivity was 58.98 ± 6.80 and specificity 71.32 ± 6.84. Using 1-to-1 matched data and combinations of variables with different classifiers, the estimated misclassification rates of the five classification algorithms tested ranged at 34.85–52.69%.
3.2. Model construction using multistage adjustment model
We attempted to improve the classification results using a multistage adjustment model. Risk factors were selected from our previous reports that used the same community-based cohort from the KoGES dataset. The selected risk factors were used for clustering the data into smaller groups with similar characteristics for pattern recognition analysis. In the alternative multistage adjustment approach, we employed simulations to test the data. In the first stage, we selected TG and BMI (p < 0.0001) as well-clustered variables related to obesity. We then clustered the data by TG and BMI values. This process gave three clusters (k = 3). We then ran 100 simulations for each group. The dataset was next divided randomly into the training set (70% of observations) and test set (30% of observations). The first stage results are shown in Supplementary Table 5.
Group 1 contained factors associated with obesity (BMI, waist-hip ratio [WHR], and body weight), blood glucose levels (fasting glucose and HbA1C), total cholesterol (TC), and homeostasis model assessment of insulin resistance (HOMA-IR) that were all within the normal ranges. Group 3 contained the highest BMI, WHR, and body weight values whereas group 2 contained the highest fasting glucose, HbA1C, TC, and HOMA-IR values.
The prevalence of T2DM (in both men and women) was similar in groups 2 and 3 but lower in group 1. In group 1 the variables related to BMI, blood glucose level, TC, and HOMA-IR were normal. The prevalence of T2DM in group 2 was significantly different than in the other two groups. In group 3 BMI, WHR, and body weight were significantly associated with T2DM.
Figure 1 plots the distribution of each group according to BMI, TG, and T2DM. Based on the results of the first stage of analysis, we further compared the classifications to assess risk factors. In Supplementary Tables 6 and 7 the range of misclassification was from 18.30% (logistic result of Supplementary Table 6) to 54.01% (SVM result of Supplementary Table 7).
Figure 1.
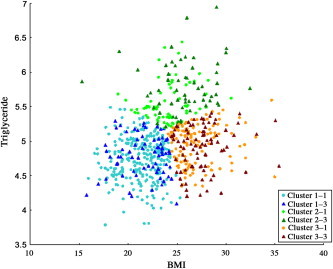
Scatter diagram for each group. Control samples are represented by Cluster 1–1, Cluster 2–1, and Cluster 3–1. T2DM samples are represented by Cluster 1–3, Cluster 2–3, and Cluster 3–3.
3.3. Model construction using various classification methods
We generated an early predictive model for T2DM based on clinical and genetic variables. However, model construction was difficult because all factors were highly correlated. We first used epidemiological and clinical variables, then constructed a model using only genetic variables (SNPs). However, if a probability existed that any subject could be normal for clinical factors but have abnormal genetic risk factors, the best model would analyze all three variables. Subsequently, we generated a model using multistage adjustments. Figure 2 shows the result of modeling using only clinical risk factors. Peripheral artery disease (PAD) is a combined variable for factors related to PAD.
Figure 2.
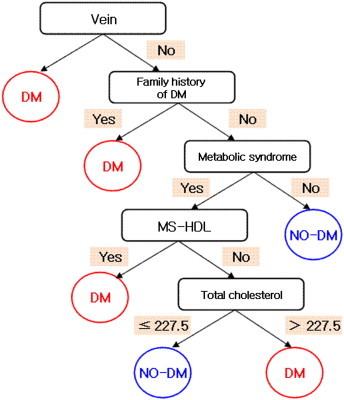
Model for epidemiological and clinical data.
Figure 3, 4 and 5 show the results of the multistage adjustment models. In Figure 3 the SNPs in TCF1_06 (HNF1 homeobox A; rs2464196) and TGFBI-01 (transforming growth factor; rs1442) are major risk factors for T2DM. Figure 4 shows that SNPs in MMP2_03 (matrix metallopeptidase 2; rs1030868) and ICAM1_01 (intercellular adhesion molecule 1; rs5491) are major risk factors for T2DM. The SNPs in SNTG1_10 (syntrophin gamma 1; rs1911830), FABP1_02 (fatty acid binding protein 1; rs2970901), and FABP4_03 (fatty acid binding protein 4; rs1054135) were also major risk factors for T2DM. For prediction using only SNPs and the multistage adjustment model (cluster 2), MMP2_03 was the first variable classified. In particular, the “T/T” form of MMP2_03 was predictive for T2DM risk.
Figure 3.
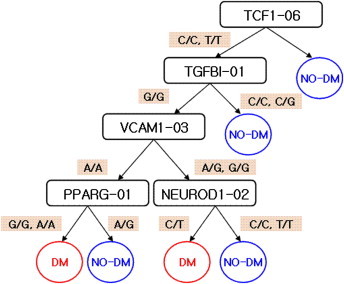
Results of classification in Cluster 1.
Figure 4.
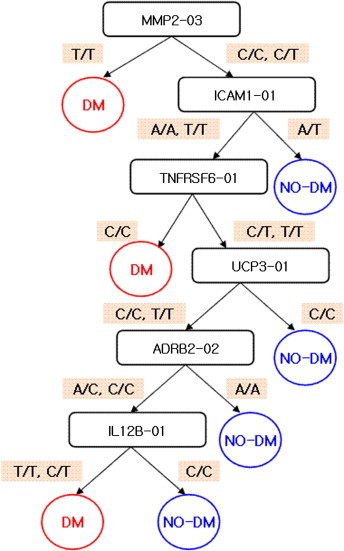
Results of classification in Cluster 2.
Figure 5.
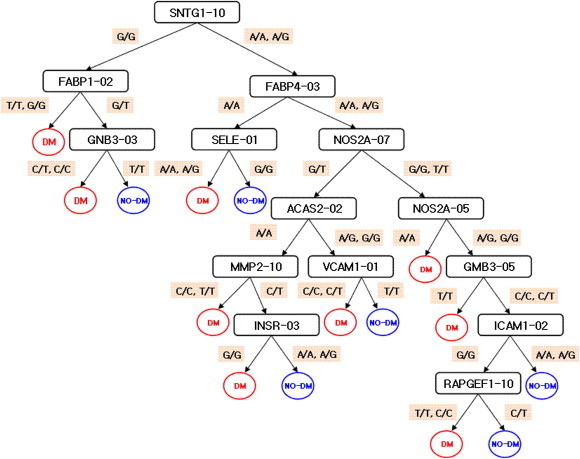
Results of classification in Cluster 3.
According to the statistical and classification analyses, the risk factors for T2DM were as follows:
-
•
Clinical information: height, BMI, waist circumference, blood pressure, HDL cholesterol, TC, TG, and fasting glucose level.
-
•
Personal and family history: hypertension, T2DM, cerebrovascular disease, and other vascular diseases.
-
•
SNPs: 499 SNPs in 87 candidate genes associated with T2DM. The selected genes included 15 genes in the insulin metabolism pathway, 8 genes in fatty acid binding/translocation, 13 genes in GLUT4 translocation, and 51 others.
Each category is represented in a Supplementary Table and corresponds to a field as shown in Figure 6.
Figure 6.
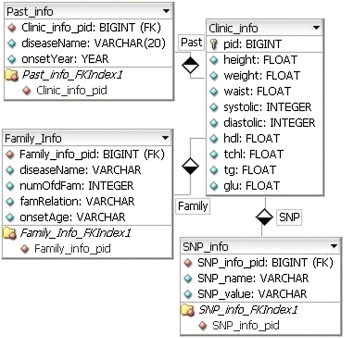
Database schema for input values.
Our system for the early prediction of T2DM is based on Object Oriented Modeling. It was developed using Fedora Core 6 and Spring’s web MVC framework, which is written in HTML, JSF, JavaScript and Java. MySQL was used as database to store and access the data. The system was implemented as a web application, and offers interfaces to input epidemiology data and SNP values and to select models. The user can select among different prediction models. The selected model analyzes the user’s input values and displays predictions about whether the input data are indicative of a person belonging to a group at risk of developing T2DM. Figure 7 shows a flowchart for this system.
Figure 7.
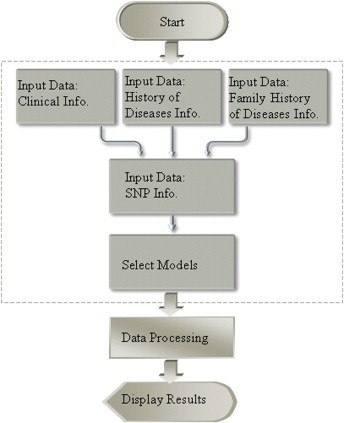
Web application flowchart for the early prediction system.
The web interfaces consist of an epidemiology information input page with a tabbed pane (Figure 8), an SNP value input page, a page for selecting models, and a page for displaying results.
Figure 8.
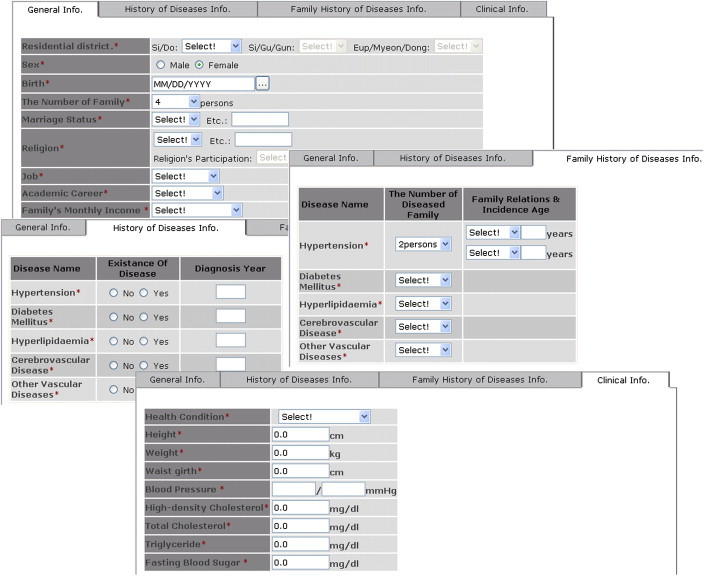
Snapshot of T2DM web application showing the interface for entering epidemiology data.
4. Discussion
To construct an optimal predictive model for T2DM we conducted various classification and clustering analyses using clinical and genotype data. We developed predictive models for T2DM using clinical and genotype data based on several classification algorithms. We first analyzed the predictive significance of SNPs by χ2 test. However, when we validated the predictive value of the models the misclassification rates were higher than expected.
The misclassification rates were 28.24–52.69% in the different models. In particular, in the SVM generated model the rates of misclassification, sensitivity, and specificity were higher than in other methods. The following reasons may explain the higher misclassification rates obtained using this model. First, the genotype distribution may contribute to the high level and wide range of misclassification rates. The genotype distribution of the 499 SNPs we investigated was 70–75% dominant genotypes (major/major allele), 5–15% recessive genotypes (minor/minor allele), and the remainder were heterozygous genotypes (major/minor allele). Hence the distance matrix calculation results could be applied to all classification algorithms. Furthermore, in analyzing the SNP pattern recognition the results were major, minor, or heterogeneous for A, T, G, C categorical data, which should be converted into numerical data to measure the distance with precision. This limitation resulted in high misclassification rates. Thus more study is needed to transform accurately such categorical data into numerical data. Second, the clinical data were variable, while the genotype data (results of the SNP association analysis) were relatively invariable. Hence important risk factors could appear as insignificant, resulting in a greater misclassification rate.
In terms of high misclassification rates, similar phenomena have been observed in predicting breast cancer [15] and rheumatoid arthritis [13]. Schwender et al [15] and Sun et al [13] tried to develop a predictive model using both clinical and genotype data. Their approach also converted the SNP allele type into numerical data and led to high misclassification rates.
The models constructed from statistical decision-tree classification algorithms and machine-learning methods did not show many differences. The classifiers could be better estimated using good quantification of SNPs. We suggest that such genetic information in molecular epidemiological data may be useful for classification of individuals into groups at high or low risk for developing T2DM. In view of our results thus far (Supplementary Table 3) we propose that subjects with group 1 characteristics can develop characteristics represented by group 3 and, subsequently, develop those represented by group 2. The risk factors for T2DM in group 1 were in normal ranges whereas group 3 exhibited an increase in values of variables related to obesity (BMI and blood glucose) and group 2 in those related to lipid, blood glucose, and cholesterol. The overall prevalence of T2DM in group 2 was higher than in the other groups.
The prevalence of T2DM observed in groups 1, 2, and 3 was 20%, 47%, and 40%, respectively. BMI and WHR were: group 1 < group 2 < group 3. HbA1C were: group 1 < group 3 < group 2. HbA1C increased in tandem with BMI (Supplementary Table 5). Thus the disease state risk factors appeared to progress from group 1 to group 3 to group 2 characteristics.
These results were validated by measuring several biological parameters. For example, fasting glucose level in group 2 was higher than in group 3. BMI values were higher in group 3 than in group 2. A comparison of group 3 versus group 2 demonstrated increases in fasting glucose levels and weight.
5. Conclusions
We developed predictive models for T2DM using clinical and genotype data. However, the misclassification rates were higher than expected. To improve the performance of the models, further studies using large sample sizes are warranted, and better research strategies to integrate genotype data classifiers need to be developed, particularly to quantify accurately the high-risk SNPs.
Authors’ contributions
JL participated of the study design, performed statistical analyses, and drafted the manuscript; BK contributed to drafting the manuscript; JYL MSP, EJJ performed statistical analyses; DBK constructed the prediction system; CSC, HJP, KBK, HLK, CHL were involved in discussion/interpretation of the data presented; BO performed the genotyping, JL, TK organized the study. All authors read and approved the final manuscript.
Acknowledgements
We thank the staff in the Division of Epidemiology and Health Index who contributed to gathering the clinical and epidemiological data. We also thank Dr. Thaler, English language editor at Bioedit, for her assistance. This study was supported by an intramural grant (2910-212-207; 2006-N72003-00) from Korea National Institute of Health.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Appendix. Supplementary Data
References
- 1.Wild S., Roglic G., Green A. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004 May;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Grant S.F., Thorleifsson G., Reynisdottir I. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006 Mar;38(3):320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 3.Sladek R., Rocheleau G., Rung J. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007 Feb;445(7130):881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 4.Saxena R., Voight B.F., Lyssenko V. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007 Jun;316(5829):1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 5.Scott L.J., Mohlke K.L., Bonnycastle L.L. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007 Jun;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeggini E., Weedon M.N., Lindgren C.M. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007 Jun;316(5829):1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meigs J.B., Shrader P., Sullivan L.M. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008 Nov;359(21):2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyssenko V., Jonsson A., Almgren P. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008 Nov;359(21):2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- 9.Alberti K.G., Zimmet P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part I, diagnosis and classification of diabetes: provisional report of a WHO consultation. Diabet Med. 1998 Jul;15(7):539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 10.Loh W.-Y., Shih Y.-S. Split selection methods for classification trees. Stat Sinica. 1997;7:815–840. [Google Scholar]
- 11.Quinlan J.R. Morgan Kaufmann; San Mateo: 1988. C4.5: programs for machine learning. [Google Scholar]
- 12.Hastie T., Tibshirani R., Friedman J.H. Springer-Verlag; New York: 2001. The elements of statistical learning. [Google Scholar]
- 13.Sun Y.V., Cai Z., Desai K. Classification of rheumatoid arthritis status with candidate gene and genome-wide single-nucleotide polymorphisms using random forests. BMC Proc. 2007;1(Suppl. 1):S62. doi: 10.1186/1753-6561-1-s1-s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allison P.D. SAS Institute Inc; Cary: 1999. Logistic regression using SAS system: theory and application. [Google Scholar]
- 15.Schwender H., Zucknick M., Ickstadt K., Bolt H.M. A pilot study on the application of statistical classification procedure to molecular epidemiological data. Toxicol Lett. 2004 June;151(1):291–299. doi: 10.1016/j.toxlet.2004.02.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.