

Abstract
Vaccination through mucosal surfaces has been shown to elicit antiviral immune responses against a number of mucosal pathogens. Here we demonstrate that both mucosal and systemic immune responses can be elicited against a model HIV-1 CN54gp140 antigen when cation-complexed plasmid DNA vaccines are applied topically to the murine pulmonary mucosa as an immune priming strategy. Furthermore, using an influenza challenge model we show that a plasmid DNA vaccine complexed to a less toxic form of PEI called dPEI (a nearly fully hydrolysed linear PEI with 11% additional free protonatable nitrogen atoms) can provide significant protection against a respiratory challenge infection in mice. Furthermore, we show that dPEI polyplexes have the potential to transfect not only mucosal epithelium, but also to enter deeper into tissues through the modulation of tight junction integrity. Taken together, these results demonstrate that less toxic forms of PEI can be effective delivery vehicles for plasmid DNAs to elicit cellular and humoral protective responses in vivo. Moreover, our observations suggest that these less toxic derivatives of PEI could be utilised for topical plasmid DNA vaccine delivery to human mucosal tissue surfaces, and that this application may permit dissemination of the immune responses through the linked mucosal network thus providing protective immunity at distal portals of pathogen entry.
Keywords: Mucosal, Vaccination, Gene delivery, Immunity, HIV-1, Influenza
Graphical abstract
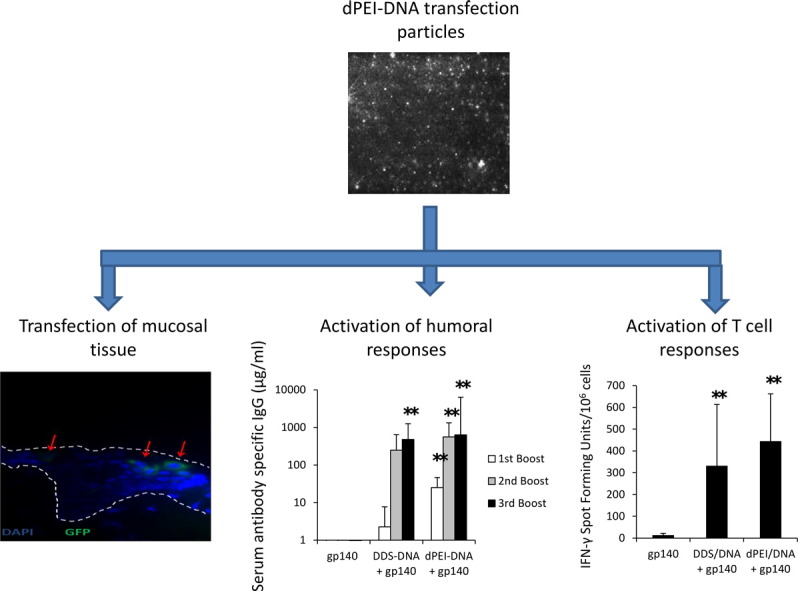
1. Introduction
The use of non-viral gene delivery systems has significant advantages for the treatment as well as prevention of a wide range of clinically relevant diseases. A number of delivery systems have been successfully employed in vitro and in vivo for the delivery of biologically relevant protein-encoding plasmids including cationic compounds based on polymers such as polyethylenimine (PEI) [1,2] or polylysine [3] and newer branched and hyperbranched derivatives of these linear molecules. Numerous lipid based emulsions have also been employed such as liposomes [4] and DOTAP/Squalene [5,6] formulations. All these cationic compounds are used for their intrinsic electrochemical property of interacting with and condensing the negatively charged DNA. These interactions allow the formation of complexes commonly referred to as polyplexes and lipoplexes depending on whether they are derived from polymer or lipid interactions. Due to its transfection efficiency and relatively low cost, PEI based gene delivery has become common place with numerous publications reporting on its merits. However the application of PEI as an in vivo gene delivery tool has been hampered by its apparent toxicity [7,8], deemed as a significant obstacle for mucosally applied vaccine applications, particularly when nasal or pulmonary delivery is required. Therefore the pursuit of DNA complexing agents with low toxicity and high transfection abilities is a primary goal for the DNA delivery field.
As a wide range of pathogens utilise mucosal surfaces as portals of entry into the body, the successful establishment of effective mucosal immunity has the potential to provide significant protection to these at risk surfaces from pathogen invasion. Mucosal delivery of vaccines can stimulate both mucosal and systemic immune responses while conventional systemic vaccinations are generally poor at activating mucosal responses [9]. Furthermore, the provision of protective responses at pathogen portals of entry, has the potential to either prevent infections altogether, eliminate the infection at the earliest time points, or at the very least, contain infections and reduce spread and the burden of infection.
Here we evaluated two different cationic compounds, Dope/Dotap/Squalene (DDS) and deacylated PEI (dPEI), for their capacity to condense plasmid DNA encoding a model trimeric gp140 vaccine candidate as a mucosal priming strategy. dPEI and DDS were chosen as mucosal transfection reagents as 1) dPEI is a nearly fully hydrolysed linear PEI with 11% additional free protonatable nitrogen atoms, enabling more efficient compacting of DNA, reduced toxicity and higher transfection rates [10], 2) while DDS has been used as a DNA delivery strategy before and has been reported to be 200 times better at transfecting than commercial liposome carriers [5]. Previous studies conducted within our laboratory have shown the mucosal delivery of unadjuvanted gp140 protein to be ineffective at generating specific immune responses [11,12]. Thus we investigated the use of DNA-cationic complexes as a mucosal priming strategy, capable of being boosted by homologous protein. In this report we show that a DNA vaccine delivered topically to mucosal surfaces can prime immune responses. In addition, we show that the DDS- and dPEI-complexed DNA vaccine formulations were capable of generating strong systemic and mucosal humoral responses as well as activating cellular responses. Finally, we show that dPEI is superior to DDS in the magnitude of elicited immune responses and that dPEI-DNA conveyed a higher degree of protection in a challenge model. Taken together, this study demonstrates the potential of dPEI as a topical mucosal delivery strategy.
2. Materials and methods
2.1. Plasmids and reagents
The HIV-1 CN54gp140 clade C Env glycoprotein expressing plasmid (gp140) was a kind gift from Roger Tatoud. The Influenza A virus (A/Aichi/2/1968(H3N2)) haemagglutinin (X31-HA) gene (GenBank accession no. CY121117.1) was synthesised and codon optomised for maximal expression in mice using the OptimumGene™ algorithm (GenScript,). The HA gene insert was subsequently cloned into the pmaxFP™-Red C vector (Lonza, UK). Large scale plasmid production was carried out using an Endo free Gigaprep kit (Qiagen, UK). Cationic dPEI (PEI Max MW 40,000 kDa), branched PEI (bPEI) and DDS emulsions were from Polysciences (Ger), Sigma Aldrich (UK) and Particle Sciences (U.S.A.) respectively. Homologous recombinant CN54-gp140 protein was purchased from Polymun (Austria) and the H3N2 A/Aichi/2/1968 was purchased from Sino Biological Inc., (China).
2.2. Plasmid cation complex formation
Cationic dPEI and bPEI were dissolved in sterile distilled DNAse free water to a final concentration of 5 mg/ml and 20 mg/ml while DDS was provided as a 24 mg/ml emulsion. Complex formation was achieved by the addition of pre-diluted cation in sterile distilled water to pre-diluted anionic DNA in sterile distilled water. For dPEI, bPEI and DDS, either 2 μg, 0.5 μg or 0.39 μl cation/0.5 μg plasmid DNA was used to generate dPEI-DNA, bPEI-DNA or DDS-DNA formulations. Resulting transfection particles were immediately vortexed at low speed for 15 s before resting for 15 min at room temperature.
2.3. Assessment of cell toxicity associated with various cationic compounds
Caco-2 cells were seeded in a 96 well flat bottomed tissue culture plate using complete RPMI (20% FCS). When the cells reached 70% confluency, the media was removed and replaced with 200 μl incomplete DMEM for 1 h. The cells were washed and DDS-DNA, dPEI-DNA, and N9 were added to the cells in a final volume of 200 μl of incomplete RPMI before 4 hour incubation at 37 °C. The cells were again washed and incubated with media containing MTT (200 μl; 0.5 mg/ml). Following incubation for 2 h at 37 °C, the media were removed and the cells were lysed using lysis buffer (98% isopropanol/2% 2 N HCL). Well contents were aspirated and adsorbance was determined using a spectrophotometer at 570 nm.
2.4. Determination of DNA-cation size using NanoSight
DNA-cation complex formation was analysed by measuring the rate of Brownian motion using a NanoSight LM10-HS system (NanoSight, Amesbury, UK) equipped with video capture and particle tracking software. Vaccine samples were prepared and pre-diluted in water accordingly. The samples were then injected into the viewing chamber and visualised using a focused laser (405 nm, 65 mW) and a Scientific CMOS Image Sensor. Particles were recorded at room temperature for 60 s, with manual shutter and gain adjustments. Samples were analysed using the NTA2.3 software.
2.5. DNA transwell studies using Caco-2 monolayers and CHO acceptor cells
The human adenocarcinoma cell line, Caco-2 and Chinese hamster ovary, CHO, cells were used as model epithelia with intact tight junction integrity and easy to transfect acceptor cells in a transwell transfection assay (see Supplementary materials for full details).
2.6. Immunisation, sampling and infections
Female BALB/c mice (Harlan, UK), 6–8 weeks old, were placed into groups of n = 6 and housed in a fully acclimatised room. All animals were handled and procedures performed in accordance with the terms of a project licence granted under the UK Home Office Animals (Scientific Procedures) Act 1986. Food and water were supplied ad libitum. Lightly anesthestised mice were placed in the supine position with all vaccinations administered as nasal drops, with delivery evenly distributed to each nare. Specifically, mice recieved3 DNA primes every 2 weeks with 20 μg/50 μl CN54-gp140 DNA-cation formulation followed by 3 protein boosts every 2 weeks with 20 μg/20 μl CN54-gp140. For influenza studies, empty vector (pmaxFP™-Red C) or pmax-X31 was used to prime followed by recombinant HA boost. A full description of sampling and infection protocols is given in the Supplementary material.
2.7. Immunohistochemistry
Penile tissue from patients undergoing gender reassignment surgery (local Research Ethics Committee approval was obtained) was used to assess polyplex transfection of model human mucosal tissues. Human penile glans tissue was cut into 3 mm by 3 mm explants while carefully removing as much stromal tissue as possible. The explants were washed then incubated in incomplete RPMI for 1 h within a 48 well tissue culture plate. The explants were then used in transfection experiments. A full description of experimental protocols used is shown in the Supplementary methods.
2.8. Antigen-specific immunoglobulin ELISA
We followed an in-house developed ELISA protocol to measure antigen-specific Immunoglobulin concentrations in serum and mucosal lavage. Full details of methods used are included in the Supplementary methods section.
2.9. Tissue processing for ELISpot and flow cytometry
Lymphocyte cultures were derived from single cell suspensions from spleens of immunised and control mice. Briefly, mice were euthanised, and their spleens were removed aseptically and placed into individual 20 ml universal tubes (Greiner, UK) containing 2 ml RPMI 1640 medium (Sigma Aldrich Ltd, UK). The spleens were then placed into Petri dishes and single cell suspensions made by disrupting the spleens. This was achieved by grinding the spleens through a 70 μm Nylon Cell Strainers (BD Falcon, UK) using forceps. The cell suspensions were then centrifuged at 350 g for 10 min. The supernatants were decanted and the pelleted cells re-suspended in ACK lysis buffer for 5 min (Gibco, UK). The cell suspensions were vortexed and centrifuged at 350 g for 10 min. The pelleted cells were decanted and re-suspended in 1 ml RPMI 1640 medium. This step was repeated three times with the cells resuspended in complete RPMI and then filtered through a 100 μm Filcon unit (BD Biosciences, UK). The cells were counted using a hemocytometer.
2.10. Antigen-specific ELISpot
Antigen-specific IgG and IgA spleen ELISpot assays (MABTECH, UK) were carried out as per the manufacturer's instructions. A full description of the experimental protocol is provided in the Supplementary methods.
2.11. Flow cytometric assessment of T cell responses in splenocytes
Isolated single cell cultures of murine splenocytes were assessed for reactivity against two pools each containing 78 peptides of 15-mers overlapping by 11 aa of HIV-1 CN54gp140 Env (Insight Biotechnology, UK) using a FACS Canto II instrument with FACS Diva software. Data analysis was performed with FlowJo (Treestar Inc., OR, USA). Full details are provided in the Supplementary methods section.
2.12. Statistical analysis
Statistical analysis of the data was carried out using a Mann–Whitney, U-test using GraphPad PRISM software.
3. Results
3.1. DDS-DNA and dPEI-DNA complexes were less toxic than bPEI-DNA and condensed DNA to form nanometer-sized transfection particles
Transfection particles were assessed for toxicity using a standard MTT assay and the epithelial cell line Caco-2. DDS-DNA transfection particles displayed no detectable toxicity as determined by MTT assay and its associated dimethylthiazol-2-yl-2,5-diphenyltetrazolium bromide reduction to formazan (Fig. 1a). The bPEI-DNA and surfactant Nonoxynol-9 (N9), positive controls both showed high levels of toxicity with cell viabilities of only 16.4% and 17% respectively, while dPEI-DNA exhibited moderate levels of cell toxicity with Caco-2 cell viability being reduced to 45.8% (± 7%). Similar results were seen with the Calu-3 respiratory cell line (data not shown). Due to the high levels of toxicity of the bPEI-DNA, it was omitted from further evaluation.
Fig. 1.
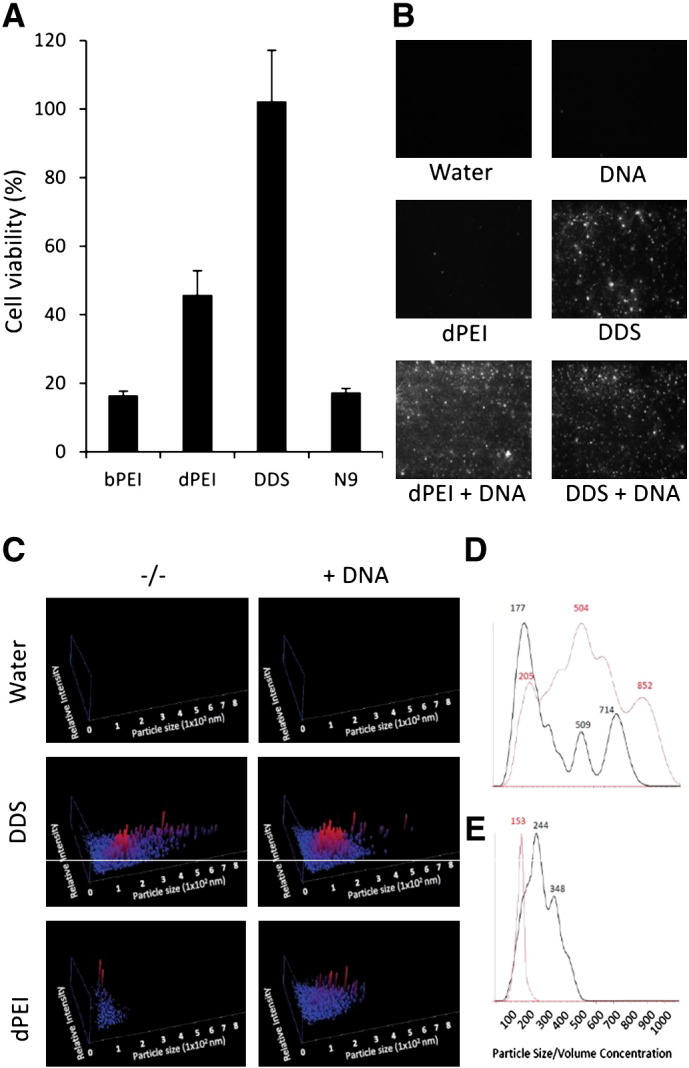
DDS and dPEI are less toxic transfection reagents than bPEI and N9 and condense DNA into nanometer-sized particles. The human adenocarcinoma cell line Caco-2 was cultured to 75% confluency before the addition of DNA-cation formulations. The complexes were incubated with the cells for 4 h before cell viability was tested using an MTT dye reduction assay (A). Polyplex and lipoplex formations were verified using a NanoSight LM10-HS system. Images represent results from nanoparticle tracking analysis (NTA) video capture (B). The particle size (x-axis) versus relative intensity (y-axis) of vaccine formulations generated by complexing DNA and cations (C). Normalised particle volume nm3 × 1012/ml versus particle size for DDS (red line) versus DDS-DNA (black line) (D) and dPEI (red line) versus dPEI-DNA (black line) (E).
Polyplex and lipoplex formations were visually verified using a NanoSight LM10-HS system (Fig. 1b). The Images are taken from the video capture of the Nanosight tracking analysis. Successful particle formation was observed for the dPEI-DNA polyplex formulation while no observable difference could be visually assessed for the DDS-DNA lipoplex formulations due to the pre-existing particulate nature of DDS (Fig. 1b). The size of resulting DNA-cation transfection particles was assessed using the nanoparticle tracking analysis (NTA), after the incubation of the cationic compounds with 20 μg DNA. Here we formulated 80 μg dPEI, or 15.6 μl DDS with 20 μg (9.52 μl) DNA in a final 50 μl volume made up in dH20. Complexing DNA with dPEI resulted in 14.96 × 108 particles/ml in the vaccine formulation with a mean diameter of 163 nm, while the complexing of DNA with DDS resulted in a vaccine formulation consisting of 6.8 × 109 particles/ml with a larger mean diameter of 206 nm (Fig. 1c). Interestingly, the addition of DNA to cation solutions resulted in a change in particle size. The addition of DNA to DDS resulted in a reduction in particle size and an increase in relative intensity of the smaller particles (Fig. 1d), while the addition of DNA to dPEI resulted in an increase in the detected particle size (Fig. 1e).
3.2. DDS-DNA and dPEI-DNA elicited antigen-specific IgG in serum and established a population of systemic antigen-specific IgG+ and IgA+ B cells
Due to toxicity issues, in vivo assessment was carried out on only the DDS-DNA and the dPEI-DNA cationic formulations. All immunisations were pulmonary applications of the complexes or recombinant protein. Antigen-specific immune responses were assessed before and after each CN54gp140 protein boost. We found that immediately after the final cation-DNA immunisation and 1st protein boost, DDS-DNA and dPEI-DNA transfection particles generated detectable humoral antibody responses in serum. The vaccine antigen-specific serum IgG responses elicited to the DDS-DNA and dPEI-DNA peaked after the third boost with 480 and 632 μg/ml of specific antibody being detected in serum respectively (Fig. 2a). No serum IgG responses were detected for the nasal gp140 protein alone vaccine regimen. Evaluation of splenocyte cultures by antigen-specific IgG ELISpot corroborated the ELISA data and demonstrated a similar response pattern. Here, mean spot forming units (SFU) of 4 (± 1.6), 154.3 (± 57.9), and 196.4 (± 43.4) were detected for gp140, DDS-DNA, and dPEI-DNA treatment groups respectively (Fig. 2b). Similar responses were seen for IgA+ B cells in splenocyte cultures where 44 (± 9.6), 141.3 (± 47.8), and 137 (± 32.3) SFU were detected for the gp140, DDS-DNA and dPEI-DNA treatment groups respectively (Fig. 2c). Furthermore, 100% of animals in the dPEI-DNA treatment regimen had antigen-specific IgG+ and IgA+ B cells in splenocyte cultures while only 66.6% of animals in the DDS-DNA treatment regimen had IgG+ and IgA+ SFU above background levels.
Fig. 2.
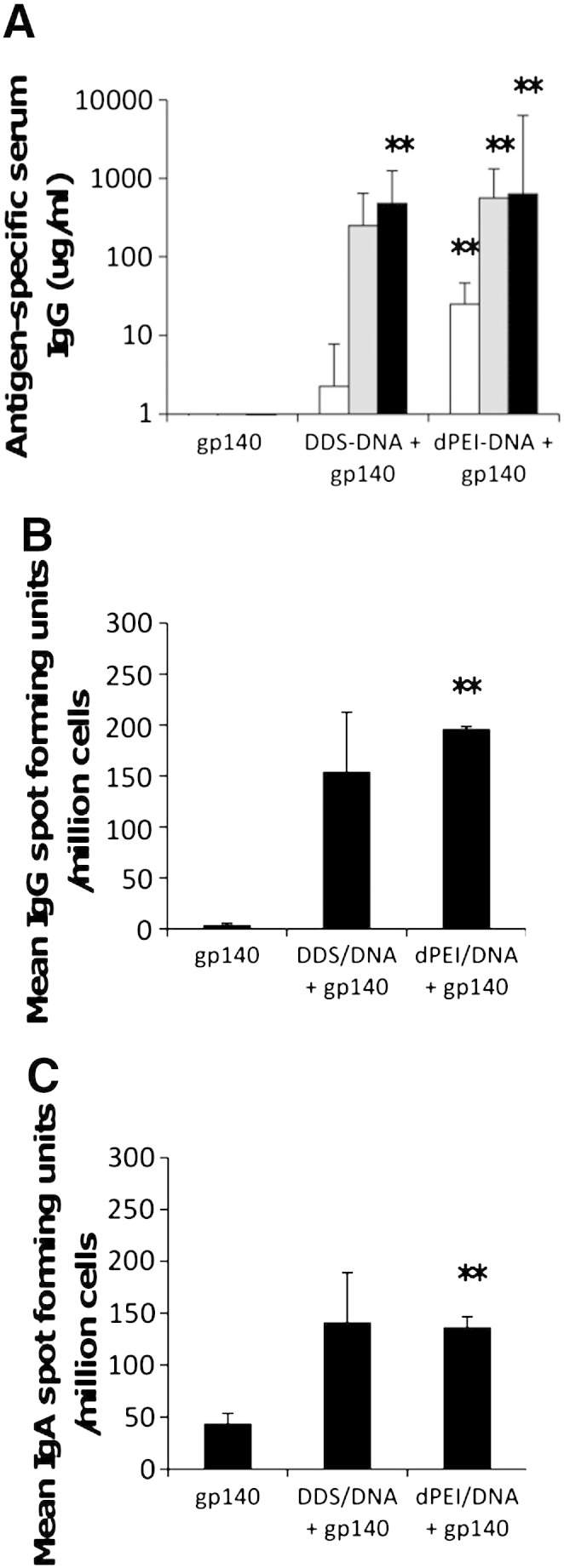
DNA prime-protein boost vaccination regimens result in antigen-specific humoral responses. Female BALB/c mice (n = 6 per group) were vaccinated 3 times at 2 week intervals with DNA followed by three protein boost vaccinations at 2 week intervals. Antigen-specific IgG antibody responses were recorded one week after each protein-boost vaccination until the end of the study, week 10 (A). Antibody results are expressed as geometric means of the group (μg/ml + SEM). Antibody secreting IgG (B) and IgA (C) cells were determined at the end of the experiment (week 10), using a commercially available direct ELISPOT assay as per the manufacturer's instructions. Results represent group means of Spot Forming Units (SFU)/million antigen stimulated cells (+ SEM). Statistical significance was assessed against the gp140 protein control group using Mann Whitney U test with *p ≤ 0.05 or **p ≤ 0.005.
3.3. DDS-DNA and dPEI-DNA transfection complexes generated antigen-specific IFN-γ, IL-2 and TNF-α producing CD4+ T cells
To characterise vaccine induced T cell responses in immunised animals, we performed intracellular cytokine staining (ICS) on isolated splenocytes using two peptide pools (15-mers overlapping by 11) covering the entire CN54gp140 Env sequence, and evaluated IFN-γ, IL-2 and TNF-α production. Splenocytes were harvested after the third CN54gp140 protein boost and the phenotype and functional response was assessed after the six hour stimulation in the presence of the pooled peptides and brefeldin A using standard ICS protocols with multi-parameter flow-cytometric analysis. With a sequential gating strategy, we removed doublets and gated on either CD4+ or CD8+ T lymphocytes. We found that CD4+ T cells in all vaccination groups produced IFN-γ, TNF-α and IL-2 in response to stimulation with the peptide pools, with the cation-DNA primed animals having a higher percentage of vaccine-antigen responsive cells than the gp140 protein alone group. The animals that had received dPEI-DNA polyplexes exhibited less variable responses than the DDS-DNA treated animals enabling the detection of significant differences from the CN54gp140 treated animals (Fig. 3a). Analysis of the CD8+ T cell response revealed that responses were almost exclusively directed toward peptides within pool 1 with little or no responses detected against peptide pool 2 (Fig. 3b). Furthermore, only low-level CD8+ T cell responses were detected in the DDS-DNA vaccine groups with only IFN-γ responses reaching significance (*p = 0.0135) compared to the gp140 control group. However, the dPEI-DNA prime boost vaccination regimen elicited higher CD8+ responses with significantly higher IFN-γ (**p = 0.008) and IL-2 (**p = 0.008) positive cells being generated than the CN54gp140 control group. The percentages of TNF-α positive CD8+ cells observed in each group were not significantly different (Fig. 3b). To support and confirm the ICS staining, an IFN-γ and IL-2T cell ELISpot assay was used (Suppl Fig. 1). Using the ELISpot assay both the DDS-DNA (**p = 0.0022) and dPEI-DNA (**p = 0.0043) formulations generated significant numbers of IFN-γ SFU/106 splenocytes with mean values of 332.3 (± 280) and 444.4 (± 217) respectively. For IL-2 producing lymphocytes, again the DDS-DNA (*p = 0.037) and dPEI-DNA (*p = 0.022) formulations elicited significantly higher SFU/106 than the CN54gp140 protein treated animals with average IL-2 SFU values of 114 (± 84.9) and 142.8 (± 94.7) (Suppl. Fig. 1). Only the dPEI-DNA prime-CN54gp140 protein boost regimen generated significant numbers of TNF-α positive cells (Fig. 3a). TNF-α production was not assessed by ELISpot due to the lack of a commercially available ELISpot kit.
Fig. 3.

Vaccine-induced T cell responses are generated after DNA prime-protein boost regimens. Vaccinated mice were sacrificed one week after the final protein boost vaccination and their splenocytes assessed for antigen-reactive T cells using 2 sets of 15mer overlapping by 11 amino acid peptide pools. Flow cytometric analysis of antigen-specific T cells was achieved by a progressive gating strategy to identify CD3+CD4+ or CD3+CD8+ T cell populations. Mean percentages of the peptide pool-reactive CD4+ (A) and CD8+ (B) T cells within vaccination groups (after exposure to stimulation) expressing IFN-γ, IL-2 and TNF-α. Statistical significance was assessed using Mann Whitney U test with *p ≤ 0.05 or **p ≤ 0.005.
3.4. DDS-DNA and dPEI-DNA both generate significant quantities of mucosal antigen-specific IgG and IgA antibody
An important hallmark of mucosal vaccines is their ability to generate effective local responses and currently most, if not all, vaccine-elicited protective responses are mediated through humoral immunity. We assessed the ability of nasal applications of DDS-DNA and dPEI-DNA complexes to generate antigen-specific antibody responses at a distal mucosal site. Vaginal lavage samples were assayed for the presence of vaccine antigen-specific IgG and IgA (Fig. 4). We found significant quantities of antigen-specific IgG antibodies in the vaginal lavage samples of animals that had received either the DDS-DNA or dPEI-DNA primes followed by CN54gp140 boost vaccination regimens, with mean concentrations of 32.8 ng/ml (**p = 0.005) and 248.2 ng/ml (**p = 0.005) respectively (Fig. 4a). This was also the case for vaccine-specific IgA production, with mean antigen-specific IgA concentrations of 108.5 ng/ml (*p = 0.0285) and 311 ng/ml (**p = 0.005) respectively being produced by the DDS-DNA and dPEI-DNA prime CN54gp140 boost regimens (Fig. 4b). Hence topical DNA vaccination using dPEI was capable of generating appreciably more antigen-specific mucosal antibody than DDS.
Fig. 4.

DDS- and dPEI-DNA induce antigen-specific antibody responses in the vaginal mucosa. Antigen-specific IgG (A) and IgA (B) from mucosal lavage were assessed (n = 6) after the final protein boost. Antibody results are expressed as group means (ng/ml + SEM). Statistical significance was assessed using Mann Whitney U test with *p ≤ 0.05 or **p ≤ 0.005.
3.5. A dPEI-DNA prime-protein boost regimen can protect against influenza challenge infection in mice
The generation of vaccine-mediated humoral and cellular immune responses is important for the protection against viral infections but a definitive test for any potential vaccine and delivery vehicle is protection against a live viral challenge. In the absence of a small animal model for HIV-1 infection, we used the dPEI-DNA and DDS-DNA complexed plasmid DNA (expressing an HA transgene) vaccine as delivery agents in an X-31 murine-adapted reassortment influenza virus infection-challenge model [13]. We first cloned the A/Aichi/2/1968(H3N2) haemagglutinin (HA) gene sequence, codon optimised for expression in mice, into the pmaxFP™-Red C destination vector to create a plasmid DNA vaccine. As multiple pulmonary protein vaccinations in mice can generate protective immune responses against influenza virus challenge, even with concentrations as low as 1 μg/100 μl dose (data not shown), we altered the vaccination regimen to incorporate a single protein HA boost. This regimen alteration would allow us to evaluate the contribution of the mucosal DNA priming by the two transfection reagents to any protection from infection with the single protein boost only serving to amplify any cation-DNA generated immune response. We found that for this DNA encoded transgene that the dPEI-DNA polyplex but not the DDS-DNA lipoplex was able to generate significant antigen-specific humoral responses against the HA protein (**p = 0.0037) (Fig. 5a). The dPEI-DNA also generated significant cellular responses against the HA protein when splenocytes were used in an IFN-γ ELISpot assay (*p = 0.03) (Fig. 5b). Nasal antibody lavage revealed the dPEI-DNA treatment regimen to significantly increase antigen-specific IgG (Fig. 5c) and IgA (Fig. 5d) compared to DDS-DNA (**p = 0.0087 and *p = 0.026 respectively) treatment group and negative controls (**p = 0.0043 and **p = 0.0037 respectively). Upon challenge of these cation-DNA primed and HA-protein boosted animals with a live virus, a significant reduction in infection was observed in dPEI-DNA vaccinated mice (**p = 0.0043) (Fig. 5f) with a significantly reduced morbidity as measured by weight loss (**p = 0.0022) observed during the challenge (Fig. 5e). The DDS-DNA priming strategy did not generate significant humoral or cellular responses compared to the negative control group but did result in significantly reduced infection-associated weight loss (*p = 0.0411) (Fig. 5e). However the infection associated weight loss for the DDS-DNA treatment group was less than that for the dPEI-DNA treatment group.
Fig. 5.
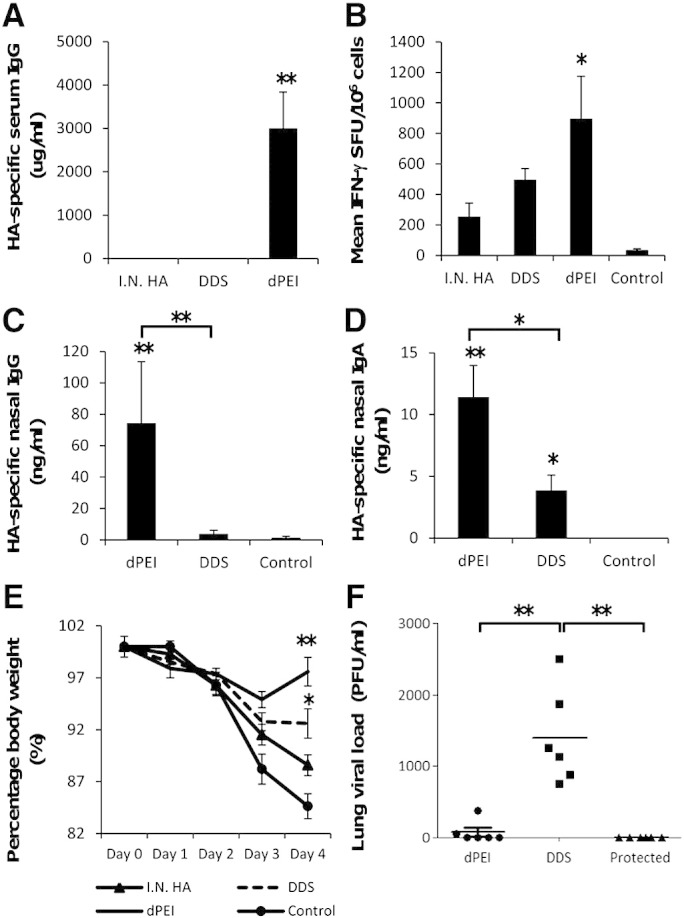
DDS- and dPEI-DNA complexes can protect against influenza infection challenge. Antigen-specific IgG antibody responses (n = 6 per group) were recorded one week after 3 cation-complexed DNA prime vaccinations (20 μg/50 μl volume) and a single recombinant HA protein boost vaccination (10 μg/50 μl). Antibody results are expressed as geometric means of the group (ng/ml + SEM) (A). Mean IFN-γ T cell responses from splenocyte cultures stimulated with whole antigen HA were assessed on day 4 post-influenza infection using a commercially available ELISPOT assay. Results represent the means of (n = 6) SFU/million splenocytes (B). Mice were infected with 5 HA units of Influenza X-31. Antigen-specific IgG (C) and IgA (D) from nasal lavage were performed. Body weights for the different vaccination groups are expressed as a percentage relative to day 0 (E). Influenza viral loads in lung samples were assessed and expressed as plaque forming units (PFU/ml) (F). Statistical significance was assessed using Mann Whitney U test with *p ≤ 0.05 or **p ≤ 0.005.
3.6. dPEI-DNA polyplexes can transfect human mucosal epithelium and traverse epithelial monolayers
To evaluate dPEI as a DNA delivery agent in people, we utilised a human derived stratified epithelial tissue model. The tissue contained a multi-layered, intact overlying epithelium (Fig. 6a) and stroma. Both, the epithelium and stroma had numerous MHC Class II positive APCs throughout (Fig. 6b). Incubation of the tissue explants with a dPEI-DNA polyplex consisting of dPEI and a GFP-encoding plasmid revealed that transfection occurred within the epithelial layer. Here both superficial epithelial cells and cells residing deeper in the epithelium can be transfected using a GFP expressing dPEI-DNA complex (Fig. 6c and d). To investigate how the polyplex formulations interacted with intact epithelium and to explain how they penetrated tissues after topical application, we utilised transwell systems. Here the transwell inserts were seeded with the human colorectal adenocarcinoma Caco-2 cell line, while the easy to transfect CHO cell line was added to the basolateral chamber to act as a transfection acceptor. Using this experimental system, we found that the transepithelial electrical resistance (TEER) changed dramatically after dPEI-DNA polyplex application to the Caco-2 containing apical transwell insert. This indicated a modulation of cellular tight junction integrity (Fig. 6h). The polyplexes were able to facilitate the transfection of apical chamber Caco-2 cells (Fig. 6e) as well as a CHO acceptor cell line at the highest concentration of polyplex delivered, when media from the basal transwell chamber were harvested and incubated with the CHO cell monolayer (Fig. 6f). Interestingly we show that the dynamics of transfection is dependent on the concentration of polyplex. As the concentration of polyplex increased (0.5 μg–2 μg DNA), the rate of apical Caco-2 transfection increased (Fig. 6i). However, at the highest concentration (5 μg DNA) tested, the polyplex ability to transfect the Caco-2 epithelium was reduced, while its ability to transfect the basolateral CHO cells was increased (Fig. 6j).
Fig. 6.
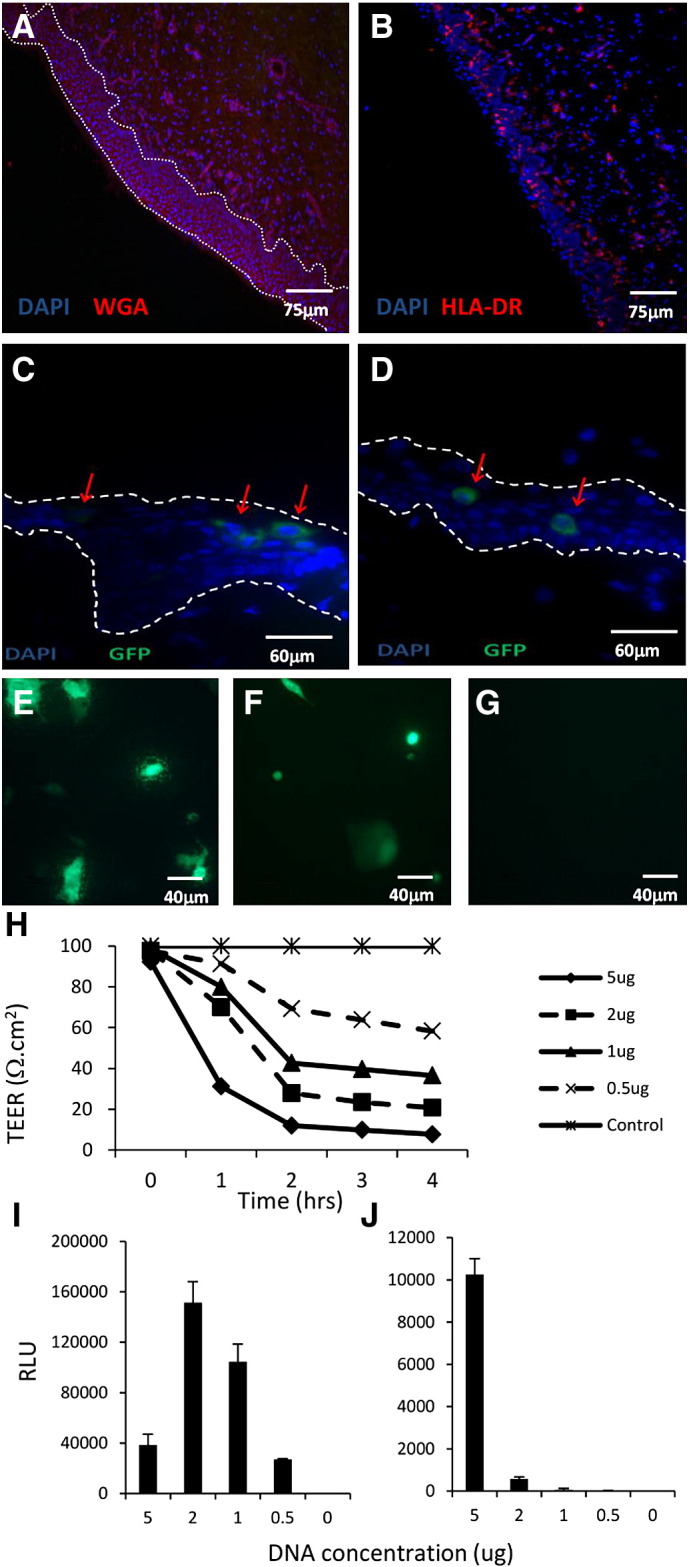
dPEI polyplexes transfect human mucosal epithelial cells and can facilitate transfection of stromal cells. Human penile glans tissue, cut into 3 mm × 3 mm explants was transfected with GFP expressing plasmid complexed to dPEI. Negative control tissue stained with DAPI and Wheat Germ Agglutinin (WGA) (A), MHC Class II (HLA-DR) expression (B) and GFP expression (C and D) was examined 48 h post transfection using a fluorescent microscope. Mucosal epithelium is indicated by the white dotted lines while GFP expressing cells (green) are indicated by red arrows. Transwell inserts containing apically cultured confluent monolayers of Caco-2 cells were transfected with dPEI-GFP polyplexes (E). After 4 h, the media in the basal chamber were removed and added to CHO cells to evaluate polyplex migration through mucosal epithelium (F). Negative control Caco-2 cells grown on transwell inserts (G). Transepithelial electrical resistance (TEER) was monitored for 4 h following application of 0–5 μg dPEI-GFP polyplexes (H). Luciferase expression from dPEI transfected cells is expressed as relative light units (RLU) for apical Caco-2 (I) and basolateral CHO (J) cell lines.
4. Discussion
Conventional vaccine strategies employing parenteral routes of administration work through the elicitation of antibody mediated responses. However, mucosally delivered vaccines offer the potential to increase vaccine efficacy through activation of not only systemic, but mucosal immune responses. The potential to establish protective immune responses against infectious agents exploiting mucosal surfaces at these portals of entry is thus an area of research receiving intense investigation. Currently there are only a few mucosal vaccines permitted for human use that target diseases such as rotavirus, influenza, polio, typhoid and cholera. However, only the cold adapted LAIV FluMist™ (MedImmune) and Nasovac (Institute of Experimental Medicine) are administered via the nasal route [14]. With the exception of the oral cholera vaccine (ORC-Vax®), all the other mucosal vaccines (including FluMist™) are live attenuated [14]. Despite attenuation, the use of live vaccines presents safety concerns, and for disease causative agents like HIV-1 will unlikely be a viable option due to the risk of reversion to virulence. Hence alternative vaccine delivery technologies, such as minimal plasmid based DNA vaccines, provide an attractive option to deliver vaccine antigens safely and easily. Despite early promising attempts to develop DNA vaccines in preclinical models, DNA based vaccines have not as yet been translated effectively to humans [15,16]. From a mucosal vaccine perspective, a major roadblock in the utility of DNA vaccines has been how to deliver them efficiently to mucosal surfaces, mainly because conventional DNA delivery strategies such as poly-l-lysine and PEI are highly toxic themselves or have suffered from low mucosal transfection rates [7,8,17]. However, over the past few years, newer transfection reagents have been generated, with reduced toxicity and increased transfection potential [8,10,18]. Here we trialled two cationic complexing reagents for their abilities to deliver a plasmid DNA encoding a model HIV CN54gp140 transgene and generate antigen-specific immune responses. We then tested the potential of these delivery agents with a second plasmid DNA construct encoding an influenza transgene for protective efficacy in a murine influenza infection-challenge model.
We compared DDS against a deacylated form of PEI for their respective abilities to prime immune responses after a topical mucosal application. DDS is a cationic lipid emulsion containing DOTAP, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and Squalene, previously shown to promote high transgene expression in the nasal passage in a luciferase reporter system [5]. The deacylated PEI is a 25 kDa PEI with its residual N-acyl moieties removed, providing it with greater in vitro and in vivo transfection potential [10]. Both complexing agents displayed reduced toxicity compared to the toxic N9 positive control in an MTT assay employing a simple columnar mucosal epithelial cell line Caco-2. Furthermore, DDS and dPEI both displayed reduced toxicity compared to 25 KDa bPEI, a common transfection reagent, suggesting that both these compounds have an improved potential for future investigations involving topical mucosal application.
DDS and dPEI plasmid DNA complexes were both able to induce systemic and mucosal humoral immune responses against gp140, as well as cellular responses in the peripheral blood and splenocyte compartments. Specific responses to the HIV CN54gp140 model antigen were generally greater and achieved significance more often for dPEI formulations than for DDS formulations. Specifically dPEI generated highly significant serum IgG, mucosal IgG and IgA antibody levels, as well as CD4 and CD8 T cell responses capable of producing IFN-γ, IL-2 and TNF-α in splenocyte cultures. On the other hand, while DDS generated significant serum IgG we observed elevated but not significantly increased IgG and IgA SFU's in splenocyte cultures and only CD4 T cells which secreted IFN-γ and IL-2 but not TNF-α. Priming with the DDS-DNA formulations generated CD8 T cells which secreted only IFN-γ. The majority of CD8 T cell responses were against the 1st peptide pool (Env 1), which contains the first 78 overlapping peptides of CN54gp140. This observation is in line with other findings which show limited vaccine-induced CD8+ T cell repertoires being generated primarily due to suppression by immunodominance mechanisms [19–22]. Certainly, within the 1st peptide pool, Env structures such as C1, V1/V2, C2, V3 and partial C3 regions are present, while in the 2nd peptide pool, the remainder of the C3 region, V4, C4, V5 and C5 exists. In the future it would be interesting to precisely map these immunodominant peptides to regions on CN54gp140. This would allow for the investigation into the possible amplification of alternative repertoires of functional T cells and if delivered as a component in a multi-construct formulation, might serve to amplify immune breadth and add further selective pressure on infecting virions, leading to more diverse and efficacious vaccine strategies. This is supported by work investigating the substitution of immunodominant epitopes in favour of weak, subdominant epitopes in a number of virus challenge models [19,23,24].
Numerous studies have shown that nasal immunisation can induce IgA and IgG antibodies as well as effector T cells in the genital tract [11,25,26]. Moreover, a number of studies have shown that mucosal vaccination has the capacity to generate long term CD8 T cell responses [27]. This is of particular interest for vaccines developed to combat sexually transmitted infections as the genital mucosa is known to be a poor site of immune induction. Within this current study we have demonstrated that a non-viral DNA strategy can facilitate strong and effective immune priming that is present in the genital tract due to mucosal immune linkage. Furthermore, it is in agreement with previous publications demonstrating that DNA vaccines administered to the respiratory tract can elicit effective cellular and humoral responses and be protective against viral infection [1,2,16]. The murine nasal mucosa has a surface area of 0.3 cm2 and consists of pseudostratified ciliated single columnar epithelium and in the lateral turbinates, M cells [28]. This makes the nasal passages an efficient antigen sampling portal which can be exploited for vaccine delivery. Here we show that polyplexes can modulate tight junction integrity within a mucosal columnar epithelium cell line. Furthermore, we show that polyplexes can facilitate the transfection of superficial epithelium, and at higher concentration, cells residing underneath the columnar epithelium. This is supported by our human mucosal explant model system, where we demonstrate that plasmid delivery using dPEI can transfect both superficial epithelium and cells deeper within the epithelium. This suggests that depending on the concentration of the delivered polyplexes, a range of cell types can be transfected when delivered via mucosal routes. It is likely that a combination of both free PEI in the polyplex formulation, a necessity for efficient gene transfer, and the intrinsic permeation enhancing properties of PEI may play a role in subepithelial transfections. In our human explant model we show that transfection can occur in areas likely to contain residing APCs such as Langerhans cells. Here no GFP positive cells were seen past the stratified epithelium. In the nasal mucosa, it is likely that ciliated epithelium and underlying stromal cells can be transfected. A recent study by Wegmann et al., 2012 demonstrated that intranasal vaccination with PEI-gp140 protein formulations resulted in up take by epithelial and microfold cells of the NALT. Furthermore they show that fluorescently labelled gp140 could be found associated with DCs in draining cervical lymph nodes 24 h post application [29]. Hence suggesting that both DNA and protein vaccines formulated with PEI can enhance vaccine penetration via the mucosal route. Our results indicate that dPEI is a superior plasmid DNA transfection reagent than DDS when administered topically via the pulmonary route and moreover, can elicit effective antigen-specific immune responses that protect mice in an influenza challenge model at mucosal portals of infection.
In conclusion we have conducted a head to head study using topically applied, pulmonary delivered dPEI or DDS complexing agents to enable efficient plasmid DNA mucosal vaccination and have shown that the resulting immune response differs quantitatively and qualitatively between the two complexing agents. Although both were protective in an influenza challenge model, dPEI was superior in terms of the magnitude of humoral and cellular responses. This has potential implications for future vaccine regimen design as we have shown that dPEI can be effectively utilised as a mucosally directed DNA priming strategy.
Acknowledgements
This work was funded in part by grants to RJS by the Bill & Melinda Gates Foundation and the Wellcome Trust, under the Grand Challenges in Global Health Initiative (grant number 37872) and by CUT'HIVAC under a European Commission FP7 Award (grant number 241904). We are also are grateful to the Foundation Dormeur for the provision of an equipment grant. Special thanks goes to FIT BIOTECH (Fin) for the provision of the GTU-GFP/Luc plasmid used in these studies, to Dr John Tregoning for the provision of the X31 virus, and to Roger Tatoud (UK HVC, Imperial College London) and Dietmar Katinger (Polymun Scientific, Aut) for the provision of plasmid and antigen. GENEART AG (Regensburg, Germany) owns a European Patent EP1240333B1 covering the complete clade C/B′ HIV-1 97CN54 coding sequence and codon optimised genes. The VRC 8400 CMV/R backbone vector used to construct the plasmid DNA has been developed by the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) (Bethesda, USA). The CN54 env plasmid was developed as part of the UK HIV Vaccine Consortium (www.ukhvc.org). The UK HVC is funded by an award from the Wellcome Trust for the provision of GMP vaccines, clinical trial support, and laboratory assays.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution–NonCommercial–No Derivative Works Licence, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2013.06.004.
Appendix A. Supplementary data
Supplementary materials.
References
- 1.Bivas-Benita M., Bar L., Gillard G.O., Kaufman D.R., Simmons N.L. Efficient generation of mucosal and systemic antigen-specific CD8 + T-cell responses following pulmonary DNA immunization. J. Virol. 2010;84:5764–5774. doi: 10.1128/JVI.02202-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bivas-Benita M., Gillard G.O., Bar L., White K.A., Webby R.J. Airway CD8(+) T cells induced by pulmonary DNA immunization mediate protective anti-viral immunity. Mucosal Immunol. 2013;6(1):156–166. doi: 10.1038/mi.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minigo G., Scholzen A., Tang C.K., Hanley J.C., Kalkanidis M. Poly-l-lysine-coated nanoparticles: a potent delivery system to enhance DNA vaccine efficacy. Vaccine. 2007;25:1316–1327. doi: 10.1016/j.vaccine.2006.09.086. [DOI] [PubMed] [Google Scholar]
- 4.Klavinskis L.S., Gao L., Barnfield C., Lehner T., Parker S. Mucosal immunization with DNA-liposome complexes. Vaccine. 1997;15:818–820. doi: 10.1016/s0264-410x(96)00278-2. [DOI] [PubMed] [Google Scholar]
- 5.Kim T.W., Chung H., Kwon I.C., Sung H.C., Shin B.C. Airway gene transfer using cationic emulsion as a mucosal gene carrier. J. Gene Med. 2005;7:749–758. doi: 10.1002/jgm.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim T.W., Kim Y.J., Chung H., Kwon I.C., Sung H.C. The role of non-ionic surfactants on cationic lipid mediated gene transfer. J. Control. Release. 2002;82:455–465. doi: 10.1016/s0168-3659(02)00138-4. [DOI] [PubMed] [Google Scholar]
- 7.Hunter A.C., Moghimi S.M. Cationic carriers of genetic material and cell death: a mitochondrial tale. Biochim. Biophys. Acta. 2010;1797:1203–1209. doi: 10.1016/j.bbabio.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 8.Beyerle A., Irmler M., Beckers J., Kissel T., Stoeger T. Toxicity pathway focused gene expression profiling of PEI-based polymers for pulmonary applications. Mol. Pharm. 2010;7:727–737. doi: 10.1021/mp900278x. [DOI] [PubMed] [Google Scholar]
- 9.Chen H. Recent advances in mucosal vaccine development. J. Control. Release. 2000;67:117–128. doi: 10.1016/s0168-3659(00)00199-1. [DOI] [PubMed] [Google Scholar]
- 10.Thomas M., Lu J.J., Ge Q., Zhang C., Chen J. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5679–5684. doi: 10.1073/pnas.0502067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mann J.F., Stieh D., Klein K., de Stegmann D.S., Cranage M.P. Transferrin conjugation confers mucosal molecular targeting to a model HIV-1 trimeric gp140 vaccine antigen. J. Control. Release. 2012;158:240–249. doi: 10.1016/j.jconrel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arias M.A., Van Roey G.A., Tregoning J.S., Moutaftsi M., Coler R.N. Glucopyranosyl Lipid Adjuvant (GLA), a Synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS One. 2012;7:e41144. doi: 10.1371/journal.pone.0041144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang S., Mozdzanowska K., Palladino G., Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J. Immunol. 1994;152:1653–1661. [PubMed] [Google Scholar]
- 14.Holmgren J., Svennerholm A.M. Vaccines against mucosal infections. Curr. Opin. Immunol. 2012;24:343–353. doi: 10.1016/j.coi.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Liu M.A. DNA vaccines: an historical perspective and view to the future. Immunol. Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- 16.Torrieri-Dramard L., Lambrecht B., Ferreira H.L., Van den Berg T., Klatzmann D. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol. Ther. 2011;19:602–611. doi: 10.1038/mt.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moghimi S.M., Symonds P., Murray J.C., Hunter A.C., Debska G. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol. Ther. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Thomas M., Lu J.J., Zhang C., Chen J., Klibanov A.M. Identification of novel superior polycationic vectors for gene delivery by high-throughput synthesis and screening of a combinatorial library. Pharm. Res. 2007;24:1564–1571. doi: 10.1007/s11095-007-9279-3. [DOI] [PubMed] [Google Scholar]
- 19.Riedl P., Wieland A., Lamberth K., Buus S., Lemonnier F. Elimination of immunodominant epitopes from multispecific DNA-based vaccines allows induction of CD8 T cells that have a striking antiviral potential. J. Immunol. 2009;183:370–380. doi: 10.4049/jimmunol.0900505. [DOI] [PubMed] [Google Scholar]
- 20.Wu L., Kong W.P., Nabel G.J. Enhanced breadth of CD4 T-cell immunity by DNA prime and adenovirus boost immunization to human immunodeficiency virus Env and Gag immunogens. J. Virol. 2005;79:8024–8031. doi: 10.1128/JVI.79.13.8024-8031.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J., Ewald B.A., Lynch D.M., Nanda A., Sumida S.M. Modulation of DNA vaccine-elicited CD8 + T-lymphocyte epitope immunodominance hierarchies. J. Virol. 2006;80:11991–11997. doi: 10.1128/JVI.01348-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yewdell J.W., Bennink J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 23.Wieland A., Riedl P., Reimann J., Schirmbeck R. Silencing an immunodominant epitope of hepatitis B surface antigen reveals an alternative repertoire of CD8 T cell epitopes of this viral antigen. Vaccine. 2009;28:114–119. doi: 10.1016/j.vaccine.2009.09.096. [DOI] [PubMed] [Google Scholar]
- 24.Im E.J., Hong J.P., Roshorm Y., Bridgeman A., Letourneau S. Protective efficacy of serially up-ranked subdominant CD8 + T cell epitopes against virus challenges. PLoS Pathog. 2011;7:e1002041. doi: 10.1371/journal.ppat.1002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Campo J., Lindqvist M., Cuello M., Backstrom M., Cabrerra O. Intranasal immunization with a proteoliposome-derived cochleate containing recombinant gD protein confers protective immunity against genital herpes in mice. Vaccine. 2010;28:1193–1200. doi: 10.1016/j.vaccine.2009.11.035. [DOI] [PubMed] [Google Scholar]
- 26.Marks E., Helgeby A., Andersson J.O., Schon K., Lycke N.Y. CD4(+) T-cell immunity in the female genital tract is critically dependent on local mucosal immunization. Eur. J. Immunol. 2011;41:2642–2653. doi: 10.1002/eji.201041297. [DOI] [PubMed] [Google Scholar]
- 27.Gallichan W.S., Rosenthal K.L. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J. Exp. Med. 1996;184:1879–1890. doi: 10.1084/jem.184.5.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim D.Y., Sato A., Fukuyama kS., Sagara H., Nagatake T. The airway antigen sampling system: respiratory M cells as an alternative gateway for inhaled antigens. J. Immunol. 2011;186:4253–4262. doi: 10.4049/jimmunol.0903794. [DOI] [PubMed] [Google Scholar]
- 29.Wegmann F., Gartlan K.H., Harandi A.M., Brinckmann S.A., Coccia M. Polyethyleneimine is a potent mucosal adjuvant for viral glycoprotein antigens. Nat. Biotechnol. 2012 doi: 10.1038/nbt.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials.