

Abstract
It has been reported that the oxidation of phosphatidylcholine (PC) is necessary for C-reactive protein (CRP) to bind to lipid membranes, but it remains elusive why CRP only binds oxidized membranes. Here we offer a new perspective on the role of membrane curvature and CRP binding using engineered lipoprotein particle (LPP) mimics. We show that CRP binds preferentially to LPP mimics with diameters of ≤ 28 nm, and binding of CRP to these mimics leads to the dissociation of native CRP into monomeric CRP, exposing CRP neo-epitopes that bind C1q. We also show that the smaller LPP mimics compete for CRP binding to oxidized low density lipoproteins (oxLDLs), suggesting that these mimics expose the same PC epitopes as those found on oxLDLs. Results from this study suggest that membrane curvature could be an additional factor influencing CRP binding of damaged membranes distinct from the oxidation of PC lipids.
Keywords: C-reactive protein, membrane curvature, low density lipoprotein, nanoparticle, phosphatiylcholine
Introduction
C-reactive protein (CRP) is an acute phase protein that plays a role in innate immunity and facilitates clearance of damaged tissues and cells 1–3. Because plasma levels of CRP can increase by as much as 1000-fold within 72 hours in response to inflammation 3, 4, it has been utilized as a biomarker for inflammatory diseases and as a predictor of cardiovascular disease (CVD) 2, 3, 5. Emerging evidence suggests that inflammation may play a key role in atherosclerosis and CRP may have a causal effect in early atherogenesis 3, 6, 7 as CRP has been found to co-localize with foam cells in atherosclerotic lesions 4, 8, 9 and apoptotic vascular cells 10, 11. Moreover, studies have shown that CRP binds to apoptotic cells 11, 12 and oxidized low density lipoproteins (oxLDLs) 4, 8, 13 but not to healthy cells or native LDL. Binding of CRP to oxLDLs can lead to an in situ conformational changes of CRP from its native pentameric structure (pCRP) to modified CRP (mCRP) 13. mCRP has different antigenicity than pCRP and is known to have pro-atherogeneic effects, including neutrophil activation14 and enhancement of cell adhesion in human coronary artery endothelial cells. 14–17 mCRP also activates the classical complement pathway of the immune system 1, 18, 19 when C1q, the first protein in the complement cascade binds to mCRP 1, 18.
While the crystal structure of pCRP reveals the calcium-dependent PC binding sites7 it remains unclear why CRP binds only to damaged or oxidized surfaces even though PC is present on all cell membranes 7. The “Lipid Whisker Model” described recognition of damaged membranes by scavenger receptor CD36 through the protrusion of oxidized phospholipids that become truncated at the fatty acid tail as a result of oxidation 20. It has been suggested that CRP may possess a similar pattern recognition property as CD36 because it participates in the innate immune response and competes for the same epitopes on oxLDL as the autoantibody EO6 oxLDL 13, 21–23. However, this model is insufficient to describe CRP recognition of damaged membranes. Although CRP does bind oxidized membranes it also binds to phosphocholine on bacterial cell walls 3, 24, 25 or phosphocholine-conjugated to keyhole limpet hemocyanine (PCh-KLH) 13 or bovine serum albumin (PCh-BSA)13, 18, and none of these phosphocoline-containing substances contain oxidizable fatty acids. Here we explore another explanation for CRP binding to damaged membranes; specifically that cryptic PC epitopes on native cells and LDL become more exposed at high curvature, thus making the PC moiety more accessible for CRP binding 13, 20, 26.
In this study, we sought to evaluate the role that membrane curvature plays in CRP recognition of damaged membranes. In order to probe for what types of nanostructure reveal the PC epitope for CRP binding, we used PC-coated gold nanoparticles (AuNPs) to mimic the exterior coating of lipoprotein particles (LPP). We observed that CRP bound LPP mimics ≤ 28 nm in diameter, and this binding resulted in a conversion from pCRP to mCRP revealed by gel electrophoresis and tryptophan fluorescence studies. The binding of CRP to these LPP mimics also exposed the C1q binding epitope on CRP and competed with CRP-oxLDL binding, as revealed by anisotropy and enzyme-linked immunosorbant assay (ELISA). These data suggest that in addition to structural modification of fatty acid tails by oxidation, membrane curvature could be another mode by which CRP recognizes damage membranes and oxLDLs.
Results
Influence of LPP mimic size on CRP binding
We have previously demonstrated binding of CRP to an LPP mimic with a 10 nm core diameter 27. In order to evaluate the influence of membrane curvature on CRP binding, here we utilized a series of LPP mimics with varying membrane curvatures. Binding of CRP to LPP mimics was evaluated using gel electrophoresis. Because of the high extinction coefficient of AuNPs, the characteristic reddish pink color of AuNPs can be easily visualized on agarose gels 28. The LPP mimics, with and without CRP, were first separated by gel electrophoresis and displayed a size-dependent electrophoretic mobility (Fig. 1A and B). A digital photograph of the gel was taken after electrophoresis (Fig. 1A and B). CRP was then transblotted onto a nitrocellulose membrane and detected by Western blot (Fig. 1A, ii). The photograph of the gel and Western blot were then superimposed to determine the binding of CRP to the LPP mimics (Fig. 1A, iii). For further clarification, refer to supporting information for a detailed description and illustration of this analysis (Fig S1 in ESI†). From the Western blot, two distinct CRP bands were observed when CRP was incubated with LPP mimics ≤ 28 nm but not with the larger LPP mimics or the PC liposome control (Fig. 1A, ii). In addition, the superimposed image of the gel and Western blot showed overlapping bands of CRP with LPP mimics ≤ 28 nm indicating strong interaction between CRP and the smaller LPP mimics (Fig 1A, iii). In contrast, no overlapping bands were observed between CRP and LPP mimics > 28 nm or PC liposomes, indicating that CRP did not bind to these LPP mimics (Fig. 1A, iii).
Figure 1.

Curvature-dependent binding of CRP to LPP mimics. CRP and LPP mimics were incubated with either (A) 250 μM CaCl2 or (B) 5 mM EDTA for 30 min before electrophoresis. Photographs of the gel with (A, i) 250 μM CaCl2 or (B, i) 5 mM EDTA after electrophoresis. Western blots of the gels with (A, ii) CaCl2 or (B, ii) EDTA. Superimposed images of the gels with (A, iii) 250 μM CaCl2 or (B, iii) 5 mM EDTA with their respective Western blots, showing the overlapping regions of LPP mimics and CRP. PC = PC liposomes.
To further confirm the CRP-LPP mimic binding, we added a 20-fold molar excess of EDTA (5 mM) to the LPP mimics before incubating with CRP (Fig. 1B). Because calcium is required for CRP to bind PC 1, the excess EDTA should chelate all the calcium in the solution, therefore preventing CRP from binding to the LPP mimics. The Western blot (Fig. 1B, ii) and superimposed image (Fig. 1B, iii) of the EDTA treated samples showed only one CRP band at the pCRP position (Fig. 1B, ii), indicating CRP did not bind to any of the LPP mimics when EDTA was present. This demonstrates that binding of CRP to the smaller LPP mimics occurred through the PC headgroups and the interaction was calcium dependent (Fig. 1A), consistent with a previous study 27.
Isoform conversion of CRP measured by tryptophan fluorescence
Because previous in vitro studies have indicated that CRP can undergo conformational changes upon binding immobilized oxLDL 13 and PCh-KLH 13 or in solution with PC liposomes containing lysoPC 4, 12, 29, 30, we sought to determine whether binding of CRP to the LPP mimics could lead to similar isoform conversion of pCRP into mCRP. After 30 min of incubation with LPP mimics in the presence of 250 μM CaCl2 (Fig. 2A) or with 5 mM EDTA (Fig. 2B), the supernatants of the CRP-LPP mimics were collected and the intrinsic tryptophan fluorescence of each sample was measured (Fig. 2). Previous studies have utilized tryptophan emission fluorescence to monitor the in situ dissociation of CRP into mCRP 12, 31, 32 as a fast and convenient method because mCRP has lower fluorescence than native CRP. As CRP dissociates from its native pentameric structure, the six tryptophan residues within each CRP protomer becomes more solvent exposed, resulting in a decrease in fluorescence intensity 32.
Figure 2.

Changes in CRP conformation by tryptophan fluorescence and PAGE. CRP and LPP mimics were incubated with (A) 250 μM CaCl2 or (B) 5 mM EDTA for 30 min, centrifuged to remove the LPP mimics and the supernatant was analyzed for tryptophan fluorescence intensity. Fluorescence emission spectra of CRP incubated with LPP mimics: 21 nm (
 ), 26 nm (
), 26 nm (
 ), 28 nm (
), 28 nm (
 ), 41 nm (
), 41 nm (
 ), 64 nm (
), 64 nm (
 ), or with PC liposome (□). Inset: Fluorescence emission spectra of the pCRP (×), and mCRP (○) controls. (C) Normalized fluorescence intensities of CRP in the supernatant incubated with either 250 μM CaCl2 (solid bars) or 5 mM EDTA (open bars). Fluorescence emission for each sample was normalized to pCRP emission and the data represents mean intensity ± SD, n = 5. ** p<0.01; ***p<0.001 compared to pCRP. (d) Isoforms of CRP in the supernatant after incubation with LPP mimics evaluated by PAGE.
), or with PC liposome (□). Inset: Fluorescence emission spectra of the pCRP (×), and mCRP (○) controls. (C) Normalized fluorescence intensities of CRP in the supernatant incubated with either 250 μM CaCl2 (solid bars) or 5 mM EDTA (open bars). Fluorescence emission for each sample was normalized to pCRP emission and the data represents mean intensity ± SD, n = 5. ** p<0.01; ***p<0.001 compared to pCRP. (d) Isoforms of CRP in the supernatant after incubation with LPP mimics evaluated by PAGE.
A significant decrease (p-value < 0.05) in tryptophan fluorescence was observed when CRP was incubated with LPP mimics ≤ 28 nm in the presence of calcium (Fig. 2C, solid bars), suggesting that either (i) mCRP was formed as a result of binding to smaller LPP mimics, or (ii) CRP remained bound to the LPP mimics in the pellet (Fig S2 in ESI†). Conversely, when 20 fold molar excess EDTA was added to the LPP mimic, no significant decrease in tryptophan fluorescence was measured (Fig. 2B and 2C). A small but significant increase (p-value < 0.05) in fluorescent intensity was observed when CRP was incubated with EDTA in the PC liposome control. This increase is likely due to light scattering of the free liposomes in the solution (Fig. 2C, open bars). Tryptophan fluorescence results confirmed that CRP only binds to AuNP-templated PC membranes within a specific size range and not to free PC liposomes, and that such interactions led to the isoform conversion of pCRP to mCRP.
The isoforms of CRP present after binding were also observed by PAGE (Fig. 2D). Both pCRP and mCRP isoforms were observed on the gel when CRP was incubated with LPP mimics ≤ 28 nm in diameter, whereas only the pCRP isoform was observed in the > 28 nm diameter LPP mimic samples (Fig. 2D). Moreover, a simultaneous decrease in mCRP band intensities and increase in pCRP band intensities is observed as the LPP size increases from 21 to 64 nm (Fig. 2D). Thus, PAGE results provided supporting evidence to the tryptophan fluorescence emission data, suggesting that membrane curvature plays a role in CRP binding and isoform conversion.
Binding of CRP-LPP mimics to complement C1q
Modified CRP has been shown to activate the classical pathway of complement via C1q binding 18, 19. Having demonstrated that binding of CRP to LPP mimics in the presence of calcium led to conversion of pCRP to mCRP, we next quantified the interactions of each CRP-LPP mimic conjugate with C1q using fluorescence anisotropy and ELISA.
Fluorescence anisotropy measures the rotational diffusion rate of a fluorescent probe, such as an aptamer, and has been used to measure the binding interactions of biomolecules 27, 33, 34. For example, the rotation rate of a free probe should be faster (lower anisotropy) compared to the rate of a probe that is bound to its target. Using a 5′Cy3-labeled mCRP-specific aptamer 35 as our probe, we detected a significant change (p-value < 0.01) in anisotropy between C1q and CRP-LPP mimics ≤ 28 nm compared to the BSA control (Fig. 3 and Table 1). The Δr values of CRP bound to LPP mimics ≤ 28 nm were comparable to or greater than the Δr for mCRP (Table 1), indicating that a considerable amount of mCRP was present in solution. Conversely, binding of C1q to CRP-LPP mimic was insignificant for the 41 and 64 nm LPP mimics and the PC liposomes, confirming that no mCRP was present (Fig. 3 and Table 1). No C1q binding was observed in the pCRP sample. The larger Δr values measured for the ≤ 28 nm LPP containing samples suggests that C1q bound to mCRP attached to the LPP mimics, therefore resulting in slower rotation rates, i.e., higher anisotropy, than the soluble mCRP (Table 1 and Fig. S3 in ESI†).
Figure 3.
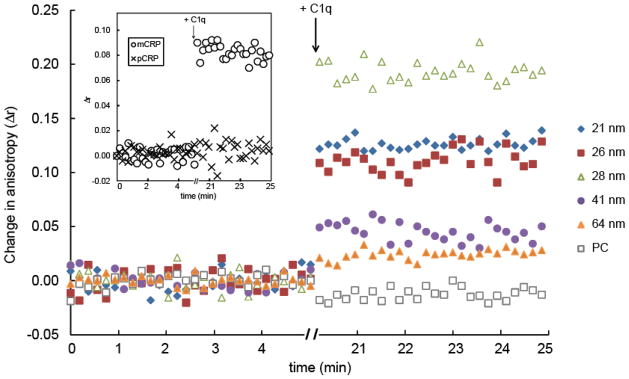
Fluid phase binding between 5′Cy3-RNA, CRP, and C1q using fluorescence anisotropy. CRP was pre-incubated for 30 min with each LPP mimic: 21 nm (
), 26 nm (
), 28 nm (
), 41 nm (
), 64 nm (
), and PC liposome (□) and the baseline anisotropy readings of CRP-LPP mimic plus 5′Cy3-RNA (0.59 μg/mL) were acquired for 5 min. After 5 min, an aliquot of C1q (5.8 μg/mL) was added to the mixture, incubated for 15 min and C1q binding was quantified by a change in anisotropy (Δr). Figure is a representative of one anisotropy measurement.
Table 1.
Binding of CRP-LPP mimics to C1q by fluorescence anisotropy. CRP, LPP mimics, and 5′Cy3-aptamer probe were incubated for 30 min and the baseline anisotropy was measured for 5 min. An aliquot of C1q (4 μg/mL) was added to the CRP-LPP mimics and incubated for 15 min before anisotropy measurements were measured for an additional 5 min. The change in anisotropy (Δr) was taken from three independent experiments and statistical significance was compared to the BSA control.
| Sample | Δr ± SD | p-value |
|---|---|---|
| 21 nm LPP + CRP | 0.118 ± 0.040 | 0.010 |
| 26 nm LPP + CRP | 0.136 ± 0.043 | 0.006 |
| 28 nm LPP + CRP | 0.166 ± 0.040 | 0.002 |
| 41 nm LPP + CRP | 0.042 ± 0.018 | 0.081 |
| 64 nm LPP + CRP | 0.025 ± 0.017 | 0.138 |
| PC liposomes + CRP | 0.022 ± 0.011 | 0.121 |
| pCRP | 0.013 ± 0.002 | 0.265 |
| mCRP | 0.081 ± 0.017 | 0.004 |
| BSA | 0.010 ± 0.001 | n.a. |
ELISA results also showed that CRP incubated with the smaller LPP mimics (≤ 28 nm) generated mCRP that bound 20 times stronger to C1q compared to the pCRP control; while CRP pre-incubated with 41 nm mimics showed moderate C1q binding (p-value < 0.01, Fig. 4). Conversely, pre-incubation of CRP with the 64 nm LPP mimic had no significant binding to C1q (Fig. 4), indicating that no CRP isoform conversion occurred. Thus, using both ELISA and anisotropy, we confirmed that the binding of CRP to smaller LPP mimics (≤ 28 nm) led to isoform conversion of pCRP into mCRP, and this mCRP isoform showed strong binding with C1q, in agreement with previous studies 18, 19.
Figure 4.

Binding of CRP-LPP mimics to immobilized C1q by ELISA. Purified human C1q (1 μg/mL) was coated on ELISA plates and the plates were incubated at increasing concentrations with each CRP-LPP mimic: 21 nm (
), 26 nm (
), 28 nm (
), 41 nm (
), 64 nm (
), and PC liposome (□). The degree of mCRP binding was detected using biotinylated polyclonal anti-CRP antibody. The measured absorbance was normalized to a BSA blank control. (a) Concentration-dependent binding of CRP-LPP mimic to C1q. Inset: C1q binding of the pCRP (×) and mCRP (○) controls. (b) Binding of CRP-LPP mimics (1.8 μg/mL CRP) to C1q. Data represents mean absorbance ±SD, n = 3. ** p<0.01; ***p<0.001 compared to pCRP.
LPP mimics inhibit CRP binding to oxLDLs
To determine whether the LPP mimics could compete for the CRP binding sites on oxLDL, a competitive ELISA was performed. It should be emphasized that although the concentrations of LPP mimics vary from size to size, the total surface area of all LPP mimic sizes used were matched to 1.1 × 10−3 m2, which corresponded to 2.6 × 10−19 moles of lipid, assuming each PC lipid has an estimated surface area between 50–80 Å2 36. Binding of CRP to plated oxLDL was significantly competed for (p-value < 0.001) when CRP was pre-incubated with the 21, 25, and 28 nm LPP mimics, with an 82, 79, and 74% reduction in CRP binding, respectively compared to the pCRP control (Fig. 5). Using PCh, a known ligand for CRP and competitor for oxLDLs 13, 18, in the form of PCh-BSA, we showed that the level of competitive binding by the smaller LPP mimics was comparable to that of PCh-BSA at 0.1 μg/mL (Fig. 5, inset). Incubation of CRP with the 41 nm LPP mimic competed for 32% (p-value < 0.05) of CRP binding to oxLDL, while pre-incubation of CRP with 62 nm LPP mimics did not compete for the CRP binding epitopes on oxLDLs (Fig. 5).
Figure 5.

Competitive binding of CRP to oxLDL using LPP mimics. Air oxidized LDLs (5.7 μg/mL) were immobilized on ELISA plates overnight at 4°C. Equal amounts of CRP (3.6 μg/mL) were pre-incubated for 30 min with LPP mimics: 21 nm, 26 nm, 28 nm, 41 nm, 64 nm, and PC liposome, or PCh-BSA, pCRP and mCRP controls. Then the CRP-LPP mimics were added to the oxLDL-coated plates. The binding of CRP to oxLDL was detected using biotinylated polyclonal anti-CRP antibody and the measured absorbance was normalized to pCRP. Inset: Competitive immunoassay using PCh-BSA. Data represents mean absorbance ±SD, n = 5. * p<0.05; ***p<0.001 compared to pCRP.
Direct immobilization leads to structural modification of native LDL
Previous studies have suggested that direct immobilization of LDL on ELISA wells could alter the structure of LDLs which resulted in significant CRP binding 13; whereas when LDL was captured using an antibody, no CRP binding was measured 13. To determine whether direct immobilization alters the structure of LDLs, we used AFM to characterize LDLs deposited on ELISA wells (Fig. 6). AFM imaging revealed that the surface of high binding ELISA wells contain fiber-like structures of approximately 40 nm in height (Fig. 6A). When LDLs were captured using an antibody, distinct individual LDL particles could be seen on the surface of the ELISA well (Fig. 6B). The intact LDL structures provided confirmation of the native antigencity of captured LDLs, in agreement with earlier studies 13. In contrast, when LDLs were directly plated on the ELISA wells, the LDLs did not retain their native structure (Fig. 6C). When LDLs were directly plated, the native structure of LDLs becomes disrupted, which led to the exposure of the other constituents of LDLs such as apoplipoprotein B, free cholesterol, cholesteryl esters and triglycerides 37. The fiber-like structures on the ELISA wells became less pronouced; suggesting that PC and the other LDL constituents may be trapped between the crevasses of the fiber-like structures and hence occupying the void spaces (Fig. 6C). In addition, direct plating of LDLs also caused clustering or aggregation of LDLs (Fig. 6C, arrows) which led to an overall height increase of the ELISA plate surface, from 50 nm to 100 nm (Fig. 6A and 6C). The visual evidence by AFM revealed that antibody-captured LDLs could retain their native structure even on a highly textured ELISA well (Fig. 6B), whereas direct plating of LDL caused a loss in membrane integrity of LDL, possibly exposing PC headgroups necessary for CRP binding.
Figure 6.

Representative topographic AFM images of LDL immobilized on ELISA well. AFM images of (A) high binding ELISA well, (B) antibody-captured LDLs and (C) direct immobilized LDLs on ELISA well. LDLs (5.7 μg/mL) were captured using a anti-Apo B polyclonal antibody or directly immobilized onto high binding ELISA well and incubated overnight at 4°C. After rinsing and drying, images were acquired in air using a tapping mode AFM. Insets show a cartoon illustrating the probable structures of the respective surfaces. Image scan size = 5 × 5μm.
Discussion
Elevated CRP and decreases in LDL size both correlate with increased risk for CVD. Clinical and epidemiologic studies have shown that individuals with predominantly small, dense LDL are at higher risk for developing coronary artery disease (CAD) 38–40. The average LDL particle is 25–26 nm in diameter 38, 41 and a 10 Å reduction in the average LDL size is correlated with a 30% increase in CAD risk 42. Small, dense LDLs are thought to be more atherogenic because they readily penetrate the arterial intima, have reduced clearance from circulation, are more susceptible to oxidation 38, and have poor affinity for LDL receptors 38, 43, 44. It has been suggested that CRP may have a causal role rather than just being an inflammatory marker for CVD 3, 4, 6 as studies have shown that CRP binds oxLDL 4, 8, 13 and co-localizes with atherosclerotic lesions in foam cells 4, 8, 9 and apoptotic vascular cells 10, 11. Although CRP circulates as a cyclic pentamer in plasma, there is evidence that binding of CRP to oxPCs and oxLDLs can cause an in situ dissociation of CRP into mCRP 12, 13. Because CRP binds oxLDLs and is found in atherosclerotic lesions 4, 8, 9, it is speculated that binding of CRP to oxLDLs may enhance inflammation and further exacerbate CVD.
The influence of truncated lipids on local membrane structure needs to be considered in any membrane model system of damaged or oxidized membrane recognition. The “Lipid Whisker Model” proposed by Hazen and co-workers describes a model in which truncated fatty acyl tails protrude outside the membrane layer as a result of lipid oxidation 20. This “lipid whiskering” is suggested to be a phenomenon seen on membranes of oxidized LPPs and apoptotic cells which provides a ligand for recognition of damaged membranes by scavenger receptors in the innate immune response 20. Oxidized lipids also have structures that resemble an inverted cone 45, 46 similar to lysolipids. The incorporation of oxidized or lysolipids within a lipid membrane has been shown to perturb membrane packing and induce areas of high curvature 47–50, hence exposing the lipid headgroups within the membrane bilayer. Therefore, in order for recognition of oxidized membrane by either CD36 or CRP to occur, a significant amount of membrane remodeling is required. Previous studies have shown that CRP bound more effectively to PC liposomes when 25–50 mol % of lysoPC was incorporated in the liposomes 4, 12, 29, 30. In addition to oxLDL, CRP also binds apoptotic cells, especially on the blebs 11 where a considerable amount of lysolipids are usually found. Incorporation of lysolipids or oxidized lipids within a lipid membrane can disrupt membrane packing and alter membrane structures, generating areas of high membrane curvature 49, 51–53.
It is unclear why CRP binds only oxLDL, although PC is present on native LDL and on all cell membranes 7, 54. The crystal structure of CRP reveals that CRP binds through the phosphate group of PC via two calcium ions, and through hydrophobic interaction with the choline moiety in a hydrophobic pocket (Phe 66, Leu 64 and Thr 76) located within each CRP protomer 7, 54, 55. In order for PC-CRP binding to occur, the PC headgroup needs to be accessible to CRP and fit within the hydrophobic binding pocket of each protomer. Taking into consideration that CRP only binds to oxLDL 13, 26, apoptotic blebs 11, lysoPC containing PC liposomes 29, and direct-plated LDL 13, we speculated that an alteration to the physical structure of native membrane such as increasing membrane curvature may be an additional mode to CRP binding.
In this work, we observed the effects of membrane curvature on CRP binding using PC-coated AuNPs. AuNP template lipids were used instead of sonicated or extruded liposomes because of the difficulty to obtain and/or to maintain < 100 nm size liposomes by the two latter methods (Fig S4 in ESI†). Using gel electrophoresis, we provided qualitative analysis of CRP binding to LPP mimics of various sizes. We showed that CRP bound to the smaller LPP mimics (≤ 28 nm), but not to the larger mimics (> 28 nm) or to PC liposomes (Fig. 1A, iii), confirming the role of membrane curvature in CRP binding. In addition, we demonstrated that the CRP-LPP mimic interactions were specific and occurred through the PC headgroups because the addition of 20 molar excess EDTA inhibited binding of CRP to all LPP mimic sizes (Fig. 1B). A cutoff at 28 nm for CRP binding is surprising and we estimate a total curvature (J) of > 0.1 nm−1 is needed in order to reveal the PC epitopes necessary for CRP binding (Fig. S4 in ESI†). However, due to the polydispersity of the synthesized LPP mimics (Table S2 in ESI†), it would be difficult to determine an exact limit for CRP binding. We speculate that because of the geometric constraints of the smaller AuNPs for lipid packing, the lipids are spread out and have more inter-lipid spacing, thereby making the PC headgroups more exposed and accessible for CRP binding (Fig. S4 in ESI†).
To examine whether the epitope created by high curvature was similar to that caused by oxidation of LDLs, we performed a CRP-oxLDL competitive assay using the LPP mimics. This showed that the smaller LPP mimics (≤ 28 nm) compete for greater than 74% of CRP binding to oxLDLs in the presence of calcium (Fig. 5). However, the 41 and 64 nm LPP mimics only competed for 30% and 5% of CRP binding to oxLDLs, respectively (Fig. 5). The competitive binding data suggests that the smaller LPP mimics have similar PC exposure and accessibility for CRP binding as those epitopes found on oxLDLs. We showed that CRP can bind LPP mimics and compete for oxLDLs in the absence of oxPCs. Using excess EDTA to chelate the calcium ions (Fig. 1B and Fig. 2B), we provided evidence that the CRP-LPP mimic interactions were specific through the PC headgroups, and not caused by arbitrary CRP insertion into defect sites on these highly curved surfaces. Because CRP binds PC headgroups regardless of the types of fatty tails (e.g., PCh-BSA 13, 18, oxPC 13, or phosphocholine on bacterial cell walls 3, 24, 25), we propose that changes to membrane curvature of LDLs caused by oxidation may suffice for CRP binding to oxLDLs rather than the chemical oxidation of LDL itself. Consequently, the membrane curvature model could provide an alternate or additional mode for CRP binding to oxLDLs that has not been previously suggested 13.
Our work also suggests a reason why highly curved membranes, as seen in small, dense LDL, could be more inflammatory through complement activation. In addition, we showed that binding of CRP to the smaller LPP mimics (≤ 28 nm) caused a dissociation of pCRP into mCRP (Fig. 2), and the resultant CRP isoform expressed epiptopes that recognize C1q (Fig. 3, Table 1), suggesting that the binding of CRP to LPP mimics ≤ 28 nm could generate a biologically relevant isoform of mCRP that activates the classical complement pathway via C1q binding (Fig. 4 and Table 1).
Because oxidation of lipids can induce curvature changes to LDLs, the membrane curvature model can explain why many conflicting reports have been made surrounding the chemical modifications of LDLs that result in CRP binding. Several studies reported that CRP binds only oxLDLs 13, 23 or enzymatically modified LDLs 8, 56, while others reported CRP binds native LDLs and very low density lipoproteins 57. However, there are some studies that showed that aggregated forms of CRP are needed in order to bind native LDLs 37, 58. Our analysis of LDL interactions with ELISA wells supports the membrane curvature model and could resolve some of the controversies surrounding CRP binding to LDLs on ELISA plates. We showed that the surface of a high binding Microlon® ELISA plate was rough and highly textured (Fig. 6A). When LDLs were captured using an anti-ApoB antibody, the structure of LDLs remained intact even on the highly textured surface (Fig. 6B), suggesting that the captured LDLs retained their native structure and lipid packing. In contrast, when LDLs were directly immobilized on the ELISA well, dissociation and aggregation of LDLs could be seen on the surface (Fig. 6C); hence these LDLs are expected to have altered membrane structure and lipid packing. As discussed earlier, in order for CRP to bind the PC headgroups within a lipid membrane, the PC must be exposed and accessible regardless of the oxidation of the lipid tails. Therefore, we speculate that membrane curvature and the PC exposure of directly captured LDLs may be sufficient to allow CRP to bind to that form of LDLs, and less dependent on whether or not the LDLs are native LDLs, oxLDLs or enzymatically modified LDLs.
Materials and Methods
AuNP synthesis
AuNP of various sizes were synthesized according to Fren’s method 59. Briefly, hydrogen tetracholroaurate (III) hydrate (0.25 mM final concentration, Strem Chemicals) was boiled under reflux in 100 mL water before trisodium citrate dihydrate (340 mM stock, JT Baker) was added (in varying amounts) to reduce the gold. The AuNP diameters were determined by dynamic light scattering (Malvern Nanosizer) and atomic force microscopy (Asylum Research), and the concentrations were measured using UV/vis spectroscopy (Perkin Elmer) (Table S1 in ESI†).
Synthesis of LPP mimics
PC lipid thin films were prepared by adding an aliquot of PC lipid stock (L-α-phosphatidylcholine, (Soy) Product No. 840054, Avanti Polar Lipids) in chloroform (500 nmoles, Sigma-Aldrich) in a glass vial and evaporating the excess chloroform overnight in vacuo. The PC thin film was rehydrated with 1 mM HEPES buffer (1 mL, pH 6.5, Sigma-Aldrich) and the PC solution was sonicated (Branson) for 1 h at 25°C before extrusion through a 100 nm polycarbonate membrane (Avanti Polar Lipids) to obtain small unilamellar liposomes. LPP mimics were synthesized as previously described with slight modification 27, 60 (Table S1 in ESI†). Briefly, AuNPs (1 mL) were incubated with 100 nmoles sodium oleate (TCI America) for 30 min at 25°C to stabilize the AuNPs. The total surface area per volume used for each AuNP size was matched to avoid bias towards smaller particles due to their greater surface area per volume. Extruded PC liposomes (100 nmoles) were then added to the oleate-stabilized AuNPs and the mixture was stirred for 1 h at 25°C. Following PC addition, decanethiol (10 nmoles, Alfa Aesar) was added to the solution to anchor the lipids around the AuNPs and the mixture was stirred for another 30 min at 25°C. The LPP mimics were buffered with HEPES (10 mM HEPES, 250 μM CaCl2, pH 7, Sigma-Aldrich) and used immediately.
Generation of modified CRP
Modified CRP was generated by treating pCRP in 8 M urea and 10 mM EDTA (Sigma-Aldrich) for 2 h at 37°C to denature the protein 61, followed by extensive dialysis against 1X PBS (Sigma-Aldrich) overnight at 4°C. The purity of mCRP was confirmed by SDS-PAGE.
Binding of CRP to LPP mimics evaluated by agarose gel electrophoresis
Purified human CRP (10 nM, Academy Biomedical Co.) was incubated with 1 mL of the LPP mimics in the presence of 250 μM CaCl2 or 5 mM EDTA for 30 min at 25°C before electrophoresis. LPP mimics, with and without CRP, were electrophoresed on a 0.8%, 0.5X TBE agarose gel (BioRad) with 0.005% SDS. Electrophoresis was carried out in a mini-sub cell GT (BioRad) for 30 min at a constant 40 V. Photographs of the gel were taken using a digital camera after electrophoresis.
Western blot
CRP was transblotted onto nitrocellulose membrane using a semi-dry transblotter (Bio Rad) for 90 min at a constant 25 V. The membrane was then blocked with 3% BSA and 0.05% Tween-20/PBS overnight at 4°C. After washing three times with 0.05% Tween-20/PBS, the membrane was probed with a biotinylated polyclonal anti-CRP antibody (1:5000, Academy Biomedical Co.) for 1 h at 25°C followed by a 30-min incubation with 1:5000 strept-IR800 (Li-COR Biotechnology) and imaged on an Odyssey imager (Li-COR Biotechnology).
Tryptophan florescence measurements of CRP
After a 30-min incubation of CRP with LPP mimics either with 250 μM CaCl2 or 5 mM EDTA, the CRP-LPP mimic mixtures were centrifuged at 21,000 g for 30 min and the supernatant was removed for fluorescence analysis. Fluorescence emission spectra of CRP were measured using FelixG software on a PTI spectrofluorometer (PTI). Fluorescence measurements were carried out in a quartz cuvette (Starna Cells) at an excitation of 295 nm and slit widths of 2 nm on both monochromators. All spectra were baseline corrected for buffer (10 mM HEPES, 250 μM CaCl2) and normalized to pCRP at an emission λmax of 345 nm (positive control).
CRP isoforms determined by polyacrylamide gel electrophoresis
Following a 30 min incubation of CRP with LPP mimics and centrifugation, the isoforms of CRP in the supernatant were separated on a 10 % (29:1 acrylamide:bis-acrylamide, BioRad) gel containing 0.005% SDS 62. Electrophoresis was carried out at 80 V for ~2 h at room temperature (BioRad) and the protein bands were visualized by silver staining.
Fluid phase binding of C1q by fluorescence anisotropy
Fluid phase C1q binding was monitored using a Cy3 labeled aptamer 35 (5′Cy3-RNA, IDT) on a Perkin Elmer LS55 fluorometer. Measurements were carried out in a quartz cuvette with excitation and emission wavelengths of 550 and 570 nm and slit widths of 2.5 and 10 nm, respectively. Changes in anisotropy (Δr) were measured after normalizing to the initial anisotropy of the 5′Cy3-RNA+CRP-LPP mimic. C1q (Calbiochem) was then added to the cuvette and incubated for 5 min at 25°C before anisotropy measurements were taken. Final concentrations were 2 μg/mL 5′Cy3-RNA, 6μg/mL CRP, and 4 μg/mL C1q.
Solid phase complement binding assay
Binding of CRP to immobilized C1q was performed as described previously 18. Purified human C1q (1 μg/mL) was diluted in coating buffer (30 mM Na2CO3, 70 mM NaHCO3, pH 9.6, Sigma-Aldrich) and incubated overnight at 4°C on high binding Microlon® ELISA plates (Santa Cruz Biotechnology). The wells were washed three times using 0.05% Tween-20/PBS and once with PBS before blocking with 3% BSA for 2 h at 25°C. Then, the CRP-LPP mimics were diluted with 1% BSA, ranging from 0–1.8μg/mL, and incubated for 1 h at 37°C, and the CRP immunoreactivity was detected using biotinylated polyclonal antibody (1:5000) for 1 h at 37°C, followed by streptavidin-HRP (1:10,000, Pierce) for 30 min at 37°C. A 3,3′5,5′-Tetramethylbenzidine (TMB) substrate (100 μL, 1-Step Slow TMB, Pierce) was added to each well and the reaction was quenched by adding 0.5 M H2SO4 (Sigma-Aldrich). The absorbance was measured at 450 nm on a microplate reader (Multiskan Ascent, Thermo-Fisher) with background subtracted at 620 nm. A blank control of 1% BSA was used and the absorbance for each sample was subtracted by this value and then normalized to pCRP.
Competitive binding assay using oxLDLs
LDLs (5.7 μg protein/mL, Calbiochem) were air-oxidized overnight at 37°C to induce mild oxidation of LDLs 63 (Fig S5 in ESI†). The mildly oxidized LDLs were then plated directly onto high binding Microlon® ELISA plates and incubated overnight at 4°C, washed three times using 0.05% Tween-20/PBS and once with PBS and blocked with 3% BSA for 2 h at 37°C. Competitive binding assays were carried out by pre-incubating CRP (3.6 μg/mL) with the various LPP mimics, or with increasing concentrations of PCh-BSA (0–10 μg/mL, Bioserach Technologies) at room temperature for 30 min followed by addition to the oxLDL-plated ELISA wells. Incubation of CRP-LPP mimics was carried out for 1 h at 37°C and CRP binding was detected using biotinylated polyclonal antibody (1:5,000) and streptavidin conjugated-HRP (1:20,000). After TMB color development, the absorbance was measured at 450 nm on a microplate reader and the binding for each sample was normalized to a pCRP positive control.
Atomic force microscopy
Individual high binding Microlon® ELISA wells were cut out from the 96-well plate, cleaned with 70% ethanol and dried with N2 gas. LDLs were immobilized by either using an anti-ApoB polyclonal antibody (Academy Biomedical Co.) for capture or directly plated onto the ELISA well. After rinsing and drying, topographical images were acquired in air using an MFP-3D SA atomic force microscope (AFM) (Asylum Research). High aspect ratio AFM cantilevers with aluminum reflex back-coating (Model: NCHR, k = 42 N/m, fo = 300 kHz, NanoWorld) were used and the images were scanned at 0.5 Hz with 512 × 512 pixels resolution. AFM images were subjected to 2nd or 3rd order polynomial flattening as needed.
Statistical analysis
Statistical analysis was evaluated using Student’s t-test and p-value of < 0.05 was considered as significant. Data was reported as mean ± SD from at least three independent experiments.
Conclusions
This study provides additional insight that shows CRP binds preferentially to highly curved membranes, suggesting the influence of membrane curvature on CRP recognition of oxidized or damaged membranes that is independent of lipid oxidation.
Supplementary Material
Acknowledgments
Support from NSF CBET-1033161 (SMR), NIH 1R15GM088960-01 (SMR) and AHA 11POST5560045 (MSW) is acknowledged. We thank the UCD Biology department for use of the Odyssey imager, and the University of Denver Physics department for use of the AFM.
Abbreviations
- CRP
C-reactive protein
- CVD
cardiovascular diseases
- mCRP
modified C-reactive protein
- LPP
lipoprotein
- LDL
low density lipoprotein
- oxLDL
oxidized low density lipoprotein
- PC
phosphatidylcholine
- PCh-KLH
phosphocholine conjugated keyhole limpet hemocyanine
- PCh-BSA
phosphocholine conjugated bovine serum albumine
- AuNPs
gold nanoparticles
- TMB
3,3′,5,5′-tetramethylbenzidine
- AFM
atomic force microscope
- CAD
coronary artery disease
References
- 1.Biro A, Rovo Z, Papp D, Cervenak L, Varga L, Fust G, Thielens NM, Arlaud GJ, Prohaszka Z. Immunology. 2007;121:40–50. doi: 10.1111/j.1365-2567.2007.02535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black S, Kushner I, Samols D. J Biol Chem. 2004;279:48487–48490. doi: 10.1074/jbc.R400025200. [DOI] [PubMed] [Google Scholar]
- 3.Pepys MB, Hirschfield GM. Journal of Clinical Investigation. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N, Qian HW, von zur Muhlen C, Hagemeyer CE, Ahrens I, Chin-Dusting J, Bobik A, Peter K. Circ Res. 2009;105:128–U155. doi: 10.1161/CIRCRESAHA.108.190611. [DOI] [PubMed] [Google Scholar]
- 5.Du Clos TW, Mold C. Immunol Res. 2004;30:261–277. doi: 10.1385/IR:30:3:261. [DOI] [PubMed] [Google Scholar]
- 6.Torzewski J, Torzewski M, Rist C, Mortensen RF, Zwaka TP, Bienek M, Waltenberger J, Koenig W, Schmitz G, Hombach V. Arterioscl Throm Vas. 2000;20:2094–2099. doi: 10.1161/01.atv.20.9.2094. [DOI] [PubMed] [Google Scholar]
- 7.Thompson D, Pepys MB, Wood SP. Struct Fold Des. 1999;7:169–177. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 8.Bhakdi S, Torzewski M, Klouche M, Hemmes M. Arterioscl Throm Vas. 1999;19:2348–2354. doi: 10.1161/01.atv.19.10.2348. [DOI] [PubMed] [Google Scholar]
- 9.Bhakdi S, Dorweiler B, Kirchmann R, Torzewski J, Weise E, Tranumjensen J, Walev I, Wieland E. J Exp Med. 1995;182:1959–1971. doi: 10.1084/jem.182.6.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, Okura T, Higaki J, Graf K, Fleck E, Hsueh WA, Law RE. Circulation. 2004;110:579–587. doi: 10.1161/01.CIR.0000136999.77584.A2. [DOI] [PubMed] [Google Scholar]
- 11.Gershov D, Kim S, Brot N, Elkon KB. J Exp Med. 2000;192:1353–1363. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji SR, Wu Y, Zhu L, Potempa LA, Sheng FL, Lu W, Zhao J. Faseb J. 2007;21:284–294. doi: 10.1096/fj.06-6722com. [DOI] [PubMed] [Google Scholar]
- 13.Chang MK, Binder CJ, Torzewski M, Witztum JL. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khreiss T, Jozsef L, Potempa LA, Filep JG. Circulation. 2004;109:2016–2022. doi: 10.1161/01.CIR.0000125527.41598.68. [DOI] [PubMed] [Google Scholar]
- 15.Edwards KM, Gewurz H, Lint TF, Mold C. Journal of Immunology. 1982;128:2493–2496. [PubMed] [Google Scholar]
- 16.Eisenhardt SU, Thiele JR, Bannasch H, Stark GB, Peter K. Cell Cycle. 2009;8:3885–3892. doi: 10.4161/cc.8.23.10068. [DOI] [PubMed] [Google Scholar]
- 17.Khreiss T, Jozsef L, Hossain S, Chan JSD, Potempa LA, Filep JG. J Biol Chem. 2002;277:40775–40781. doi: 10.1074/jbc.M205378200. [DOI] [PubMed] [Google Scholar]
- 18.Ji SR, Wu Y, Potempa LA, Liang YH, Zhao J. Arteriosclerosis Thrombosis and Vascular Biology. 2006;26:935–941. doi: 10.1161/01.ATV.0000206211.21895.73. [DOI] [PubMed] [Google Scholar]
- 19.Jiang HX, Siegel JN, Gewurz H. Journal of Immunology. 1991;146:2324–2330. [PubMed] [Google Scholar]
- 20.Greenberg ME, Li XM, Gugiu BG, Gu XD, Qin J, Salomon RG, Hazen SL. Journal of Biological Chemistry. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- 21.Hazen SL, Chisolm GM. Proc Natl Acad Sci U S A. 2002;99:12515–12517. doi: 10.1073/pnas.212532799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A, Witztum JL. Nature Medicine. 2002;8:1218–1226. doi: 10.1038/nm1102-1218. [DOI] [PubMed] [Google Scholar]
- 23.van Tits L, de Graaf J, Toenhake H, van Heerde W, Stalenhoef A. Arterioscl Throm Vas. 2005;25:717–722. doi: 10.1161/01.ATV.0000157979.51673.2c. [DOI] [PubMed] [Google Scholar]
- 24.Singh SK, Suresh MV, Prayther DC, Moorman JP, Rusinol AE, Agrawal A. Clin Chim Acta. 2008;394:94–98. doi: 10.1016/j.cca.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agrawal A, Simpson MJ, Black S, Carey MP, Samols D. J Immunol. 2002;169:3217–3222. doi: 10.4049/jimmunol.169.6.3217. [DOI] [PubMed] [Google Scholar]
- 26.Hazen SL. Journal of Biological Chemistry. 2008;283:15527–15531. doi: 10.1074/jbc.R700054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackiewicz MR, Hodges HL, Reed SM. J Phys Chem B. 2010;114:5556–5562. doi: 10.1021/jp911617q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanauer M, Pierrat S, Zins I, Lotz A, Sonnichsen C. Nano Letters. 2007;7:2881–2885. doi: 10.1021/nl071615y. [DOI] [PubMed] [Google Scholar]
- 29.Volanakis JE, Narkates AJ. Journal of Immunology. 1981;126:1820–1825. [PubMed] [Google Scholar]
- 30.Volanakis JE, Wirtz KW. Nature. 1979;281:155–157. doi: 10.1038/281155a0. [DOI] [PubMed] [Google Scholar]
- 31.Potempa LA, Maldonado BA, Laurent P, Zemel ES, Gewurz H. Mol Immunol. 1983;20:1165–1175. doi: 10.1016/0161-5890(83)90140-2. [DOI] [PubMed] [Google Scholar]
- 32.Chou C, Hsu HY, Wu HT, Tseng KY, Chiou A, Yu CJ, Lee ZY, Chan TS. J Biomed Opt. 2007;12 doi: 10.1117/1.2714029. [DOI] [PubMed] [Google Scholar]
- 33.Li W, Wang KM, Tan WH, Ma CB, Yang XH. Analyst. 2007;132:107–113. doi: 10.1039/b614138b. [DOI] [PubMed] [Google Scholar]
- 34.McCauley TG, Hamaguchi N, Stanton M. Anal Biochem. 2003;319:244–250. doi: 10.1016/s0003-2697(03)00297-5. [DOI] [PubMed] [Google Scholar]
- 35.Wang MS, Black JC, Knowles MK, Reed SM. Analytical and Bioanlytical Chemistry. 2011;401:1309–1318. doi: 10.1007/s00216-011-5174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagle J, Tristram-Nagle S. Current Opinion in Structural Biology. 2000;10:474–480. doi: 10.1016/s0959-440x(00)00117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Beer FC, Soutar AK, Baltz ML, Trayner IM, Feinstein A, Pepys MB. J Exp Med. 1982;156:230–242. doi: 10.1084/jem.156.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chait A, Brazg RL, Tribble DL, Krauss RM. Am J Med. 1993;94:350–356. doi: 10.1016/0002-9343(93)90144-e. [DOI] [PubMed] [Google Scholar]
- 39.Austin MA. Int J Clin Lab Res. 1994;24:187–192. doi: 10.1007/BF02592460. [DOI] [PubMed] [Google Scholar]
- 40.Ohmura H, Mokuno H, Sawano M, Hatsumi C, Mitsugi Y, Watanabe Y, Daida H, Yamaguchi H. Metabolism. 2002;51:1081–1087. doi: 10.1053/meta.2002.34695. [DOI] [PubMed] [Google Scholar]
- 41.Austin MA, Breslow JL, Hennekens CH, Buring JE, Willett WC, Krauss RM. Jama. 1988;260:1917–1921. [PubMed] [Google Scholar]
- 42.Austin MA. Curr Atheroscler Rep. 2000;2:200–207. doi: 10.1007/s11883-000-0021-4. [DOI] [PubMed] [Google Scholar]
- 43.Sacks FM, Campos H. J Clin Endocrinol Metab. 2003;88:4525–4532. doi: 10.1210/jc.2003-030636. [DOI] [PubMed] [Google Scholar]
- 44.Grundy SM. Clin Chem. 1995;41:139–146. [PubMed] [Google Scholar]
- 45.Hermetter A, Moumtzi A, Trenker M, Flicker K, Zenzmaier E, Saf R. J Lipid Res. 2007;48:565–582. doi: 10.1194/jlr.M600394-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Rhode S, Grurl R, Brameshuber M, Hermetter A, Schutz GJ. Journal of Biological Chemistry. 2009;284:2258–2265. doi: 10.1074/jbc.M807591200. [DOI] [PubMed] [Google Scholar]
- 47.Schutz GJ, Rhode S, Grurl R, Brameshuber M, Hermetter A. J Biol Chem. 2009;284:2258–2265. doi: 10.1074/jbc.M807591200. [DOI] [PubMed] [Google Scholar]
- 48.Hazen SL, Zhang RL, Shen ZZ, Wu WJ, Podrez EA, MacPherson JC, Schmitt D, Mitra SN, Mukhopadhyay C, Chen YH, Cohen PA, Hoff HF, Abu-Soud HM. Circ Res. 1999;85:950–958. doi: 10.1161/01.res.85.10.950. [DOI] [PubMed] [Google Scholar]
- 49.McMahon HT, Gallop JL. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 50.Cui Q, Yoo J. Biophys J. 2009;97:2267–2276. doi: 10.1016/j.bpj.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Visser AJWG, Borst JW, Visser NV, Kouptsova O. Bba-Mol Cell Biol L. 2000;1487:61–73. doi: 10.1016/s1388-1981(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 52.Megli FA, Russo L. BBA-BIOMEMBRANES. 2008;1778:143–152. doi: 10.1016/j.bbamem.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 53.Schutz GJ, Plochberger B, Stockner T, Chiantia S, Brameshuber M, Weghuber J, Hermetter A, Schwille P. LANGMUIR. 2010;26:17322–17329. doi: 10.1021/la1026202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volanakis JE. Molecular Immunology. 2001;38:189–197. doi: 10.1016/s0161-5890(01)00042-6. [DOI] [PubMed] [Google Scholar]
- 55.Shrive AK, Cheetham GMT, Holden D, Myles DAA, Turnell WG, Volanakis JE, Pepys MB, Bloomer AC, Greenhough TJ. Nature Structural Biology. 1996;3:346–354. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 56.Agrawal A, Singh SK, Suresh MV, Voleti B. Annals of Medicine. 2008;40:110–120. doi: 10.1080/07853890701749225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rowe IF, Soutar AK, Trayner IM, Thompson GR, Pepys MB. Clinical and Experimental Immunology. 1984;58:237–244. [PMC free article] [PubMed] [Google Scholar]
- 58.Ji SR, Wu Y, Potempa LA, Qiu Q, Zhao J. International Journal of Biochemistry & Cell Biology. 2006;38:648–661. doi: 10.1016/j.biocel.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Basu S, Pande S, Jana S, Bolisetty S, Pal T. Langmuir. 2008;24:5562–5568. doi: 10.1021/la8000784. [DOI] [PubMed] [Google Scholar]
- 60.Sitaula S, Mackiewicz MR, Reed SM. Chem Commun. 2008:3013–3015. doi: 10.1039/b801525b. [DOI] [PubMed] [Google Scholar]
- 61.Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotte-Taylor K, Horkko S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V, Witztum JL. Atherosclerosis Vi. 2001;947:214–223. doi: 10.1111/j.1749-6632.2001.tb03943.x. [DOI] [PubMed] [Google Scholar]
- 62.Taylor KE, van den Berg CW. Immunology. 2007;120:404–411. doi: 10.1111/j.1365-2567.2006.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin-Nizard F, Furman C, Delerive P, Kandoussi A, Fruchart JC, Staels B, Duriez P. J Cardiovasc Pharmacol. 2002;40:822–831. doi: 10.1097/00005344-200212000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.