

Abstract
Two substituted oxines, nitroxoline (5) and 5-chloroquinolin-8-yl phenylcarbamate (22), were identified as hits in a high-throughput screen aimed at finding new anti-angiogenic agents. In a previous study, we have elucidated the molecular mechanism of antiproliferative activity of nitroxoline in endothelial cells, which comprises of a dual inhibition of type 2 human methionine aminopeptidase (MetAP2) and sirtuin 1 (SIRT1). Structure–activity relationship study (SAR) of nitroxoline offered many surprises where minor modifications yielded oxine derivatives with increased potency against human umbilical vein endothelial cells (HUVEC), but with entirely different as yet unknown mechanisms. For example, 5-nitrosoquinolin-8-ol (33) inhibited HUVEC growth with sub-micromolar IC50, but did not affect MetAP2 or MetAP1, and it only showed weak inhibition against SIRT1. Other sub-micromolar inhibitors were derivatives of 5-aminoquinolin-8-ol (34) and 8-sulfonamidoquinoline (32). A sulfamate derivative of nitroxoline (48) was found to be more potent than nitroxoline with the retention of activities against MetAP2 and SIRT1. The bioactivity of the second hit, micromolar HUVEC and MetAP2 inhibitor carbamate 22 was improved further with an SAR study culminating in carbamate 24 which is a nanomolar inhibitor of HUVEC and MetAP2.
Introduction
Angiogenesis has been implicated in tumor growth and metastasis as well as other human disorders such as age-related macular degeneration and rheumatoid arthritis. Inhibitors of angiogenesis are starting to emerge as a new type of therapeutic agents against cancer and age-related macular degeneration. The known inhibitors of angiogenesis currently used in the clinic and at various stages of preclinical and clinical development target different aspects of endothelial cell biology. While drugs such as bevacizumab1 and sunitinib2 act by blocking angiogenesis signaling cascade, other drug candidates including endostatin3 and meta-stat4 disrupt both the processes of extracellular matrix breakdown and endothelial cell migration. Another group of anti-angiogenic agents in clinical development, including cilengitide,5 ombrabulin,6 squalamine,7 and lodamin,8 directly inhibit the growth of endothelial cells, which has also proven to be a viable strategy for inhibiting angiogenesis.
One of the most potent classes of inhibitors of endothelial cell proliferation is the natural products fumagillin and ovalicin along with their synthetic derivatives like TNP-470.9 After disappointing results in phase II clinical trials, the late Folkman and colleagues successfully resurrected TNP-470 using a nanomedicinal approach and the new product named lodamin was demonstrated to be not only efficacious but also orally bioavailable. TNP-470 is a potent inhibitor of human umbilical vein endothelial cell (HUVEC) proliferation with an IC50 value below 1 nM. The principal molecular target of the fumagillin family of angiogenesis inhibitors was elucidated to be methionine aminopeptidase type 2 (MetAP2).10 The constitutively expressed isoform MetAP1 and the inducible isoform MetAP2 are both ribosomally-associated metalloproteases performing the vital function of cotranslational removal of initiator methionine from the growing polypeptides.11 Despite a significant overlap of substrates between the two isoforms, a specific set of substrates appear to exist that are processed solely by MetAP2.12 But why exactly endothelial cells are so uniquely sensitive to MetAP2 inhibition is still an unanswered question. Small-molecules like TNP-470 work by inducing transcriptional activation of p53, which in turn leads to the expression of p21 and the latter inhibits cyclinE·Cdk2 setting forth a cell cycle arrest in the late G1 phase.13 Besides the understanding of these final downstream events, the steps in between MetAP2 dysfunction and p53 activation are as yet unknown.
From a drug development perspective, one unwelcome feature of fumagillin analogs is their irreversible mode of inhibition, where the reactive spirocyclic epoxide covalently modifies the His-231 side-chain of MetAP2.14 We have thus actively pursued the discovery of new reversible inhibitors of MetAP2. Through modification of fumagillin, we identified an analog named fumarranol15 which was found to interact with MetAP2 reversibly. In a complementary approach, we performed high-through-put screen (HTS) of a 175 000-compound library (acquired from ASDI, Inc., Newark, DE) using MetAP2 as a target and identified two different chemotypes of oxines, 5-substituted-8-hydroxyquinoline (5, Chart 1) and 8-quinolinyl carbamate (22, Chart 1). Coincidentally, nitroxoline (5) was also a hit when the Johns Hopkins Drug Library (JHDL), comprising of drugs in clinical use, was screened for inhibition of HUVEC. Nitroxoline is prescribed to treat acute microbial urinary tract infections16 in several countries including the European Union. We recently published our findings on the unique mode of anti-angiogenic activity of nitroxoline and demonstrated its potential for application in cancer chemotherapy using mouse xenograft models of human breast and bladder cancers.17 We showed that unlike fumagillin and TNP-470, nitroxoline inhibits MetAP2 (IC50 = 55 nM) and sirtuin 1 (SIRT1, IC50= 20.2 μM) in a synergistic fashion to induce premature senescence in endothelial cells. Sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases and they play a crucial role in many cellular processes including gene silencing and DNA repair.18 Among the seven human sirtuins (SIRT1–7), it has been shown that SIRT1 plays a role in cellular senescence and angiogenesis19 and thus, SIRT1 inhibition leads to the suppression of angiogenesis.20
Chart 1.

Synthesis of oxine derivatives.
Oxines are historically known for their extraordinary metal chelating abilities. Notwithstanding a continued use in analytical applications and the most notable utilization in the manufacturing of organic light-emitting diodes (OLEDs),21 substituted oxines such as cloxyquin22 and clioquinol23 besides nitroxoline have also been in use as antimicrobial agents. The metal chelation property is thought to be the underlying cause for the bioactivity of cloxyquin and clioquinol, and it has been shown that nitroxoline shares that feature to manifest its antibacterial activity.24 Herein, we report the structure–activity relationship (SAR) studies of the two oxines we identified as hits in the HTS by obtaining and evaluating different analogs through either purchasing from commercial source or synthesizing in-house. We characterized each analog in HUVEC proliferation assay using [3H]-thymidine incorporation, and enzymatic assays with MetAP1 and MetAP2. In addition, we also determined the effects of a subset of oxine derivatives on MetAP and SIRT1 activities in endothelial cells by assessing the processing of N-terminal methionine of 14-3-3γ, a common substrate of both MetAP1 and MetAP2; and by measuring acetylation status of lysine-382 on p53, a known SIRT1 substrate. A selection of the potent inhibitors of HUVEC proliferation were screened against two other malignant cell lines, HeLa and Jurkat T, and a human primary cell line, human foreskin fibroblast (HFF), to evaluate any selectivity the compounds possessed. We were able to identify new oxine derivatives that exhibited improved inhibitory activity against endothelial cell proliferation. Furthermore, we made the surprising finding that subtle structural alterations can lead to dramatic changes in the mechanism of action of the resulting derivatives.
Results and discussion
Library screen and identification of substituted oxines as hits
A target-based HTS of a library of 175 000 compounds provided by ASDI, Inc. was performed to find novel inhibitors of MetAP2 using a coupled enzyme assay with Met-Pro-pNA as the substrate.25 Among 294 hits that exhibited at least 50% inhibition of MetAP2 activity at a final concentration of 10 μM, nitroxoline (5, Chart 1), 5-chloroquinolin-8-yl phenylcarbamate (22, Chart 1), and 5-chloro-8-allyloxyquinoline (27, Chart 1) stood out for their potency of inhibition (>90%). We validated the hits by measuring their IC50 values for MetAP2 and found them to be 55 nM and 5.26 μM for quinolines 5 and 22 respectively. The IC50 value for allyloxyquinoline 27 was found to be higher than 50 μM. In a HUVEC proliferation assay, the IC50 values for nitroxoline, 22 and 27 are 1.9 μM, 1.2 μM and >50 μM, respectively. Since, allyloxyquinoline 27 could not be validated in both the enzyme and cellular assays; it was not pursued further in ensuing SAR studies. Divalent manganese has been demonstrated to be the physiologically relevant metal cofactor of human MetAP2;26 however, this issue has not been settled in the case of human MetAP1. Cobalt(II) is by far the best metal ion activator of the human MetAP1 catalytic reaction in vitro, and hence we and others have been carrying out MetAP1 assays using Co2+ as the cofactor.27 Thus, we used 10 μM Mn2+ and Co2+ in the MetAP2 and MetAP1 assays, respectively. In the cases where an oxine derivative was found to be active against both the isoforms (e.g., 4, 18, and 34), a likelihood of the coupling enzyme inhibition in the MetAP assay was ruled out by performing an additional control experiment (see ESI†).
Synthesis of oxine derivatives
Substituted 8-hydroxyquinolines
Commercially available 8-hydroxyquinolines 1 through 11 were acquired and used as such except nitroxoline (5), which was of technical grade and hence was recrystallized before use (see ESI†). The 7-aminomethyl-8-hydroxyquinolines 12–19 were synthesized using Mannich condensation between an 8-hydroxyquinoline and an aldehyde by following the procedure of Chi et al.28 with some modifications (Chart 1, eqn 1).
Quinolin-8-yl carbamates
The carbamates 20–25 were prepared by a simple addition reaction of the commercially available isocyanates with 8-hydroxyquinolines (Chart 1, eqn 2).
8-Substituted quinolines
Quinolin-8-yl phosphate 26 was made by reacting cloxyquin (4) with diethyl chlorophosphate using a combination of triethylamine and DMAP as the base. The quinolines 27, 28, and 30 were synthesized by carrying out allylation of quinolin-8-ol (3, 4 or 9) under phase-transfer conditions (Chart 1, eqn 3). The phenoxide anion generated from quinolinol 4 was reacted with ethylene carbonate at 120 °C to force a nucleophilic ring opening to provide quinoline 29 in 78% yield (Chart 1, eqn 4).
Commercially available 8-aminoquinoline (31) was converted to 8-quinolinyl p-toluenesulfonamide (32) by treating the amine (31) with p-toluenesulfonyl chloride in dichloromethane using triethylamine as the base.
Modifications to the nitro group of nitroxoline
Nitrosoquinolinol 33 was prepared according to the reported procedure by treating 8-hydroxyquinoline 1 with nitrous acid.29 Aminoquinolines 34 and 45 were prepared by subjecting nitroquinolines 5 and 44, respectively, to catalytic hydrogenation.30
Initially we attempted monoalkylation of aminoquinoline 34 using 1.2 equivalents of an aldehyde as shown in Chart 1, eqn 5; but we obtained a mixture of mono and dialkylated products. Determined to press on with these examples, we added additional equivalents of aldehyde and drove the reaction to completion forming the dialkylated products. The dialkylamino-quinolines 35–37 were obtained by sodium triacetoxyborohydride mediated reductive alkylation of the dihydrochloride salt of aminoquinoline 34 with an appropriate aldehyde (Chart 1, eqn 5).31
The dihydrochloride salt of 5-aminoquinolin-8-ol (34) was dissolved in pyridine and treated with different acid chlorides to yield oxines 38–42 (Chart 1, eqn 6).32 Following a literature procedure33 quinaldine 2 and cloxyquin (4) were nitrated using a mixture of concentrated HNO3–H2SO4 to afford quinolinols 43 and 44, respectively.
8-Hydroxy derivatized nitroxolines
To probe the role of the 8-hydroxy group in nitroxoline, we synthesized ester and ether derivatives. Due to the presence of the strongly electron-with-drawing nitro group in the para position, we could only make pivalate ester 46, phosphate 47, and sulfamate 48, all of which were found to be stable. The esters were prepared by treating nitroxoline (5) with the corresponding acid chloride and DMAP/Et3N or DIEA (in the case of 48) in dichloromethane (Chart 1, eqn 7).
The 8-alkoxy derivatives were synthesized by reacting nitroxoline with either ethyl bromoacetate and potassium carbonate in the case of 49, or iodoacetamide and caesium carbonate in the case of 50; where, in both the cases the reaction mixture in DMSO was held at 60 °C for 14 h (Chart 1, eqn 7).
8-Aminoquinoline derivatives
Aminoquinolines 51–53 were obtained from NCI/DTP repository. Nitrosoquinoline 54 was prepared from sulfonamide 32, in the same manner as the synthesis of nitrosoquinoline 33. 5-Nitroquinolin-8-yl sulfonamide 55 was obtained by oxidizing the corresponding nitrosoquinoline 54 using concentrated nitric acid in glacial acetic acid.
Oxine derivatives: MetAP and HUVEC assays
In our initial campaign on the SAR of nitroxoline, we decided to replace the electron-withdrawing nitro group with other electron-donating or electron-withdrawing groups and measure their activities against HUVEC proliferation and the two isoforms of MetAP (see Table 1).
Table 1.
Oxine derivatives: MetAP and HUVEC assay results
| IC50,a μM |
|||||
|---|---|---|---|---|---|
| Entry | Substituents (when not specified, R# = H) | hMetAP1 | hMetAP2 | HUVEC | |
| 1 |
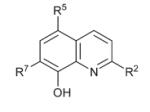
|
— | >15 | 2.03 ± 0.3 | 6.21 ± 1.1 |
| 2 | R2 = Me | >15 | >15 | 2.70 ± 0.61 | |
| 3 | R2 = CN | >15 | >15 | >50 | |
| 4 | R5 = Cl (Cloxyquin) | 12.9 ± 1.0 | 1.27 ± 0.6 | 1.41 ± 0.1 | |
| 5 | R5 = NO2 (Nitroxoline) | >15 | 0.055 ± 0.02 | 1.90 ± 0.3 | |
| 6 | R5 = SO3H | >15 | >15 | >50 | |
| 7 | R5 = Cl, R7 = I (Clioquinol) | >15 | 2.39 ± 0.31 | 2.42 ± 0.5 | |
| 8 | R5 = Cl, R7 = Br | >15 | >15 | 2.21 ± 0.6 | |
| 9 | R5, R7 = Cl | >15 | >15 | 3.64 ± 0.53 | |
| 10 | R5, R7 = Br | >15 | >15 | 2.53 ± 0.71 | |
| 11 | R5, R7 = I | >15 | >15 | 2.62 ± 0.08 | |
| 12 | R7 = CH2N(CH2CH2)2O | >50 | >15 | >20 | |
| 13 |
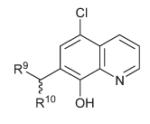
|
R9 = N(CH2CH2OH)2 | >50 | >15 | 10.73 ± 0.2 |
| 14 | R9 = N(CH2CH2)2CH2 | >50 | 7.68 ± 1.2 | 13.51 ± 1.4 | |
| 15 | R9 = N(CH2CH2)2O | >50 | 2.66 ± 0.2 | 5.57 ± 0.16 | |
| 16 | R9 = N(CH2CH2)2N–Me | >50 | 3.81 ± 0.74 | 9.56 ± 0.8 | |
| 17 | R9 = N(CH2CH2)2NPh(4-OMe) | >15 | >15 | 1.46 ± 0.33 | |
| 18 | R9 = N(CH2CH2)2O, R10 = Ph | 14.01 ± 0.8 | 0.33 ± 0.05 | 1.92 ± 0.6 | |
| 19 | R9 = NH-2-thiazolyl, R10 = 2-furanyl | >50 | 2.75 ± 0.4 | 2.53 ± 0.13 | |
| 20 |
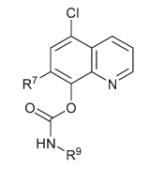
|
R9 = CH2CH2Cl | >15 | 0.365 ± 0.04 | 2.11 ± 0.25 |
| 21 | R7 = Cl, R9 = CH2CH2Cl | >15 | >15 | 2.62 ± 0.5 | |
| 22 | R9 = Ph | >15 | 5.26 ± 0.6 (b = 24%) | 1.21 ± 0.51 | |
| 23 | R9 = −(3,5-difluoro)Ph | >15 | >15 | 2.20 ± 0.1 | |
| 24 | R9 = −(3,5-dichloro)Ph | >15 | 0.024 ± 0.007 | 0.324 ± 0.06 | |
| 25 | R7 = Cl, R9 = −(4-fluoro)Ph | >15 | >15 | 3.58 ± 0.9 | |
| 26 |
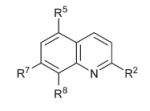
|
R5 = Cl, R8 = OPO(OEt)2 | >15 | >15 | >50 |
| 27 | R5 = Cl, R8 = OCH2CH=CH2 | >15 | >50 | >50 | |
| 28 | R5, R7 = Cl, R8 = OCH2CH=CH2 | >15 | >15 | 2.32 ± 0.23 | |
| 29 | R5 = Cl, R8 = OCH2CH2OH | >15 | >15 | 2.40 ± 0.3 | |
| 30 | R2 = CN, R8 = OCH2CH=CH2 | >15 | >15 | >50 | |
| 31 | R8 = NH2 | >15 | >15 | >50 | |
| 32 | R8 = NHSO2-(4-Me)Ph | 6.97 ± 0.25 (b = 21%) | >15 | 0.308 ± 0.04 | |
IC50 values are the average of at least three independent experiments consisting of triplicates and ± standard deviation is given. b= the bottom value in the dose–response curve, noted in parentheses only when it was above 15%.
Except for the cyanoquinoline 3 and quinoline-5-sulfonic acid 6, oxines 1 to 11 inhibited HUVEC proliferation with IC50 values in the single-digit micromolar range. Among those that inhibited HUVEC proliferation, only the known antimicrobials cloxyquin (4), nitroxoline (5), and clioquinol (7) affected MetAP2 activity in vitro. The lack of inhibition of either HUVEC or MetAPs by oxines 3 and 6 may be attributed to the poor coordination ability of the pyridine nitrogen. The strongly electron-withdrawing cyano group would decrease the electron density on the pyridine nitrogen in quinolinol 3 and the strongly acidic sulfonic acid in quinolinol 6, under our assay conditions, would lock-up that nitrogen in the form of a zwitterion. In contrast with quinolinol 3, quinolinol 2 carrying an electron-donating methyl group, did inhibit HUVEC with an IC50 of 2.7 μM.
Clioquinol (7) has been administered to Alzheimer’s disease (AD) patients in clinical trials34 and studies have also shown clioquinol to exhibit anticancer activity by several different mechanisms.35 That both cloxyquin and clioquinol have been used in the clinic raised the possibility that their paths to the clinic may be shortened through expedient clinical evaluation. The MetAP isoform-selectivity exhibited by cloxyquin, nitroxoline, and clioquinol (Table 1) is notable. Cloxyquin, nitroxoline, and clioquinol are known to chelate metal ions including Mn2+ and Co2+ ions with varying avidities but they only inhibited MetAP2 with Mn2+ as the cofactor (Table 1), and none of them inhibited either metalloform of MetAP1 (data not shown, IC50s: 12.9 μM for 4 with Mn2+; and >50 μM for 4 with Co2+, 5 and 7 with Mn2+ or Co2+). In fact nitroxoline also inhibited the cobalt (II) form of MetAP2 (data not shown, IC50 = 1.48 μM). Even though oxines 8–13 and 17 are equipped with the same LX type36 chelating motif like 4, 5, and 7, they did not inhibit either of the metalloforms of MetAP enzymes.
The anomalies pointed out here cannot be adequately explained by a mere metal chelation model. While the HUVEC inhibition by cloxyquin (4) and clioquinol (7) can be ascribed to their activity against MetAP2 as one of the cellular targets, how swapping the halogens in clioquinol for other halogens (8–11) leads to the loss of inhibition of MetAP enzymes but not the HUVEC activity remains to be elucidated.
Taking advantage of the Mannich reaction that is known to be facile with oxines,37 we prepared several analogs bearing substitutions at C-7 (12 to 19) and measured their IC50 values for HUVEC, MetAP1, and MetAP2 inhibition. Among the Mannich adducts tested, only 12 bearing a proton at C-5 turned out to be inactive; however, the remaining adducts carrying a 5-chloro substituent did show inhibition of HUVEC. Except 13 and 17 with polar diethanolamino and bulky phenylpiperazino groups respectively, all the other Mannich adducts (14, 15, 16, 18, and 19) inhibited MetAP2, where the adduct 18 was the most potent with an IC50 of 330 nM. It is noteworthy that none of the hydroxyquinolines (1–32) showed any significant inhibition of MetAP1, exhibiting isoform specificity. In the case of these Mannich adducts, MetAP2 inhibition can at least partly account for their antiproliferative activity in HUVEC. As a side note, the oxines 4, 14, and 15 were also found to reactivate latent HIV-1 without inducing global T-cell activation, and thus they might serve as leads in the development of a therapeutic strategy for the eradication of AIDS.38
Quinolinyl carbamate 22 was the other validated hit in our HTS and a limited SAR was performed with this structure as well. All the carbamates (20–25) turned out to be active against HUVEC proliferation. Three of the six carbamates showed little inhibitory activity towards either of the MetAPs (IC50 >15 μM). Two of them are derived from 5,7-dichloroquinolin-8-ol (21 and 25) and the third non-MetAP inhibitor is the 3,5-difluorophenyl-carbamate 23. Carbamate 24 is the most potent MetAP2 inhibitor (IC50 = 24 nM) among all the oxines we have tested in this study, and the enzyme activity is also reflected in its potency against HUVEC (IC50 = 324 nM).
In order to interrogate the role of the 8-hydroxy group in the oxine inhibitors, we prepared quinoline derivatives 26–30, and 32. Only the derivatives 28, 29, and 32 inhibited HUVEC and all were inactive in the MetAP2 assay. Sulfonamide 32 was actually quite potent at annihilating HUVEC (308 nM), however, it did not achieve that through inhibition of either MetAP1 or MetAP2 and the real target needs to be elucidated.
Nitroxoline analogs: MetAP and HUVEC assays
In the second SAR campaign (see Table 2) at probing the nitroxoline structure (5), we first made the nitroso derivative 33. When assayed for the ability to inhibit HUVEC proliferation, the nitrosoquinoline (33) was about five times more potent, and the potency improved with complete reduction of the nitro group (34). The potency for HUVEC inhibition was retained even after dialkylation (quinolines 35–37) of the amino group on 34. Alkylated amines 35–37 showed weak inhibition of MetAP1 and MetAP2, but the trend has been towards MetAP1 selectivity. In the case of 37, the selectivity totally switched over to MetAP1. With long-chain alkyl substitution, 36 did not affect either of the MetAP isoforms and hence it must be acting on a different target altogether to inhibit HUVEC proliferation.
Table 2.
Nitroxoline analogs: MetAP and HUVEC assays
| IC50,a μM |
|||||
|---|---|---|---|---|---|
| Entry | Substituents (when not specified, R# = H) | hMetAP1 | hMetAP2 | HUVEC | |
| 5 |
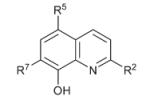
|
R5 = NO2 (Nitroxoline) | >15 | 0.055 ± 0.02 | 1.90 ± 0.3 |
| 33 | R5 = NO | >15 | >50 | 0.364 ± 0.2 | |
| 34 | R5 = NH2 | 8.92 ± 1.2 | 11.88b ± 1.1 (b = 26%) | 0.218 ± 0.04 | |
| 35 | R5 = N(CH2CH3)2 | 7.21 ± 1.3 (b = 29%) | 20.13b ± 2.2 (b = 25%) | 0.329 ± 0.17 | |
| 36 | R5 = N([CH2]5CH3)2 | >40 | >40 | 0.789 ± 0.21 | |
| 37 | R5 = N(CH2(4-OMe)Ph)2 | 1.46 ± 0.3 (b = 32%) | >40 | 0.551 ± 0.08 | |
| 38 | R5 = NHCOCH3 | >40 | >40 | 10.2 ± 1.1 (b = 28%) | |
| 39 | R5 = NHCO(CH2)4CH3 | >40 | >40 | 8.45 ± 0.91 | |
| 40 | R5 = NHCOPh | 6.45b ± 1.1 | >40 | 0.789 ± 0.11 | |
| 41 | R5 = NHSO2CH(Me)2 | >40 | >40 | 12.94 ± 0.13 | |
| 42 | R5 = NHCON(CH2CH2)2O | >40 | >40 | 1.88 ± 0.3 | |
| 43 | R2 = CH3, R5, R7 = NO2 | 17.31 ± 1.7 | >15 | 15.74 ± 1.3 | |
| 44 | R5 = Cl, R7 = NO2 | >15 | >15 | 7.73 ± 0.77 | |
| 45 | R5 = Cl, R7 = NH2 | >40 | >15 | 0.273 ± 0.07 | |
| 46 |
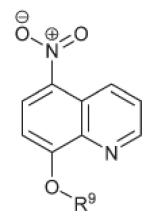
|
R9 = COC(Me)3 | >15 | 0.434 ± 0.1 | 1.6 ± 0.05 |
| 47 | R9 = PO(OPh)2 | >15 | 0.117b ± 0.07 | 2.01 ± 0.06 | |
| 48 | R9 = SO2N(Me)2 | >15 | 0.057 ± 0.015 | 0.77 ± 0.1 | |
| 49 | R9 = CH2CO2Et | >15 | >15 | 7.3 ± 0.81 | |
| 50 | R9 = CH2CONH2 | >15 | 1.057 ± 0.5 | 6.3 ± 1.4 | |
| 51 |
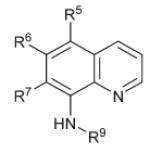
|
R5 = Cl | >40 | >15 | 0.721 ± 0.3 |
| 52 | R7 = Cl | >40 | >15 | 1.66 ± 0.06 | |
| 53 | R5 = Cl, R6 = OMe | >40 | >15 | >20 | |
| 54 | R5 = NO, R9 = SO2(4-Me)Ph | 10.51 ± 0.9 (b = 18%) | >15 | 0.204 ± 0.02 | |
| 55 | R5 = NO2, R9 = SO2(4-Me)Ph | 21.05b ± 1.8 (b = 19%) | >15 | 0.305 ± 0.07 | |
IC50 values are the average of at least three independent experiments with each consisting of triplicates and ± standard deviation is given.
The curve was non-sigmoidal. b = the bottom value in the dose-response curve, given in parentheses only when it was above 15%.
On the other hand, acylated compounds (38–42) derived from aminoquinoline 34, produced mixed results in HUVEC proliferation experiments. Only benzamide 40 showed an inhibition of HUVEC below 1 μM. Unlike nitroxoline, the derivatives 33–45 either lost their ability to inhibit the MetAP isoforms completely or became weak inhibitors. A second nitro group on the nucleus (43) or scrambling of the position of the nitro group (44) resulted in a loss of potency against HUVEC. Despite the amino group being in a scrambled position in relation to aminoquinoline 34, quinolinol 45 inhibited HUVEC proliferation with very high potency but in a manner that is independent of either of the MetAP isoforms as targets.
An often invoked role of the 8-hydroxy group in nitroxoline is its participation in chelation besides a well documented intramolecular hydrogen bonding with the pyridine nitrogen. We decided to fathom the importance of the 8-hydroxy group in nitroxoline using ester derivatives 46–48. All the three esters (46–48) retained the MetAP2 inhibitory activity of nitroxoline, with sulfamate ester 48 displaying improved potency against HUVEC. The alkylation of nitroxoline (49, 50) resulted in a substantial loss of potency against HUVEC. Except the aminoquinoline 53, all the quinolines 51–55 inhibited HUVEC proliferation. Interestingly, they did not affect the activity of MetAP2.
Assessment of cell type selectivity of oxine analogs
A subset of compounds representing different chemotypes of oxines with high potency for inhibition of HUVEC proliferation were further examined in proliferation assays using two malignant cell lines, HeLa and Jurkat T cells. We deemed it necessary to assess these compounds for selectivity against other human primary cell lines and we chose HFF for this purpose. Since thymidine uptake in HFF is known to be very feeble, we performed cell-viability assays on HUVEC and HFF side-by-side, using calcein-AM (see ESI†). Of the derivatives tested, only the aminoquinoline 34 showed a good selectivity profile; it was nearly 30 times more selective for HUVEC inhibition than HFF, and about 8 times more potent against HUVEC than HeLa and Jurkat T cell lines (Table 3). The lack of selectivity in other cases may be indicative of a target or process that is very fundamental to the four cell types that we examined.
Table 3.
Evaluation of a subset of oxine derivatives: selectivity against other cell lines and in vivo (HUVEC) activity on MetAPs and SIRT1
| IC50,a μM; 3H-T Uptake |
IC50,a μM; Calcein-AM |
IC50,a μM; hMetAPs |
Immunoblotb |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | HUVECc | HeLa | Jurkat-T | HUVEC | HFF | hMetAP1c | hMetAP2c | Ac-p53 | Met-14-3-3γ |
| 17 | 1.46 | ND | ND | ND | ND | >15 | >15 | + | − |
| 18 | 1.92 | ND | ND | 3.55 ± 0.9 | 3.66 ± 0.7 (b = 18%) | 14 | 0.33 | ND | ND |
| 24 | 0.324 | ND | ND | 0.746 ± 0.19 | 2.69 ± 0.5 (b = 17%) | >15 | 0.024 | ND | ND |
| 32 | 0.308 | 1.12 ± 0.2 | 0.79 ± 0.08 | 1.47 ± 0.2 | 0.909 ± 0.12 (b = 21%) | 6.97 | >15 | − | − |
| 5 | 1.90 | 3.97 ± 0.9 | 1.58 ± 0.3 | 5.13 ± 1.2 | 3.69 ± 1.0 (b = 21%) | >15 | 0.055 | ++ | + |
| 33 | 0.364 | 0.64 ± 0.2 | 0.36 ± 0.07 | 3.72 ± 0.8 | 9.08 ± 1.2 (b = 17%) | >15 | >50 | + | − |
| 34 | 0.218 | 2.36 ± 0.5 | 1.28 ± 0.3 | 1.84d ± 0.15 (b = 19%) | 37.3 ± 2.1 (b = 38%) | 8.92 | 11.88d | − | − |
| 40 | 0.789 | ND | ND | ND | ND | 6.45d | >40 | +++ | + |
| 45 | 0.273 | ND | ND | ND | ND | >40 | >15 | − | − |
| 46 | 1.6 | ND | ND | ND | ND | >15 | 0.434 | ++ | + |
| 47 | 2.01 | ND | ND | ND | ND | >15 | 0.117 | ++ | + |
| 48 | 0.77 | 2.41 ± 0.8 | 1.73 ± 0.2 | 4.21 ± 1.4 | 2.26 ± 0.3 (b = 23%) | >15 | 0.057 | +++ | + |
| 51 | 0.721 | ND | ND | 3.51 ± 1.1 | 4.77 ± 1.0 (b = 21%) | >40 | >15 | − | − |
| 54 | 0.204 | 0.59 ± 0.1 | 0.45 ± 0.05 | 1.32 ± 0.2 | 0.782 ± 0.13 (b = 22%) | 10.51 | >15 | − | + |
| 55 | 0.305 | 1.03 ± 0.2 | 0.82 ± 0.15 | 1.46 ± 0.21 | 0.783 ± 0.11 (b = 21%) | 21.05d | >15 | − | − |
Note: IC50 values are the average of at least three independent experiments, each one consisting of triplicates, and ± standard deviation is given.
Immunoblotting of compound treated HUVEC lysates (see Fig. 1): The inducers of p53 acetylation are grouped into four categories (−, +, ++, and +++) based on the immunoblot signals of negative control (DMSO, 1.0) and the positive control (nitroxoline at 5 μM, 11.6), thus −, <2.0; +, 2.0–7.0; ++, 7.1–11.6; +++, >11.6. In the case of Met-14-3-3γ the compounds are grouped into two categories (− and +) based on the immunoblot signals relative to the control (DMSO, 1.0), hence, −, >1.5; and +, ≥1.5.
Dose-response curve was non-sigmoidal; b = bottom value of the curve.
Effect of substituted oxines on MetAP and SIRT1 activities in HUVEC
As we have demonstrated earlier, nitroxoline inhibits HUVEC by a concurrent inhibition of two targets: MetAP2 and sirtuins (SIRT1 and SIRT2). Furthermore, inhibition of either of these two enzymes alone is sufficient to arrest the growth of HUVEC. To evaluate the activity of a selected subset of substituted oxines in a cellular context, we treated HUVEC with different oxines at the same dose (5 μM) at which nitroxoline elicited strong signals on the immunoblot. And in the case of nitroquinoline 48, which is more potent than nitroxoline, the cells were treated with a range of concentrations (0.5–10 μM) of the compound. After a 24 h treatment, the levels of Met-14-3-3γ (the marker for MetAP inhibition), 14-3-3γ, Ac-p53 (the marker for suppression of SIRT1 function), and total p53 in the cells were probed and visualized using immunoblotting (Fig. 1).
Fig. 1.

Effect of substituted oxines on SIRT1 and MetAP activities in HUVEC. A representative immunoblot is shown and the relative pixel densities (Ac-p53 band versus p53 and Met-14-3-3γ versus 14-3-3γ band in every lane) were calculated using ImageJ software. The signals are being compared to the control (relative density of DMSO = 1.0).
When compared to nitroxoline, nitrosoquinoline 33 at the same concentration (5 μM) had very little effect on the acetylation status of p53, and neither did it exert any effect on MetAP activity in cells (Fig. 1). The latter result is in agreement with the enzyme assay where we had seen insignificant inhibition of MetAP1 or MetAP2 by 33 at concentrations up to 15 or 50 μM, respectively. Hence, we surmise this nitroso derivative is distinct from nitroxoline in its mechanism of action, interacting with as yet unidentified target(s) in HUVEC. The esters of nitroxoline 46–48 all had retained high inhibitory potency against MetAP2 in vitro, but are more potent HUVEC inhibitors than nitroxoline, which was reaffirmed by the immunoblot analyses. With sulfamate ester 48, we observed a dose-dependent effect on the level of Ac-p53. It is clear from the immunoblot that the signal for Ac-p53 in cells treated with 2 μM sulfamate 48 is comparable to that obtained from cells treated with 5 μM nitroxoline and perhaps this also explains why sulfamate 48 is superior to nitroxoline at inhibiting HUVEC proliferation. Similarly to nitroxoline, a biphasic mode of sirtuin inhibition was also seen with sulfamate 48 with the maximal inhibition peaking at around 2 μM (for nitroxoline it does at 5 μM). Mannich adduct 17 at 10 μM did increase the level of Ac-p53 when compared to the DMSO treated cells. Although 17 is a weak inhibitor of SIRT1, it is interesting because it has a novel structure with elaborate appendage on the 7th position of quinolinol and thus it is amenable to further structural refinements. Benzamidoquinolinol 40 appears to be a good inhibitor of SIRT1 and MetAP in the cell. Curiously, in the enzyme assays it did not inhibit MetAP2 (IC50 >40 μM), but did inhibit MetAP1 moderately (6.45 μM). Benzamide 40 inhibited HUVEC proliferation with an IC50 of 789 nM, and this compound deserves further study. Unlike benzamide 40, the nitrosoquinoline 54 whose IC50 for HUVEC proliferation was 204 nM, did not much alter the levels of Ac-p53 or Met-14-3-3γ, suggesting that its inhibition of endothelial cell proliferation is independent of SIRT1 and MetAP enzymes. Up to 10 μM of the substituted oxines 32, 34, 45, 51, and 55 had no effect on the levels of Ac-p53 or Met-14-3-3γ (data not shown), and the latter is also in accordance with the MetAP enzyme inhibition data. Due to the cytotoxicity of carbamate 24, not enough cells were left even after treatment with only 0.5 μM of the compound for 24 h (this concentration is a log unit lower than the concentration we set out to use to compare against cellular activity of nitroxoline), and thus we did not attempt further experiments with carbamate 24.
Metal complexation studies
In order to gain some insight into the metal complexation properties, we recorded UV–vis spectra of selected oxine derivatives (5, 24, 33, 40, 48, and 54) at 10 μM in MetAP assay buffer in the absence and presence of Co2+ and Mn2+ ions (see ESI†). The choice of metal ion concentration and the buffer was very deliberate on our part to at least emulate the enzyme assay conditions. Carbamate 24 exhibited one absorption maximum at 393 nm, and this peak neither shifted nor a new peak emerged upon incubation with Mn2+ or Co2+ ions; suggesting a lack of complexation. This result is in full agreement with an earlier report where a carbamate similar to 24 used as a prochelator (targeting acetylcholine esterase and monoamine oxidase; a project on AD therapy) was demonstrated to have very little affinity for Fe2+, Cu2+, or Zn2+ ions.39 Carbamate 24 happens to be the strongest inhibitor of MetAP2 (24 nM) in this SAR study and it is obvious now that metal ion sequestration by chelation is not the modus operandi at least in this case.
Nitroxoline is known to form a chelate with Mn2+ (21 μM 5 + 10 mM MnCl2 was used in this study; bathochromic and hypsochromic shifts were seen, λmax = 448 nm shifted to 430 nm),24 and we also observed a measurable shift even at 10 μM (both 5 and MnCl2). However, under the same conditions CoCl2 caused a huge hypochromicity coupled with a bathochromic shift (450 nm to 435 nm). Interestingly, nitroxoline inhibited both Mn2+- and Co2+-metalloforms of MetAP2 (0.055 and 1.48 μM, respectively), but inhibited neither metalloforms of MetAP1. Sulfamate 48 also registered similar shifts as nitroxoline did, except that 48 exhibited two absorption maxima (λmax = 319, 448 nm) and both of them were changed in the presence of Mn2+ and Co2+. Although, we cannot judge the stabilities of these complexes with sulfamate 48, we can confirm that it does form chelates with Mn2+ and Co2+. Interestingly nitrosoquinoline 33, showed very little affinity for Mn2+, because the spectral traces of 33 with or without Mn2+ completely overlapped. Perhaps this provides a clue as to why nitroxoline can inhibit MetAP2 and nitroso derivative 33 cannot. On the other hand, Co2+ did cause both a bathochromic and a hyperchromic shift in the UV–vis spectrum of 33. However, the ability of oxine 33 to chelate Co2+ has not translated into inhibition of either isozyme of MetAP. Benzamide 40 on the other hand exhibited signs of complex formation (hypsochromic shifts) with both Mn2+ and Co2+, but it only inhibited MetAP1 moderately. Addition of Mn2+ or Co2+ chloride to nitroso derivative 54 gave rise to huge hypsochromic shifts suggesting the formation of chelates, but this has not made sulfonamides 54 or 55 into strong inhibitors of MetAPs. The only conclusion we can draw from all these results is that metal chelation or lack thereof is not a dependable parameter to predict or adequately explain MetAP or cellular activities of the oxine derivatives.
Implications of the SAR
There are ongoing clinical trials on clioquinol (7) for relapsed or refractory hematological malignancies, eczema, and Parkinson’s disease. Our finding that clioquinol is anti-angiogenic through inhibition of MetAP2 could prompt a close scrutiny of clioquinol’s anti-angiogenic effects.
Mannich adducts (12–19) in general inhibited HUVEC proliferation through specifically affecting MetAP2 activity. Mannich adduct 18 was the most potent inhibitor while maintaining high potency against MetAP2, and hence this should serve as a starting point for further SAR. An outlier among the Mannich adducts, 17, did not inhibit MetAPs and weakly inhibited SIRT1. Further studies are needed to reveal the molecular mechanism of 17 in HUVEC. The second hit in our HTS, carbamate 22 was improved upon by SAR to yield highly cytotoxic 24. Given the number of chlorine atoms on the structure and its indiscriminate cytotoxicity, carbamate 24 does not appear to be an attractive candidate for further development.
In the second SAR campaign at probing the nitroxoline structure (5), a minor modification from nitro to nitroso (33) resulted in five-fold enhancement in the potency against HUVEC proliferation, but MetAP2 inhibitory activity was abolished. As we have demonstrated earlier, nitroxoline inhibits HUVEC by concurrent inhibition of two targets: MetAP2 and sirtuins (SIRT1 and SIRT2). Furthermore, inhibition of either of these two enzymes alone is sufficient to arrest the growth of HUVEC. Nitroso compound 33 showed a weak inhibition of SIRT1 and no inhibition of MetAP in vivo (HUVEC). Thus, nitro to nitroso functional group conversion (5 → 33); which is, as such a small structural change, led to a complete change in the mechanism of action.
In an earlier study, Strobl et al. showed that nitrosoquinoline 33 inhibits the proliferation of MCF-7 human breast cancer cell line through induction of differentiation and apoptosis;40 importantly, the cellular activity of 33 was attributed to the formation of reactive oxygen species (ROS). However, much higher concentrations of 33 (10 μM) were required to observe ROS response in comparison to its IC50 for HUVEC inhibition (364 nM). We also performed dihydrodichlorofluorescein diacetate (DCF-DA) assay in HUVEC treated with 10 μM 33, but failed to observe any significant increase in ROS levels (data not shown). We surmise that nitrosoquinoline 33 acts through a different mechanism to inhibit HUVEC proliferation. When we continued along the reduction path all the way to aminoquinoline 34 (i.e. 5 → 33 → 34), it became even more potent against HUVEC proliferation. Although the aminoquinoline 34 regained weak activity against MetAPs in vitro, it did not affect MetAP activity in HUVEC or SIRT1 (data not shown). The fact that the HUVEC inhibitory activity was not significantly sacrificed by the long-chain alkyl substitution on amine 34 offers a clue for the design of molecular probes to facilitate target identification pertaining to this series of compounds. Among the acylated analogs of aminoquinoline 34, most were inactive towards MetAPs or HUVEC proliferation, except the benzamide 40 and urea 42. The most interesting compound in this set is 40 which inhibited SIRT1 slightly better than nitroxoline and also caused N-terminal methionine retention on 14-3-3γ due to the inhibition of MetAP1. Despite a modest in vitro MetAP1 inhibition, benzamide 40 showed a two-fold higher potency against HUVEC proliferation than nitroxoline. Since, MetAP1 and SIRT1 activities do not appear to be synergistic (Zhang, F. unpublished results: MetAP1 knockdown in HUVEC in conjunction with a SIRT1 inhibitor, EX-527 treatment did not increase EX-527’s IC50), benzamide 40 is likely to be acting on a separate target besides SIRT1.
Based on structural intuition, we were expecting that derivatization of 8-hydroxy group of nitroxoline to its ester would lead to inactive analogs due to altered metal ion biding properties. On the contrary, all the three nitroxoline esters (46–48) retained the activity against HUVEC and MetAP2. Sulfamate 48 was about twice as potent as nitroxoline at HUVEC inhibition and its IC50 for MetAP2 inhibition was nearly identical to that of nitroxoline. The UV–vis studies also revealed that sulfamate 48 can chelate both Mn2+ and Co2+. Sulfamate 48 was very effective in blocking SIRT1 and MetAP2 activities in endothelial cells, and in fact 48 was much more potent at inhibiting SIRT1. Fortuitously, this SAR establishes that the only readily amenable functional group in nitroxoline can be utilized for derivatization without being detrimental to nitroxoline’s bioactivity.
Another structural variation where the 8-hydroxy group in the nitroxoline structure was replaced with an amino group (54, 55) yielded a different type of endothelial cell inhibitors. Quinoline 55 potently inhibited HUVEC proliferation without affecting MetAPs or SIRT1. Further SAR and target identification studies are warranted to demystify the mechanism of action of these 8-aminoquinoline derivatives. To recapitulate the SAR of nitroxoline, we have uncovered three mechanistically divergent chemotypes: (1) 5-aminoquinolinols (e.g.34 and 35); (2) 5-amidoquinolinols (e.g.40); and (3) 5-nitro-8-amidoquinolines (e.g.55).
Conclusions
In this SAR study of nitroxoline, we found that incremental functional group alterations can lead to dramatic changes in the mechanism of action of the analogs. As summarized in Fig. 2, conversion from 5 to 48 led to preservation and bolstering of the existing enzymatic and cellular activities of nitroxoline. Simply removing a single oxygen atom from the structure, that is transformation of 5 to 33, completely abolished the MetAP2 inhibitory activity and decreased activity towards SIRT1 as well. Reduction of the nitro group to an amino group (34) gave rise to a mechanistically new inhibitor of endothelial cells. Amazingly, further acylation of the amino group in 34 (40), restored most of the SIRT1 and MetAP inhibitory activity. If the 8-hydroxy group of 5 was transformed into sulfonamido derivative 55, all the bioactivities against SIRT1 and MetAPs vanished, and yet the antiproliferative activity against HUVEC increased. Interestingly, a number of analogs were uncovered with enhanced potency against HUVEC proliferation without affecting MetAP2 and SIRT1, suggesting that a rather fragment sized (in a fragment-based drug discovery approach, a typical fragment has an MW ≤250; nitroxoline, MW = 190) nitroxoline-based pharmacophore transmutes into a putative ligand for other distinct targets upon structural modifications. This SAR phenomenon may be attributable to the small sizes of nitroxoline and the analogs. Nitroxoline contains only 14 heavy atoms while the analogs except sulfonamide 55 possess no more than 20 atoms. As previously suggested by Reynolds et al.,41 this is the linear range for the ‘maximal affinity’ whereby each heavy atom can make a high impact. Thus, even minor manipulations of the functional groups could have a huge impact on the overall ligand surface characteristics and target specificity. In addition to identifying more potent but mechanistically distinct inhibitors of endothelial cell proliferation as novel leads for the development of anti-angiogenic agents, the many surprises we encountered in the medicinal chemistry of nitroxoline serves as a reminder that while dealing with the SAR of small bioactive molecules, minor changes in structure may lead to major changes in bioactivities and mechanisms of action.
Fig. 2.

Nitroxoline, summary of SAR.
While each one of the three chemotypes exemplified by 34, 40 and 55 will be the subject of future studies, a much improved version of nitroxoline, sulfamate 48 is a prime candidate for in vivo angiogenesis and xenograft studies in mice.
Experimental section
General
Unless stated otherwise, all non-aqueous reactions were carried out under ambient atmosphere in oven-dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (rt) is noted as 25 °C. All solvents were of reagent grade purchased from Fisher Scientific or VWR and used as received. Commercially available starting materials and reagents were purchased from Acros, Aldrich, or TCI America and were used as received, unless stated otherwise.
Analytical thin layer chromatography (TLC) was performed using Analtech Uniplates (silica gel HLF, W/UV254, 250 μm). Visualization was achieved using 254 nm UV light or additionally by staining with iodine, or ceric ammonium molybdate stain. The retention factor is reported as follows: Rf (eluent system). Crude products were purified by air-flashed column chromatography over silica gel (0.06–0.2 mm, 60 Å, from Acros) using indicated eluents. Melting points were recorded on a Mel-Temp II apparatus and are uncorrected. NMR data were collected on a Varian Unity-400 (400 MHz 1H, 100.6 MHz 13C) or Bruker-Spectrospin 400 MHz spectrometers. Chemical shifts are reported in ppm (δ) with the solvent resonance or 0.1% tetramethylsilane contained in the deuterated solvent as the internal reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet, br = broad, app = apparent, exch = exchangeable), coupling constants (J, reported in Hertz, Hz), and number of protons. Low resolution mass spectra were acquired on a Thermo-Finnigan MAT, LCQ Classic ESI-mass spectrometer or Voyager DE-STR, MALDI-TOF (Applied Biosystems) instruments. The MALDI-samples were prepared by mixing the droplets of the sample solutions in chloroform or methanol and 2,5-dihydroxybenzoic acid solution in acetone, where the latter served as the matrix. Data are reported in the form m/z (molecular ion).
When necessary, products were purified using a JASCO HPLC fitted with a semi-preparative VyDAC C18 column (7 micron, 10 × 250 mm). A mixture of water, acetonitrile, and 2-propanol was used as a mobile phase while eluting at a flow rate of 3 mL min−1. Samples were analyzed for purity on a JASCO HPLC equipped with a Rainin Microsorb-MV C18 column (5 μm, 4.6 × 250 mm). The mobile phase set at a flow rate of 0.8 mL min−1 was programmed for a gradient elution starting from a 4 : 4 : 2 mixture of H2O–MeCN–MeOH to a final less polar ratio of 0 : 9 : 1 over six minutes, maintained at that ratio for three more minutes, and in the next 0.2 min the ratio was reverted back to 4 : 4 : 2 and the column was equilibrated over 0.8 min, making each run a total of 10 min long. Purity of the final compounds was determined to be >95%, using a 10 μL injection (approximately 1 mM in acetonitrile, most of the time from a DMSO stock solution) with quantitation by area under the curve (AUC) at 256 and 277 nm (JASCO Diode Array Detector). The retention time (tR) is reported in minutes with the AUC given in parentheses.
General procedure for the synthesis of 7-(2-aminomethyl)-8-hydroxyquinolines
The Mannich bases 12 through 19 were prepared by modifying a known procedure.26 To a solution of 8-hydroxy-quinoline (1 mmol) in absolute ethanol (15 mL), triethylamine (1.2 mmol, 170 μL) and paraformaldehyde (1.2 mmol, 36.2 mg) were added and the reaction mixture was refluxed for 12 h and cooled to room temperature. The reaction mixture was filtered and the filtrate was concentrated. Recrystallization of the residue from 1 : 1 EtOH–H2O was the way of purification unless stated otherwise.
5-Chloro-7-((4-(4-methoxyphenyl)piperazin-1-yl)methyl)-quinolin-8-ol (17)
Yield: 244 mg (91%). Rf: 0.26 (7 : 3 hexanes–EtOAc); mp: 129 °C; tR: 6.347 min (98%); 1H-NMR (400 MHz, CDCl3): δ 8.88 (dd, J = 3.7, 1.2 Hz, 1H, H-2), 8.51 (dd, J = 8.3, 1.4 Hz, 1H, H-4), 7.56 (dd, J = 8.3, 3.7 Hz, 1H, H-3), 7.38 (s, 1H, H-6), 6.91 (dd, J = 9.2, 1.6 Hz, 2H, Ph), 6.85 (dd, J = 9.2, 1.6 Hz, 2H, Ph), 3.92 (s, 2H, CH2-7), 3.77 (s, 3H, OMe), 3.19 (app t, J = 4.8 Hz, 4H, piperazine), 2.82 (app t, J = 4.8 Hz, 4H, piperazine). 13C-NMR (100.6 MHz, CDCl3): δ 154.32, 149.97, 146.72, 141.87, 139.28, 132.19, 128.23, 126.54, 125.56, 122.07, 121.14, 115.20, 115.51, 56.11, 53. 45, 52.34, 50.03. MALDI-TOF: m/z 384 (M + H)+, 406 (M + Na)+.
General procedure for the synthesis of 7-(2-amino-2-arylmethyl)-8-hydroxy-quinolines
To a solution of 5-chloro-8-hydroxyquinoline (1 mmol, 180 mg) in absolute ethanol (15 mL), triethylamine (1 mmol, 140 μL) and aromatic aldehyde (1 mmol) were added and the reaction mixture was stirred at room temperature for 2 days. The reaction mixture was concentrated and the residue was recrystallized from 1 : 1 EtOH–H2O.
5-Chloro-7-(morpholino(phenyl)methyl)quinolin-8-ol (18)
42 Yield: 315 mg (89%). Rf: 0.35 (7 : 3 hexanes–EtOAc). mp: 89 °C. tR: 6.080 min (97%). 1H-NMR (400 MHz, acetone-d6): δ 8.91 (pair of dd’s, J = 3.4, 1.2 Hz, 1H, H-2), 8.53 (pair of dd’s, J = 6.5, 1.4 Hz, 1H, H-4), 7.71 (pair of dd’s, J = 6.5, 4.4 Hz, 1H, H-3), 7.61 (pair of d’s, J = 8.0 Hz, 2H, 2,6-Ph), 7.27 (pair of t’s, J = 8.0 Hz, 2H, 3,5-Ph), 7.15 (d, 1H, 4-Ph), 3.71 (br t, J = 4.8 Hz, 2H, 2,6-morpholine), 2,88 (br s, 3H, 2,6-morpholine, OH), 2.85 (br s, 1H, −N–CH(Ph)-), 2.47 (br s, 4H, 3,5-morpholine). 13C-NMR (100 MHz, acetone-d6): δ 153.35, 150.43, 149.97, 149.87, 142.38, 139.99, 133.73, 133.54 (C-4), 129.56 & 128.97 (3,5-Ph), 128.54 & 128.20 (2,6-Ph), 127.26 (4-Ph), 125.92, 125.42, 124.00 (C-3), 123.69 (C-2), 120.86, 111.28, 69.09 & 67.55 (2,6-morpholine), 53.51 (3,5-morpholine). ESI-MS: m/z 355 (M + H)+, 377 (M + Na)+.
5-Chloro-7-(furan-2-yl(thiazole-2-ylamino)methyl)quinolin-8-ol (19)
42 Yield: 258 mg (72%). Rf: 0.32 (7 : 3 hexanes–EtOAc). mp: 126 °C. tR: 6.147 min (97%). 1H-NMR (400 MHz, CDCl3): δ 8.85 (dd, J = 4.0, 1.6 Hz, 1H, H-2), 8.51 (dd, J = 8.4, 1.6 Hz, 1H, H-4), 7.69 (s, 1H, H-6), 7.57 (dd, J = 8.4, 4.4 Hz, 1H, H-3), 7.41 (dd, J = 2.8, 0.8 Hz, 1H, furyl-H-5), 7.15 (d, J = 3.6 Hz, 1H, thiazole-H-5), 6.55 (d, J = 3.6 Hz, 1H, thiazole-H-4), 6.40 (s exch., 1H, −OH), 6.34 (dd, J = 3.2, 2.0 Hz, 1H, furyl-H-3), 6.27 (dd, J = 2.8, 0.8 Hz, 1H, furyl-H-4), 1.62 (br s, 1H, −NH−). 13C-NMR (100 MHz, CDCl3): δ 168.93, 152.62, 149.11, 148.72, 142.47, 138.84, 138.60, 133.13, 126.32, 125.85, 122.53, 121.55, 120.61, 110.42, 107.74, 107.11, 52.02. MALDI-TOF: m/z 358 (M + H)+, 380 (M + Na)+.
General procedure for the synthesis of 8-quinolinyl carbamates
Triethylamine (3 drops) was added to a suspension of 8-hydroxyquinoline (1 mmol) and an isocyanate (1 mmol) in dichloromethane (15 mL). The reaction mixture was stirred for 2 days at room temperature and the solvent was removed using a rotary evaporator and the residue was subjected to flash-column chromatography over silica gel (eluent: hexanes or 5% EtOAc in hexanes) to afford the respective quinolinyl carbamate.
5,7-Dichloroquinolin-8-yl 2-chloroethylcarbamate (21)
42 Yield: 233 mg (73%). Rf: 0.65 (7 : 3 hexanes–EtOAc). mp: 112 °C. tR: 4.747 (96%); 1H-NMR (400 MHz, acetone-d6): δ 9.00 (dd, J = 4.0, 1.2 Hz, 1H, H-2), 8.60 (dd, J = 8.4, 1.1 Hz, 1H, H-4), 7.88 (s, 1H, H-6), 7.75 (dd, J = 8.4, 4.0 Hz, 1H, H-3), 7.54 (br s, 1H, NH), 3.76 (t, J = 6 Hz, 2H, H-2′), 3.61 (app. q, J = 6 Hz, 2H, H-1′). 13C-NMR (100 MHz, acetone-d6): δ 154.29, 152.90, 150.87, 139.78, 133.98, 133.61, 129.00, 127.97, 126.62, 124.08, 44.16, 43.72. MALDI-TOF: m/z 319 (M + H)+, 341 (M + Na)+.
5-Chloroquinolin-8-yl 3,5-difluorophenylcarbamate (23)
42 Yield: 188 mg (56%). Rf: 0.67 (7 : 3 hexanes–EtOAc). mp: 152 °C. tR: 6.333 min (96%). 1H-NMR (400 MHz, CDCl3): δ 8.98 (dd, J = 4.4, 1.3 Hz, 1H, H-2); 8.56 (dd, J = 5.5, 1.3 Hz, 1H, H-4); 8.32 (br s, 1H, NH); 7.59 (dd, J = 5.5, 4.4 Hz, 1H, H-3); 7.53 (d, J = 5.8 Hz, 1H, H-7); 7.41 (d, J = 6.3 Hz, 2,6-aniline); 7.01 (d, J = 5.8 Hz, 1H, H-6); 6.42 (d, J = 6.3 Hz, 1H, 4-aniline). 13C-NMR (100 MHz, CDCl3): δ 165.01 (d), 153.11, 151.23, 150.89, 140.31, 139.46, 132.92, 128.45, 126.27, 122.30, 121.98, 112.47, 109.07 & 108.78 (pair of d’s), 103.02 (t). MALDI-TOF: m/z 335 (M + H)+, 357 (M + Na)+.
5-Chloroquinolin-8-yl 3,5-dichlorophenylcarbamate (24)
Yield: 300 mg (82%). Rf: 0.61 (7 : 3 hexanes–EtOAc). mp: 159 °C. tR: 8.480 min (96%). 1H-NMR (400 MHz, acetone-d6): δ 9.88 (br s, 1H, NH), 8.99 (dd, J = 4.4, 1.6 Hz, 1H, H-2), 8.65 (dd, J = 8.6, 1.4 Hz, 1H, H-4), 7.81 (d, J = 8.2 Hz, 1H, H-7), 7.75 (dd, J = 8.6, 3.8 Hz, 1H, H-3), 7.70 (app. t, J = 2.0 Hz, 2H, H-2,6-aniline), 7.65 (d, J = 8.2 Hz, 1H, H-6), 7.20 (app. t, J = 2.0 Hz, H-4-aniline). 13C-NMR (100 MHz, acetone-d6): δ 152.13, 143.35, 142.40, 135.91, 133.61, 128.96, 127.88, 127.34, 124.02, 123.46, 122.85, 117.63. MALDI-TOF: m/z 367 (M + H)+, 389 (M + Na)+.
5,7-Dichloroquinolin-8-yl 4-fluorophenylcarbamate (25)
Yield: 312 mg (89%). Rf: 0.68 (7 : 3 hexanes–EtOAc). mp: 148 °C. tR: 5.973 min (96%). 1H-NMR (400 MHz, CDCl3): δ 8.85 (br s, 1H, NH), 7.71 (dd, J = 5.1, 3.3 Hz, 1H, H-2), 7.53 (dd, J = 5.7, 3.5 Hz, 1H, H-4), 7.43 (dd, J = 8.8, 4.8 Hz, 1H, H-3), 7.35 (s, 1H, H-6), 7.28 (t, J = 8.3 Hz, 2H, H-2,6-aniline), 7.01 (t, J = 8.8, 2H, H-3,5-aniline). 13C-NMR (100 MHz, CDCl3): δ 161.14 (d), 149.05, 133.93, 131.99, 131.90, 131.16, 129.07, 122.90, 122.82, 118.03, 117.98, 116.12, 115.90. MALDI-TOF: m/z 351 (M + H)+, 373 (M + Na)+.
Synthesis of 5-chloroquinolin-8-yl diethyl phosphate (26)
Diethyl chlorophosphate (1 mmol, 145 μL) was added to a mixture of DMAP (1 mmol, 122 mg), Et3N (2.5 mmol, 350 μL), and 5-chloro-8-hydroxyquinoline (1 mmol, 180 mg) in dichloromethane (20 mL) and stirred at rt for 24 h. The reaction mixture was washed with saturated NaHCO3 (2 × 20 mL) and brine (1 × 15 mL) and concentrated to dryness. The crude mixture was purified using flash-column chromatography over silica gel (eluent: 20% EtOAc in CH2Cl2). Yield: 291 mg (92%). Rf: 0.43 (7 : 3 hexanes–EtOAc). tR: 7.107 min (98%). 1H-NMR (400 MHz, CDCl3): δ 9.06 (d, J = 1.6 Hz, 1H, H-2), 8.57 (d, J = 4.8 Hz, 1H, H-4), 7.66 (dd, J = 4,8, 1.6 Hz, 1H, H-3), 7.59 (app. t, J = 5.2 Hz, 2H, H-6,7), 4.38 (q, J = 4.6 Hz, 4H, OEt), 1.41 (t, J = 4.6 Hz, 6H, Et). 13C-NMR (100 MHz, CDCl3): δ 154.3, 153.2, 138.7, 132.5, 128.2, 127.6, 127.4, 122.9, 115.1, 51.4, 12.7. MALDI-TOF: m/z 316 (M + H)+, 338 (M + Na)+.
General procedure for the synthesis of 8-allyloxyquinolines
A solution of 8-hydroxyquinoline (1 mmol) in aqueous NaOH (0.25 M, 8 mL; 2 mmol) containing tetrabutylammonium bromide as the phase-transfer catalyst (0.1 mmol, 33 mg) was stirred for 30 min., and allyl bromide (1 mmol, 85 μL) and benzene (8 mL) were added and the reaction mixture was stirred vigorously at 50 °C for 14 h. The reaction mixture was partitioned between water (15 mL) and EtOAc (15 mL) after cooling to room temperature and the extraction was repeated twice. The organic layers were combined and washed with 1 M NaOH solution (15 mL) and concentrated. The crude product was purified by flash-column chromatography over silica gel using 1 : 2 hexanes–ether as eluent to afford the allyloxyquinolines.
8-(Allyloxy)-5,7-dichloroquinoline (28)
Yield: 218 mg (86%). Rf: 0.59 (7 : 3 hexanes–EtOAc). mp: 64 °C. tR: 5.427 min (97%). 1H-NMR (400 MHz, CDCl3): δ 9.03 (d, J = 1.7 Hz, 1H, H-2), 8.54 (dd, J = 4.8, 1.7 Hz, 1H, H-4), 7.63 (s, 1H, H-6), 7.53 (dd, J = 4,8, 1.7 Hz, 1H, H-3), 6.22 (m, 1H, CH=CH2), 5.39 and 5.22 (two dd’s, J = 14.3, 1.2 and J = 6.9, 1.2 Hz; 2H, CH=CH2), 4.98 (dd, J = 6.5, 1.1 Hz, 2H, CH2–CH=CH2). 13C-NMR (100 MHz, CDCl3): δ 155.2, 152.8, 140.7, 133.8, 133.6, 129.1, 127.9, 126.6, 129.1, 114.5, 109.8, 72.5. MALDI-TOF: m/z 255 (M + H)+, 277 (M + Na)+.
8-(Allyloxy)quinoline-2-carbonitrile (30)
Yield: 172 mg (82%). Rf: 0.51 (7 : 3 hexanes–EtOAc). tR: 5.707 min (100%). 1H-NMR (400 MHz, CDCl3): δ 8.27 (d, J = 6.8 Hz, 1H, H-4), 7.73 (d, J = 6.8 Hz, 1H, H-3), 7.61 (t, J = 6.9 Hz, 1H, H-6), 7.43 (dd, J = 6.9, 1.2 Hz, 1H, H-5), 7.18 (dd, J = 6.9, 1.2 Hz, 1H, H-7), 6.11 (m, 1H, CH=CH2), 5.56 and 5.41 (pair of dd’s, J = 15.2, 1.4; 11.0, 1.4 Hz, 2H, CH=CH2), 4.91 (dd, J = 5.5, 1.1 Hz, 2H, CH2CH=CH2). 13C-NMR (100 MHz, CDCl3): δ 156.2, 137.1, 136.9, 133.6, 132.5, 130.2, 129.7, 123.3, 120.3, 116.6, 116.2, 108.9, 71.5. MALDI-TOF: m/z 211 (M + H)+, 233 (M + Na)+.
General procedure for the alkylation of 5-amino-8-hydroxyquinoline
The reductive amination procedure we used was adapted from the one described by Röhrig et al.31 5-Amino-8-hydroxyquinoline dihydrochloride salt (1 mmol, 233 mg) was suspended in CH2Cl2 (20 mL) and AcOH (0.75 mL) and an aldehyde (2.5 mmol) were added and stirred vigorously at rt for 15 min. Sodium triacetoxyborohydride (4 mmol, 850 mg) was added in one portion and the mixture was stirred overnight at rt. The reaction mixture was diluted with water (30 mL) and extracted with CH2Cl2 (2 × 25 mL) and the residue left after evaporation of CH2Cl2 was slurried with silica gel and the desired products were obtained after flash-column chromatography.
5-(N,N-Diethyl)amino-8-hydroxyquinoline (35)
Acetaldehyde (2.5 mmol, 150 μL) was used, and the column was eluted with 5% MeOH–CH2Cl2 to obtain quinoline 35 as a dark-green wax. Yield: 207 mg (96%). tR: 3.867 min (96%). 1H-NMR (400 MHz, CDCl3): δ 8.80 (d, J = 4.0 Hz, 1H, H-2), 8.65 (d, J = 8 Hz, 1H, H-4), 7.45 (d, J = 8.0 Hz, 1H, H-3), 7.20 (d, J = 8.0, 4.0 Hz, 1H, H-6), 7.15 (d, J = 8.0 Hz, 1H, H-7), 3.10 (q, J = 7.0 Hz, 4H, CH2), 1.01 (t, J = 7.0 Hz, 6H, CH3). 13C-NMR (100 MHz, CDCl3): δ 157.1, 153.7, 148.2, 133.3, 132.2, 131.1, 121.3, 120.3, 109.2, 44.1, 13.5. MALDI-TOF: m/z 217 (M + H)+, 239 (M + Na)+.
5-(N,N-Di-n-hexyl)amino-8-hydroxyquinoline (36)
Hexanal (2.5 mmol, 307 μL) was used, and the column was eluted with 20% EtOAc–CH2Cl2 to obtain quinoline 36 as a yellow-green syrup. Yield: 310 mg (94%). tR: 8.613 min (96%). 1H-NMR (400 MHz, CDCl3): δ 8.78 (dd, J = 4.0, 2.0 Hz, 1H, H-2), 8.68 (d, J = 8.0 Hz, 1H, H-4), 7.44 (dd, J = 4.0, 8.0 Hz, 1H, H-3), 7.22 (d, J = 8.0, 4.0 Hz, 1H, H-6), 7.14 (d, J = 8.0 Hz, 1H, H-7), 3.03 (t, J = 5.5 Hz, 4H, NCH2), 1.25 (m, 16H, CH2), 0.84 (t, J = 5.0 Hz, 6H, CH3). 13 C-NMR (100 MHz, CDCl3): δ 148.6, 147.7, 138.8, 133.2, 127.2, 121.1, 120.3, 109.4, 55.6, 31.7, 27.3, 27.1, 22.7, 14.1. MALDI-TOF: m/z 329 (M + H)+, 351 (M + Na)+.
5-(N,N-Di[(4-methoxy)benzyl])amino-8-hydroxyquinoline (37)
p-Methoxybenzaldehyde (2.5 mmol, 305 μL) was used, and the column was eluted with 10% EtOAc–CH2Cl2 to obtain quinoline 37 as a lemon yellow syrup. Yield: 264 mg (91%). tR: 7.080 min (96%). 1H-NMR (400 MHz, CDCl3): δ 8.81 (d, J = 4.0 Hz, 1H, H-2), 8.76 (d, J = 8 Hz, 1H, H-4), 7.37 (d, J = 8.0 Hz, 1H, H-3), 7.28 (d, J = 7.0 Hz, 2H, PMB), 7.21 (d, J = 8.0 Hz, 1H, H-6), 6.95 (d, J = 8.0 Hz, 1H, H-7), 6.83 (d, J = 7.0 Hz, 2H, PMB), 4.15 (s, 3H, OCH3), 3.77 (s, 2H, CH2). 13 2 C-NMR (100 MHz, CDCl3): δ 158.6, 147.7, 144.6, 138.9, 138.7, 132.8, 130.1, 129.2, 121.3, 120.9, 120.3, 114.1, 110.1, 109.1, 55.2, 48.8. MALDI-TOF: m/z 401 (M + H)+, 422 (M + Na)+.
General procedure for the synthesis of 5-acylamino-8-hydroxyquinolines
5-Amino-8-hydroxyquinoline dihydrochloride salt was dissolved in pyridine and treated with carbonyl halides given below according to the procedure described by Ghedini et al.32 The crude products were purified by flash-column chromatography over silica gel.
N-(8-Hydroxyquinolin-5-yl)hexanamide (39)
Yield: 56%. tR: 6.067 min (96%). 1H-NMR (400 MHz, CDCl3) δ: 8.82 (d, J = 4.0 Hz, 1H, H-2), 8.59 (br s, 1H, OH), 8.31 (d, J = 8 Hz, 1H, H-4), 7.67 (d, J = 8.0 Hz, 1H, H-6), 7.45 (dd, J = 8.0, 4.0 Hz, 1H, H-3), 7.54 (br s, 1H, NH), 7.37 (d, J = 8.0 Hz, 1H, H-7), 2.74 (t, J = 8 Hz, 2H, COCH2), 1.72 (m, 2H, COCH2CH2), 1.34 (m, 4H), 0.86 (t, J = 8.0 Hz, 3H, CH3). 13C-NMR (100 MHz, CDCl3) δ: 173.4, 152.4, 143.6, 134.5, 138.2, 130.1, 122.1, 120.1, 116.6, 112.0, 34.7, 28.4, 26.3, 22.1, 17.8. MALDI-TOF: m/z 259 (M + H)+, 281 (M + Na)+.
N-(8-Hydroxyquinolin-5-yl)propane-2-sulfonamide (41)
Yield: 87%. tR: 4.373 min (97%). 1H-NMR (400 MHz, CDCl3): δ 8.84 (dd, J = 8.0, 4.0 Hz, 1H, H-2), 8.05 (dd, J = 8.0, 4.0 Hz, 1H, H-4), 7.43 (d, J = 8.0 Hz, 1H, H-6), 7.25 (dd, J = 8.0, 4.0 Hz, 1H, H-3), 6.57 (d, J = 8.0 Hz, 1H, H-7), 4.61 (br s, 2H, NH, OH), 3.91 (sept, J = 8 Hz, 1H, iPr), 1.62 (d, J = 8 Hz, 6H, Me). 13C-NMR (100 MHz, CDCl3): δ 150.5, 142.2, 141.7, 136.9, 130.3, 123.9, 120.2, 119.1, 108.2, 53.4, 17.0. MALDI-TOF: m/z 267 (M + H)+.
N-(8-Hydroxyquinolin-5-yl)morpholine-4-carboxamide (42)
Yield: 72%. tR: 3.613 min (100%). 1H-NMR (400 MHz, CDCl3): δ 8.91 (d, J = 4.0 Hz, 1H, H-2), 8.31 (d, J = 8.0 Hz, 1H, H-4), 7.38 (dd, J = 8.0, 4.0 Hz, 1H, H-3), 7.19 (d, J = 8.0 Hz, 1H, H-7), 7.05 (d, J = 8.0 Hz, 1H, H-6), 3.80 (br s, 2H), 3.65 (m, 4H, OCH2), 3.45 (m, 4H, NCH ). 13C-NMR (100 MHz, CDCl3): δ 158.1, 150.4, 142.9, 138.7, 134.5, 130.3, 123.8, 120.5, 118.1, 113.1, 66.7, 48.6. MALDI-TOF: m/z 274 (M + H)+, 296 (M + Na)+.
Synthesis of 5-nitroquinolin-8-yl pivalate (46)
Pivaloyl chloride (2 mmol, 250 μL) was added to an ice-cold mixture of nitroxoline (2 mmol, 380 mg), DMAP (0.8 mmol, 98 mg), and Et3N (8 mmol, 1.1 mL) in CH2Cl2 (20 mL), and the mixture was stirred vigorously for 14 h at rt. The reaction mixture was washed with 10% aqueous NaHCO3 and the CH2Cl2 layer was concentrated. The crude residue was subjected to flash-column chromatography (eluent: hexanes–CH2Cl2 = 7 : 3) to afford the ester 46 as a yellow solid. Yield: 465 mg (85%). Rf: 0.67 (2 : 3 EtOAc–hexanes). tR: 7.613 min (99%). 1H-NMR (400 MHz, CDCl3): δ 8.99 (d, J = 8.8 Hz, 1H, H-4), 8.93 (d, J = 3.6 Hz, 1H, H-2), 8.38 (d, J = 8.4 Hz, 1H, H-6), 7.59 (dd, J = 8.8, 3.6 Hz, 1H, H-3), 7.46 (d, J = 8.8 Hz, 1H, H-7), 1.49 (s, 9H, t-Bu). 13C-NMR (100 MHz, CDCl3) δ: 178.0, 153.7, 151.4, 142.9, 141.2, 132.4, 125.5, 124.6, 122.8, 120.0, 39.7, 27.6. MALDI-TOF: m/z 275 (M + H)+, 297 (M + Na)+.
Synthesis of 5-nitroquinolin-8-yl dimethylsulfamate (48)
To an ice-cold mixture of nitroxoline (1 mmol, 190 mg) and DIEA (2 mmol, 190 μL) in CH2Cl2 (15 mL), N,N-dimethylamino-sulfonyl chloride (1 mmol, 107 μL) was added and the stirring was continued at rt for 16 h. The reaction mixture was washed with 10% aqueous NaHCO3 (1 × 15 mL) and the CH2Cl2 layer was concentrated to dryness. The crude residue was loaded onto a silica gel column and eluted with 1% EtOH in CH2Cl2 to yield sulfonamide 48 as a yellow solid. Yield: 222 mg (88%). Rf: 0.27 (EtOH–CHCl3 = 1 : 19). tR: 5.600 min (100%). 1H-NMR (400 MHz, CDCl3): δ 9.10 (d, J = 4.1 Hz, 1H, H-2), 8.45 (d, J = 7.9 Hz, 1H, H-4), 7.90 (d, J = 8.2 Hz, 1H, H-6), 7.75 (dd, J = 7.9, 4.1 Hz, 1H, H-3), 7.20 (d, J = 8.2 Hz, 1H, H-7), 3.15 (s, 6H, NMe2). 13C-NMR (100 MHz, CDCl3): δ 162.3, 152.1, 143.7, 138.4, 131.0, 126.2, 125.4, 122.5, 109.9, 42.8. MALDI-TOF: m/z 254 (M + H)+, 276 (M + Na)+.
Synthesis of ethyl 2-(5-nitroquinolin-8-yloxy)acetate (49)
Nitroxoline (5.3 mmol, 1 g) was heated at 60 °C with ethyl bromoacetate (5.3 mmol, 0.6 mL) and potassium carbonate (7.3 mmol, 1 g) in DMSO (20 mL) for 18 h, cooled to rt, diluted with water (100 mL) and extracted with a mixture of 2 : 3 EtOAc–Et2O. Solvent was evaporated and the crude product was purified by column chromatography on silica gel (eluent: 1 : 3 EtOAc–CH2Cl2). Yield: 980 mg (67%). Rf: 0.38 (EtOAc–hexanes = 2 : 3). tR: 8.893 min (98%). 1H-NMR (400 MHz, CDCl3): δ 9.15 (d, J = 8.8 Hz, 1H, H-4), 9.01 (d, J = 3.5 Hz, 1H, H-2), 8.42 (d, J = 8.8 Hz, 1H, H-6), 7.67 (dd, J = 8.8, 3.5 Hz, 1H, H-3), 6.93 (d, J = 8.8 Hz, 1H, H-7), 5.03 (s, 2H, OCH2CO), 4.25 (q, J = 7.5 Hz, 2H, Et), 1.25 (t, J = 7.5 Hz, 3H, Et). MALDI-TOF: m/z 277 (M + H)+, 299 (M + Na)+.
Synthesis of 2-(5-nitroquinolin-8-yloxy)acetamide (50)
Acetamide 50 was also prepared according to the procedure described above for making the acetate 49, except the reaction was run with 1 mmol of iodoacetamide and potassium carbonate was supplanted with caesium carbonate. Yield: 193 mg (78%). Rf: 0.31 (2 : 3 EtOAc–hexanes). tR: 5.802 min (98%). 1H-NMR (400 MHz, CDCl3): δ 9.40 (d, J = 8.8 Hz, 1H, H-4), 8.81 (d, J = 3.5 Hz, 1H, H-2), 8.55 (d, J = 8.8 Hz, 1H, H-6), 7.68 (dd, J = 8.8, 3.5 Hz, 1H, H-3), 7.51 (d, J = 8.8 Hz, 1H, H-7), 6.80 (br s, 2H, NH2), 4.20 (s, 2H, OCH2CO). MALDI-TOF: m/z 248 (M + H)+, 270 (M + Na)+.
Synthesis of 4-methyl-N-(5-nitrosoquinolin-8-yl)benzene-sulfonamide (54)
The sulfonamide derivative of 8-aminoquinoline (32) was subjected to nitrosation (2 mmol scale) in a manner similar to the synthesis of nitrosoquinoline 33, and sulfonamide 54 was collected as a beige colored solid after filtration from the reaction mixture which was spectroscopically pure and used as such. Yield: 595 mg (91%). Rf: 0.37 (EtOH–CHCl3 = 1 : 19). tR: 6.840 min (100%). 1H-NMR (400 MHz, CDCl3) δ: 9.40 (s, 1H, H-2), 8.76 (d, J = 3.0 Hz, 1H, H-4), 8.15 (d, J = 7.6 Hz, 1H, H-6), 7.87 (s, 1H, H-3), 7.81 (d, J = 8.0 Hz, 2H, Ts), 7.48 (m, 2H, H-7), 7.15 (d, J = 8.0 Hz, 2H, Ts), 2.29 (s, 3H, Me). 13C-NMR (100 MHz, CDCl3) δ: 156.1, 150.3, 149.4, 142.5, 137.2, 136.9, 136.4, 129.5, 127.6, 122.8, 121.3, 120.6, 118.7, 26.6. MALDI-TOF: m/z 327 (M + H)+, 350 (M + Na)+.
Synthesis of 4-methyl-N-(5-nitroquinolin-8-yl)benzene-sulfonamide (55)
Concentrated nitric acid (0.32 mL) was added dropwise to a suspension of nitrosoquinoline 51 (1 mmol, 326 mg) in glacial acetic acid (6 mL) at rt. The reaction mixture was stirred at rt for 2 h and then cooled to 0 °C and basified with the addition of 10% NaOH to pH 10–11. The resulting brown precipitate was filtered, resuspended in water, and acetic acid was added to adjust the pH again to 5–6. The light-brown precipitate was finally filtered, washed with water and air-dried. Yield: 264 mg (77%). Rf: 0.39 (EtOH–CHCl3 = 1 : 19). tR: 6.827 min (100%). 1H-NMR (400 MHz, CDCl3): δ 9.33 (s, 1H, H-2), 8.76 (s, 1H, H-4), 8.13 (d, J = 7.2 Hz, 1H, H-6), 7.82 (s, 1H, H-3), 7.80 (d, J = 7.6 Hz, 2H, Ts), 7.46 (s, 1H, H-7), 7.15 (d, J = 7.6 Hz, 2H, Ts), 2.29 (s, 3H, Me). 13C-NMR (100 MHz, CDCl3): δ 150.3, 149.8, 142.5, 141.1, 137.2, 136.9, 136.4, 129.5, 127.6, 126.9, 124.9, 121.3, 118.7, 26.6. MALDI-TOF: m/z 344 (M + H)+, 366 (M + Na)+.
Acknowledgements
We would like to thank Dr. Jon Lorsch for providing us with some oxine derivatives he had in turn procured from NCI/DTP repository. This work was supported by National Cancer Institute (CA78743).
Footnotes
Electronic supplementary information (ESI) available: Synthesis and analytical data for the substituted quinolines 12–16, 20, 22, 27, 29, 32–34, 38, 40, 43–45, and 47. Expression, purification, and assay protocols pertaining to human MetAPs. Experimental details for the cell-based assays: (1) 3H-thymidine incorporation in HUVEC, HeLa, and Jurkat T cells and (2) Calcein-AM in HUVEC and HFF cell lines. Immunoblotting with compound treated HUVEC lysates. Representative dose–response curves of MetAP, MetAP2, HUVEC, and HFF inhibition. Representative UV–vis spectra of compounds with or without metal ions. 1H-NMR and HPLC traces of the previously unknown compounds. See DOI: 10.1039/c2ob06978d
Notes and references
- 1.Ferrara N, Hillan KJ, Gerber H, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discovery. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 2.Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST) Ther. Clin. Risk Manag. 2007;3:341–348. doi: 10.2147/tcrm.2007.3.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkman J. Antiangiogenesis in cancer therapy–endostatin and its mechanisms of action. Exp. Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 5.Reardon DA, Nabors LB, Stupp R, Mikkelsen T. Cilengitide: an integrin-targeting arginine-glycine-aspartic acid peptide with promising activity for glioblastoma multiforme. Expert Opin. Invest. Drugs. 2008;17:1225–1235. doi: 10.1517/13543784.17.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delmonte A, Sessa C. AVE8062: a new combretastatin derivative vascular disrupting agent. Expert Opin. Invest. Drugs. 2009;18:1541–1548. doi: 10.1517/13543780903213697. [DOI] [PubMed] [Google Scholar]
- 7.Connolly B, Desai A, Garcia CA, Thomas E, Gast MJ. Squalamine lactate for exudative age-related macular degeneration. Ophthalmol. Clin. North Am. 2006;19:381–391. doi: 10.1016/j.ohc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Benny O, Fainaru O, Adini A, Cassiola F, Bazinet L, Adini I, Pravda E, Nahmias Y, Koirala S, Corfas G, D’Amato RJ, Folkman J. An orally delivered small-molecule formulation with antiangiogenic and anticancer activity. Nat. Biotechnol. 2008;26:799–807. doi: 10.1038/nbt1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9(a).Lefkove B, Govindarajan B, Arbiser JL. Fumagillin: an anti-infective as a parent molecule for novel angiogenesis inhibitors. Expert Rev. Anti. Infect. Ther. 2007;5:573–579. doi: 10.1586/14787210.5.4.573. [DOI] [PubMed] [Google Scholar]; (b) Mazitschek R, Huwe A, Giannis A. Synthesis and biological evaluation of novel fumagillin and ovalicin analogues. Org. Biomol. Chem. 2005;3:2150–2154. doi: 10.1039/b503163j. [DOI] [PubMed] [Google Scholar]
- 10(a).Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997;4:461–471. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]; (b) Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradshaw RA, Yi E. Methionine aminopeptidases and angiogenesis. Essays Biochem. 2002;38:65–78. doi: 10.1042/bse0380065. [DOI] [PubMed] [Google Scholar]
- 12.Warder SE, Tucker LA, McLoughlin SM, Strelitzer TJ, Meuth JL, Zhang Q, Sheppard GS, Richardson PL, Lesniewski R, Davidsen SK, Bell RL, Rogers JC, et al. Discovery, identification, and characterization of candidate pharmacodynamic markers of methionine aminopeptidase-2 inhibition. J. Proteome Res. 2008;7:4807–4820. doi: 10.1021/pr800388p. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Griffith EC, Sage J, Jacks T, Liu JO. Cell cycle inhibition by the anti-angiogenic agent TNP-470 is mediated by p53 and p21WAF1/CIP1. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6427–6432. doi: 10.1073/pnas.97.12.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science (Washington, D. C.) 1998;282:1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, Chong CR, Hu X, Liu JO. Fumarranol, a rearranged fumagillin analogue that inhibits angiogenesis in vivo. J. Med. Chem. 2006;49:5645–5648. doi: 10.1021/jm060559v. [DOI] [PubMed] [Google Scholar]
- 16.Wagenlehner FME, Pilatz A, Naber KG, Perletti G, Wagenlehner CM, Weidner W. Anti-infective treatment of bacterial urinary tract infections. Curr. Med. Chem. 2008;15:1412–1427. doi: 10.2174/092986708784567699. [DOI] [PubMed] [Google Scholar]
- 17.Shim JS, Matsui Y, Bhat S, Nacev BA, Xu J, Bhang HC, Dhara S, Han KC, Chong CR, Pomper MG, So A, Liu JO. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J. Natl. Cancer Inst. 2010;102:1855–1873. doi: 10.1093/jnci/djq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guarente L. Diverse and dynamic functions of the Sir silencing complex. Nat. Genet. 1999;23:281–285. doi: 10.1038/15458. [DOI] [PubMed] [Google Scholar]
- 19.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 20(a).Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat. Rev. Cancer. 2009;9:123–128. doi: 10.1038/nrc2562. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–1705. doi: 10.1158/0008-5472.CAN-08-3365. [DOI] [PubMed] [Google Scholar]
- 21.Albrecht M, Feige M, Osetska O. 8-Hydroxyquinolines in metallo-supramolecular chemistry. Coord. Chem. Rev. 2008;252:812–824. [Google Scholar]
- 22.Hongmanee P, Rukseree K, Buabut B, Somsri B, Palittapongarnpim P. In vitro activities of cloxyquin (5-chloroquinolin-8-ol) against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007;51:1105–1106. doi: 10.1128/AAC.01310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohde W, Mikelens P, Jackson J, Blackman J, Whitcher J, Levinson W. Hydroxyquinolines inhibit ribonucleic acid-dependent deoxyribonucleic acid polymerase and inactivate Rous sarcoma virus and herpes simplex virus. Antimicrob. Agents Chemother. 1976;10:234–240. doi: 10.1128/aac.10.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelletier C, Prognon P, Bourlioux P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob. Agents Chemother. 1995;39:707–713. doi: 10.1128/AAC.39.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Guo XC, Yi T, Yoshimoto T, Pei D. Two continuous spectrophotometric assays for methionine aminopeptidase. Anal. Biochem. 2000;280:159–165. doi: 10.1006/abio.2000.4513. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- 27(a).D’souza VM, Bennett B, Copik AJ, Holz RC. Divalent metal binding properties of the methionyl aminopeptidase from Escherichia coli. Biochemistry. 2000;39:3817–3826. doi: 10.1021/bi9925827. [DOI] [PubMed] [Google Scholar]; (b) Hu X, Dang Y, Tenney K, Crews P, Tsai CW, Sixt KM, Cole PA, Liu JO. Regulation of c-Src nonreceptor tyrosine kinase activity by bengamide A through inhibition of methionine aminopeptidases. Chem. Biol. 2007;14:764–774. doi: 10.1016/j.chembiol.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu X, Addlagatta A, Matthews BW, Liu JO. Identification of pyridinylpyrimidines as inhibitors of human methionine aminopeptidases. Angew. Chem., Int. Ed. 2006;45:3772–3775. doi: 10.1002/anie.200600757. [DOI] [PubMed] [Google Scholar]; (d) Addlagatta A, Hu X, Liu JO, Matthews BW. Structural basis for the functional differences between type I and type II human methionine aminopeptidases. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]; (e) Hu X, Zhu J, Srivathsan S, Pei D. Peptidyl hydroxamic acids as methionine aminopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2004;14:77–79. doi: 10.1016/j.bmcl.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 28.Chi K, Ahn YS, Shim KT, Park TH, Ahn JS. Synthesis of Mannich bases using substituted aromatic alcohols with secondary amines: relative reactivity and regioselectivity depending on substrates. J. Korean Chem. Soc. 2001;45:51–60. [Google Scholar]
- 29.Isaev AA, Lomovskii II, Korolev KG, Karimov RK. Technology of preparing 8-hydroxy-5-nitroquinoline. Chem. Heterocycl. Compd. 2005;41:1027–1030. [Google Scholar]
- 30.Sleath PR, Noar BJ, Eberlein GA, Bruice TC. Synthesis of 7,9-didecarboxymethoxatin (4,5-dihydro-4,5-dioxo-1H-pyrrolo[2,3-f]quinoline-2-carboxylic acid) and comparison of its chemical properties with those of methoxatin and analogous o-quinones. Model studies directed toward the action of PQQ requiring bacterial oxidoreductases and mammalian plasma amine oxidase. J. Am. Chem. Soc. 1985;107:3328–3338. [Google Scholar]
- 31.Röhrig UF, Awad L, Grosdidier A, Larrieu P, Stroobant V, Colau D, Cerundolo V, Simpson AJG, Vogel P, Van den Eynde BJ, Zoete V, Michielin O. Rational design of indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 2010;53:1172–1189. doi: 10.1021/jm9014718. [DOI] [PubMed] [Google Scholar]
- 32.Ghedini M, La Deda M, Aiello I, Grisolia A. Fine-tuning the luminescent properties of metal-chelating 8-hydroxyquinolines through amido substituents in 5-position. Inorg. Chim. Acta. 2004;357:33–40. [Google Scholar]
- 33.Majerz-Maniecka K, Musiol R, Skórska-Stania A, Tabak D, Mazur P, Oleksyn BJ, Polanski J. X-ray and molecular modelling in fragment-based design of three small quinoline scaffolds for HIV integrase inhibitors. Bioorg. Med. Chem. 2011;19:1606–1612. doi: 10.1016/j.bmc.2011.01.045. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Branicky R, Stepanyan Z, Carroll M, Guimond M, Hihi A, Hayes S, McBride K, Hekimi S. The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J. Biol. Chem. 2009;284:314–323. doi: 10.1074/jbc.M807579200. [DOI] [PubMed] [Google Scholar]
- 35.Yu H, Lou JR, Ding W. Clioquinol independently targets NF-kappaB and lysosome pathways in human cancer cells. Anticancer Res. 2010;30:2087–2092. [PubMed] [Google Scholar]
- 36.Green MLH. A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem. 1995;500:127–148. [Google Scholar]
- 37.Bronson RT, Bradshaw JS, Savage PB, Fuangswasdi S, Lee SC, Krakowiak KE, Izatt RM. Bis-8-hydroxyquinoline-armed diazatrithia-15-crown-5 and diazatrithia-16-crown-5 ligands: possible fluorophoric metal ion sensors. J. Org. Chem. 2001;66:4752–4758. doi: 10.1021/jo0017584. [DOI] [PubMed] [Google Scholar]
- 38.Xing S, Bhat S, Shroff N, Zhang H, Lopez J, Margolick J, Liu JO, Siliciano RF. Novel structurally-related compounds reactivate latent HIV-1 in a bcl-2 transduced primary CD4+ T cell model without inducing global T cell activation. J. Antimicrob. Chemother. 2012;67:398–403. doi: 10.1093/jac/dkr496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng H, Youdim MBH, Fridkin M. Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer’s therapy. ACS Chem. Biol. 2010;5:603–610. doi: 10.1021/cb900264w. [DOI] [PubMed] [Google Scholar]
- 40(a).Martirosyan A, Leonard S, Shi X, Griffith B, Gannett P, Strobl J. Actions of a histone deacetylase inhibitor NSC3852 (5-nitroso-8-quinolinol) link reactive oxygen species to cell differentiation and apoptosis in MCF-7 human mammary tumor cells. J. Pharmacol. Exp. Ther. 2006;317:546–552. doi: 10.1124/jpet.105.096891. [DOI] [PubMed] [Google Scholar]; (b) Strobl JS, Seibert CW, Li Y, Nagarkatti R, Mitchell SM, Rosypal AC, Rathore D, Lindsay DS. Inhibition of toxoplasma gondii and plasmodium falciparum infections in vitro by NSC3852, a redox active antiproliferative and tumor cell differentiation agent. J. Parasitol. 2009;95:215–223. doi: 10.1645/GE-1608.1. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds CH, Bembenek SD, Tounge BA. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007;17:4258–4261. doi: 10.1016/j.bmcl.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 42.Liu JO, Shim JS, Chong CR, Bhat S. Quinoline compounds as inhibitors of angiogenesis, human methionine aminopeptidase, and SIRT1, and methods of treating disorders. World Pt. 2010;042163:112. [Google Scholar]