

Abstract
Tumor-stroma interactions play a crucial role in cancer progression by eliciting factors that promote proliferative, angiogenic and invasive supports to the tumor microenvironment. Mesenchymal stromal/stem cells (MSC) contribute to stroma in part as cancer-associated fibroblasts (CAF), but a complete understanding of how MSC contribute to the tumor stroma is lacking. In this study, we show how CAF phenotypes rely upon MSC expression of the multifunctional cell surface glycoprotein CD44, a putative stem cell marker. Through bone marrow transplantation experiments in a transgenic mouse model of cancer, we determined that CD44 deficiency leads to a relative reduction in the contribution of bone marrow-derived cells to tumor stroma. CD44 attenuation in MSC limited their expression of CAF markers induced by tumor conditioning, and these MSC migrated poorly and provided weak angiogenic support compared to wild-type MSC. These defects were linked to deficiencies in the ability of CD44-attenuated MSC to transcriptionally upregulate Twist expression. Together our results establish that CD44 expression contributes critical functions in the tumor stroma.
Keywords: MSC:mesenchymal stem cell/multipotent stromal cell, tumor microenvironment, migration, CD44, Twist, tumor/carcinoma associated fibroblasts (TAF/CAF), FAP, FSP, α-SMA
Introduction
Mesenchymal stem/stromal cells (MSC) are a population of multipotent stem cells that exist in most tissues of the body. Under circumstances of growth or repair, these resident progenitor cells repopulate the tissue and provide structural support and paracrine stimulation. They are known to migrate towards inflamed/injured environments, including tumors (1-3) and can be induced to express markers associated with tumor/carcinoma associated fibroblasts (TAF/CAF) and myofibroblasts.(4-6)
Stromal cells of mesenchymal phenotype fill the interstitial space of tumors and provide growth factors, matrix remodeling factors and other tumorigenic supportive factors. (7, 8) They are often referred to as activated (myo)fibroblasts or TAF and have been shown to originate from multiple sources including local tissue-derived fibroblasts, local tissue-derived MSC (9), bone marrow-derived MSC (4), and cancer cells through the epithelial to mesenchymal transition (EMT)(10, 11). Regardless of their origin, we and others have shown the pro-tumorigenic (4, 12, 13) and pro-metastatic(14) effects of these stromal cells within the tumor microenvironment. Furthermore, the presence of stromal cells is not restricted to a specific tumor type as TAF, myofibroblasts, MSC, and fibroblasts have been isolated from several different tumor types, including breast,(15) pancreatic,(16) gastric,(17) and lung.(18)
The TAF population is a heterogeneous population of cells intertwined throughout the tumor parenchyma. We now divide this population into at least 2 subpopulations: one that is defined by a pericytic role, termed an activated myofibroblast and defined by the expression of α smooth muscle actin (α-SMA) and the chondroitin sulfate neural glial proteoglycan (NG2); the other that is defined by a fibroblastic role, termed an activated fibroblast and defined by the expression of the seprase, fibroblast activation protein (FAP) and the calcium binding protein, fibroblast specific protein (FSP).(5)
While our TAF population is loosely defined by a selection of markers, all of the stromal cells express CD44. This multifunctional glycoprotein is expressed on many cell types from progenitor populations to lineage-specific cells and commonly regulates cell-cell adhesion and migration. Its most noted ligands include hyaluronan (HA) and osteopontin (19-21). Both ligands have been implicated as prognostic markers of cancer progression and metastases in breast, prostrate, lung, and ovarian cancers.(22-26)
In this study, we examined the deficiencies of tumor stromal mesenchyme lacking CD44 using murine tumor models. We found that CD44-/- MSC form stroma-deficient tumor microenvironments: first, because of their inability to fully differentiate into a TAF subpopulation, the activated fibroblast, as identified by the expression of FAP and FSP; second, because of impaired migratory capacity; and third, because of their diminished pro-angiogenic supportive capabilities.
Materials and methods
Cell culture
Primary human bone marrow MSC, WT murine MSC and transgenic CD44-/- murine MSC (Jackson Laboratory, Bar Harbor, ME) were isolated as previously described (Kidd et al., 2009) and grown in minimum essential medium, alpha (α-MEM) with 20% fetal bovine serum (FBS) and 10% penicillin/streptomycin/gentamicin (Gibco/Invitrogen, Carlsbad, CA). All primary MSC were used between passages 1 and 6. Expression of cell surface markers CD105, CD90, CD44, CD73, CD140b, CD146, and CD166 and lack of expression of CD31, CD45 and CD34 were validated in human MSC by flow cytometry. MSC were also subjected to colony formation assays and differentiation assays (Figures S3 and S4), including assays of adipocyte, osteoblast and chondrocyte differentiation as previously described (Kidd et al., 2009). Tumor cell lines were purchased from ATCC (Manassas, VA) and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS and 10% penicillin/streptomycin/gentamicin.
Antibodies
Antibodies to CD31, α-SMA, FAP, CD44, PDGFR-β, vimentin and GFP (Abcam Inc, Cambridge, MA), β-gal (Novus Biologicals, Littleton, CO), RFP (Rockland, Gilbertsville, PA), FSP (Dako, Glostrup, Denmark), NG2 (Millipore, Billerica, MA) and osteopontin (R&D Systems, Minneapolis, MN) and secondary Alexafluor antibodies 647, 594, and 488 (Invitrogen, Carlsbad, CA)were purchased and used according to manufacturer's instructions.
Immunohistochemistry/Immunofluorescence
Tumor sections were deparaffinized in a series of xylene and ethanol gradient incubations before being subjected to boiling sodium citrate buffer. Sections were blocked in 3% BSA, 2% FBS for 2 h before being stained with primary antibody (consecutively for multiple primaries) for 2 h at room temperature or overnight at 4°C and secondary antibody for 2 h at room temperature. Slides were stained with DAPI (Sigma, St. Louis, MO) before coverslip application over Dako Fluorescent Mounting Medium. Multispectral data image analysis was performed using an Olympus Ix81 microscope with a Nuance camera attachment and InForm (Perkin Elmer, Waltham, MA, USA). All images were taken and analyzed with a single acquisition algorithm utilizing the Nuance and InForm software (Perkin Elmer, Waltham, MA, USA). Fluorescent intensity was calculated per cell based on nuclear dapi stain.
Fluorescence activated cell sorting
Cells were subjected to trypsinization, washed, filtered and resuspended in ice-cold PBS supplemented with 2% FBS. Cells were then sorted into GFP+ and GFP- sub-populations (BD FACS Aria).
Western blotting
Cells were lysed in protein lysis buffer containing 50mM HEPES (238.3g/mol), 300mM NaCl (58.4 g/mol), 2mM EDTA (372.24 g/mol), 50mM NaF (41.99 g/mol), 2mM sodium orthovandate (183.91 g/mol), 10% (v/v) glycerol, 2% (v/v) NP40, 1% (v/v) Triton X- 100 and a cocktail of protease and phosphatase inhibitors (Sigma,. After 1-2 hours on ice, lysates were spun down for 30 min at 13,000rpm, lysate supernatant was quantified by Bradford assay (Bio-Rad, Hercules, CA) and then lysate was boiled with loading buffer and loaded onto a 12% SDS-PAGE gel (Bio-Rad, Hercules, CA). After electrophoresis, proteins were transferred onto Hybond-P membranes (Amersham Pharmacia Biotech, Piscataway, NJ), or nitrocellulose odyssey membranes (Licor Biosciences, Lincoln, NE) which were subjected to immunoblotting. Signals were detected using ECL-Plus (Bio-Rad, Hercules, CA). Alternatively, if a fluorophore-conjugated secondary antibody was used, the immunoblot signals were detected on the Odyssey infrared imaging system (Licor Biosciences).
Chromatin immunoprecipitation
CD44 (Abcam), Stat3 and p300 (Cell Signaling) antibodies were used in the ChIP assay performed using the EZ-ChIP kit (Millipore) according to the manufacturer's instructions. Briefly, MSC were first cross-linked with 1% formaldehyde for 10 min. The reaction was stopped by adding 2.5 M glycine at a 1/20 volume. Cell lysis and sonication were followed by overnight antibody incubations at 4 °C. Finally, cross-linking was reversed by a 3 hr incubation at 65°C and purified DNA analyzed by quantitative PCR. Twist1 proximal promoter F: 5′-CGGGGGAGGGGGACTGGAAAGC-3′; Twist 1R: 5′- AGGCCTCCTGGAAACGGTGCCG-3′; Twist1 distal promoter F: 5′-TACTCCAGCGCGGTGCACAAAACT-3′; Twist1 distal promoter R: 5′- AACGAAGAGCCCCAAAGAGGGTGT-3′. (Yang et al., 2008)
Viral transduction
Adenoviral vectors
Soluble CD44 adenoviral vector was (27), amplified in 293T cells, and concentrated on a cesium chloride gradient.
Lentiviral vectors
Five GIPZ shRNA lentiviral constructs against CD44 (clone ID: 1-V2LHS_111680; 2-V2LHS_111682; 3- V2LHS_111684; 4-V3LHS_334831; 5-V3LHS_334830) and one negative control shRNA (RHS4346) (Open Biosystems, Huntsville, AL) were transfected into HEK-293T cells with pPax and pMD2 plasmids using JetPrime Transfection reagent or 50mM BES solution. Supernatants were collected and filtered after 72 h for a total of 40mL. Virus was concentrated in 8.5% polyethylene glycol (8.4mL PEG- from 50% stock solution) and 0.3M sodium chloride (3.75mL NaCl- from 4M stock solution) and balanced with PBS to equal volume. The solution was placed on a shaker at 4°C for 90 min and then centrifuged at 4000rpm at 4°C at for 45min. The resulting pellet was resuspended in 300μl PBS, snap frozen, and stored at -80°C. For transduction, 20μl of each concentrate was combined with 5μl of 5mg/ml polybrene stock in 10mL of fresh 20% serum medium. The medium was changed 24 h after transduction. Cells were sorted by FACS for GFP positivity. ffLuc/cGFP co-expressing vector was made by subcloning the ffLuc from pGL4.51[luc2/CMV/Neo] Vector (Promega, Madison, WI) into pCDH-CMV-MCSEF1- copGFP_CD511B-1 (System Biosciences, Mountain View, CA).
Migration assay
MSC migration was assayed by using 8μm pore, 6.5mm-transwell inserts in a 24-well plate. MSC serum-starved for 18h were plated at 4×104 cells per well, with TCM, serum medium, or serum free medium for 24 h. Transwell inserts were analyzed by manual cell counts from captured images (Olympus BX41 microscope with an Olympus DP70 camera attachment) and by quantitative counts obtained by solubilizing stained transwell membranes in 300μl 2% deoxycholic acid for 4 h then measuring optical density at a fixed wavelength of 595nm for each sample (Beckman Du 640 Spectrophotometer). The following migratory inhibitors used were: 1- MMP inhibitor V (Calbiochem, Rockland, MA; #444285) this MMP inhibitor is selective for MMP2 at <20nM, MMP9 at <200nM and MMP14 at <1μM. We used multiple concentrations because our target inhibition was of MMP14, however migration is also inhibited when MMP2 and MMP9 are inhibited at concentrations lower than 1μM; 2- oHA(28); 3- s44, an adenoviral vector expressing soluble CD44, was transfected into MSC or into Skov-3 tumor cells. Secreted s44 was present in the medium 48-72 hours post-transfection and was used for subsequent experiments.
Tube formation assay
HUVEC (American Type Culture Collection) were cultured in EGM2 (Clonetics, Workingham, UK) and plated (3×104 per well) into a 24-well plate in triplicate. Medium was replaced 12 h later with MSC-conditioned medium and the cells were imaged every 12 h for 12 days. Wells were imaged and analyzed with an Olympus Ix81 microscope with a DSU confocal attachment and Slidebook software (Center Valley, PA). 4 images were taken per well. Tubes were manually counted and averaged among groups.
PCR
Maxima Hot Start 10× buffer, 25mM MgCl2, 25mM dNTP and 5U Taq was purchased from Fermentas Life Sciences (Glen Burnie, MD). Primers purchased from Integrated DNA Technologies (Coralville, IA) were prepared as 10μM stock solutions and are listed in Table S1.
Real time PCR
RNA (1μg) was reverse transcribed using a kit from Signalway Biotechnology (Pearland, TX). cDNA (20μL) was diluted with 100μL of distilled RNAse/DNase free water. Sybr-green master mix was purchased from Applied Biosystems (Foster City, CA): 1.0μL cDNA was used per reaction. 0.5μL forward and reverse primer stocks, 10μL distilled water, and 12μL Sybr-green master mix was added per well for a total volume of 25μL. All reactions were done in triplicate and carried out in a 96well fast plate in an ABI7900HT Thermocycler (Applied Biosystems). Relative expression was calculated by normalizing samples to GAPDH (human) or 18s (murine) and then compared between control and sample sets using the delta-delta Ct method. All data are presented as fold change over control (2-ΔΔCt).
In vivo murine tumor models
All transgenic mice used were female between the ages of 5-12 weeks. All mice weighed an average of 25g.
Human xenograft model
Nod/SCID mice (Jackson Laboratory strain NOD.CB17-Prkdcscid/J; stock #001303) were housed and treated according to protocols approved by MDACC Institutional Animal Care and Use Committee. Mice (n=20) were anesthetized and were injected subcutaneously with 5×105 Renilla-luciferase (rLuc)-labeled Skov-3 cells suspended in 100μL of PBS into both hind limbs. 56 days after injection, BLI (IVIS-Xenogen 100 system; Caliper Lifesciences, Hopkinton, MA) confirmed tumor engraftment. Then, each mouse received ffLuc-labeled MSC (1×106) in 100μl PBS by tail vein injection. The mice were divided into four equal groups of five mice each: two groups received an intratumoral injection of the soluble CD44-expressing adenovirus (Ad-s44) in the right hindlimb 48 h prior to intravenous administration of MSC. BLI was conducted daily for 5 consecutive days after MSC administration. The ffLuc substrate, d-luciferin (100μL of 4mg/mL in PBS; Biosynth International Inc, Itasca, IL) or rLuc substrate coelenterazine (100μL of 200ng/mL in PBS; Biotium, Inc., Hayward, CA) was injected i.p. 5 minutes prior to imaging. Images were analyzed with the Living Image Software (IVIS-Xenogen).
Murine models
WT and transgenic CD44-knockout murine MSC were admixed with 1×106 4T1 tumor cells and injected into the mammary fat pad of Nod-SCID mice (n=20). Three weeks following injection, mice were sacrificed and tumors and organs removed for immunohistochemical analysis. These tumors were weighed in order to compare the growth differences between the tumors in each group, however upon analysis, the weights were not taken into account because we acquired, at random, the same number of images per tumor which were then used for comparative, quantitative analysis.
EO771 murine mammary carcinoma cells (2×105) were injected into the mammary fat pad of C57/B6 CD44 transgenic mice (Jackson Laboratory strain B6.129(Cg)-Cd44tm1Hbg/J; stock #005085) and control WT C57/B6 mice (Jackson Laboratory strain C57BL/6J; stock #000664) (n=20). Tumor growth was followed for 4 weeks with daily caliper measurements and mice were sacrificed just prior to reaching excessive tumor burden, in compliance with IACUC guidelines. Excised tumors were weighed and suspended in formalin in preparation for immunohistochemical analysis. It is important to note that we were unable to observe lung metastases in any of the tumor model groups.
Bone marrow transplant model
The tumors from the RFP+ BMT mice (n=15) were analyzed for co-expression of RFP (to label the BMT cells) or GFP (to label the host cells) and the TAF markers FAP, FSP, and α-SMA; and the endothelial marker CD31. Likewise, tumors from the CD44-knockout BMT mice (n=15) were analyzed for co-expression of LacZ (to label the BMT cells) or GFP (to label the host cells) and FAP, FSP, α-SMA, and CD31. Tumor size, by weight was not significantly different between analyzed samples. Co-expression was characterized by InForm software to identify immunostaining co-localization. Briefly, each cell was identified based on nuclear DAPI stain (average of 2000 cells per image). The fluorescence intensity of each fluorescent label per cell was then enumerated. The fluorescence intensities were plotted (488 label on the Y axis and 594 on the X axis), and co-stained cells were quantified.
Percentages displayed in Figures 6A-C of GFP + or RFP + or β-gal co-stained cells in tumors from BMT mice were calculated averages of co-stained (double positive staining) cells per total number of cells per tumor image among five mice (10 images per tumor). Statistical significance was determined by the one tailed Student t-test with 90% CI.
Figure 6. Ectopic expression of Twist in CD44 knockdown MSC partially restores phenotype under tumor conditioned mediain vitro.
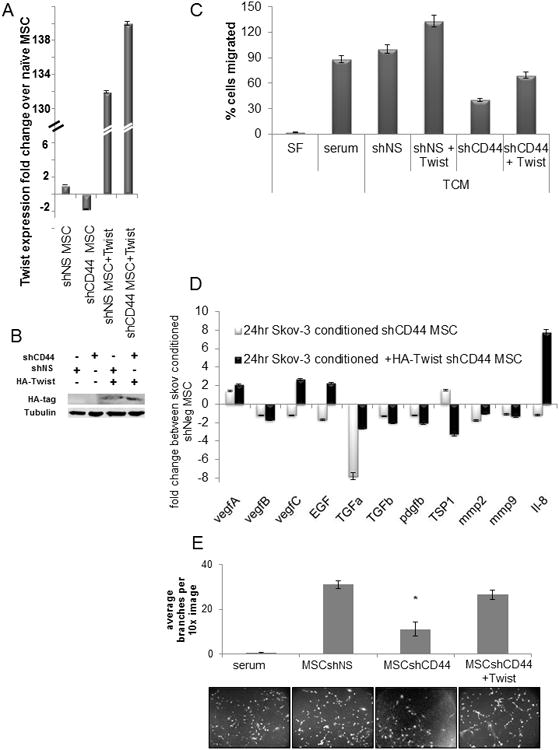
Validation of HA-tagged, ectopic Twist expression by (A) RT-PCR and (B) western blot. (C) Transwell migration assay showed that ectopic expression of Twist in sh44MSC increased the migration capacity 2-fold toward TCM and that this effect was not inhibited by oHA. (D) Ectopic Twist expression in sh44MSC showed an increase in angiogenesis-associated growth factor expression by RT-PCR compared to sh44MSC. Fold changes relative to shNSMSC expression levels. (E) HUVEC tube formation potential was significantly decreased (p<0.05) when conditioned with medium from shCD44MSC compared to medium from naïve MSC or shNS MSC but ectopic Twist expression restored pro-angiogenic potential in sh44MSC. Graph represents the number of branch points counted per image.
Data analysis
Analysis of the bioluminescent imaging was carried out by creating standard regions of interest around the right and left hind-legs to calculate the flux (photons per second) of the target signal in the given area. Relative quantities of MSC and tumor sizes were then calculated by normalizing the MSC flux against the tumor flux to achieve comparable numbers of migrated MSC toward each tumor individually. Statistical significance was determined by the Student t-test.
Results
CD44-/- mice show delay in tumor progression
We engrafted EO771 murine breast carcinoma cells (2×105) into the mammary fat pads of transgenic CD44-knockout (CD44-/-), LacZ-expressing mice and wild-type (WT), RFP-expressing mice. Despite 100% tumor engraftment in both groups, the CD44-/- mice survived significantly longer (p<0.05; log-rank test) than the WT mice (Figure 1A). Because mice were sacrificed according to institutional/IACUUC standards, mice were euthanized when tumor burden because too great, therefore, our survival curve suggests that CD44-/- mice bearing tumors develop slower than tumors in WT mice. Immunofluorescent staining for tumor stromal markers FAP, FSP and αSMA revealed that tumors from WT mice had significantly more accumulative stroma (p<0.01) than tumors from CD44-/- mice (Figure 1B). Eosinophilic structures including large, elongated stromal cytoplasms and collagen fibers were more readily visualized by H&E staining in the WT group than in the CD44-/- group revealing general differences in stromal architecture between the two groups (Figure 1C).
Figure 1. Tumor stroma incorporation is diminished in CD44-/- mice compared to controls.
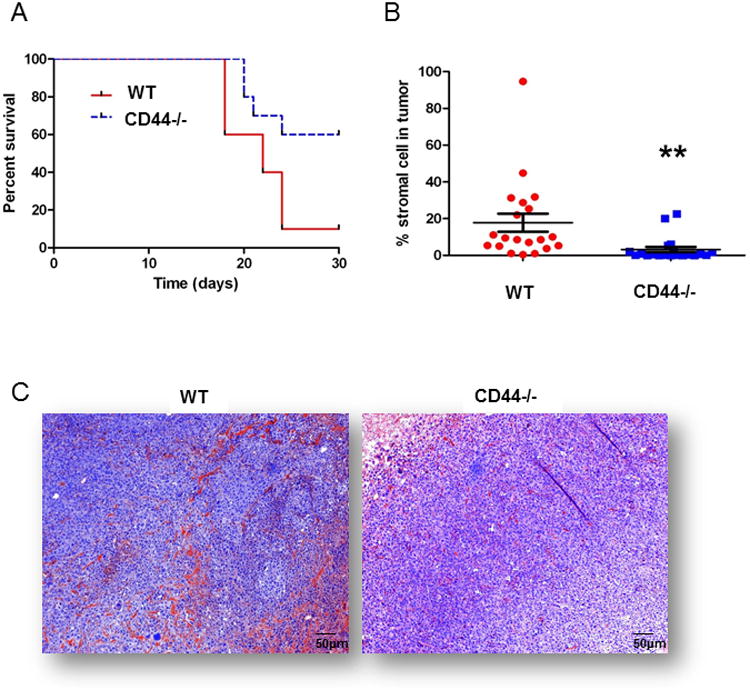
(A) EO771 tumor burdened CD44-/- mice survived significantly longer (p<0.05) than WT mice. Mice were euthanized due to increased burden of primary tumor in accordance with institutional standards. (B) Incorporation of host stroma into the EO771 tumors was significantly less (p<0.01) in tumors engrafted in CD44-/- (β-gal+) mice than in tumors engrafted in WT (RFP+) mice. Stroma % was measured based on the averaged accumulation of αSMA, FAP and FSP expression in both tumor models based on the immunofluorescent score algorithm using Nuance software. (C) H&E tumor section from CD44-/- mice shows that stromal cells were evenly dispersed with tumor cells whereas stromal cells formed clusters of linear structural patterns in WT mice.
Endogenous tumor incorporation of BM derived stromal components
Having observed diminished tumor stroma incorporation within CD44-/- mice compared to WT mice, we questioned whether the reduction in tumor associated stromal cells was niche-specific. There are two major sources of tumor stroma: bone marrow and local neighboring tissue. We performed bone marrow transplant (BMT) studies to examine the contribution of CD44-/- bone marrow (BM) compared to WT BM in WT recipient mice. Briefly, irradiated WT GFP+ mice underwent bone marrow transplantation with 2×106 whole BM cells from either CD44-/- donor mice (henceforth defined as CD44-/-BMTWT) or WT RFP+ donor mice (WT BMTWT; Figure S1A-B). Four weeks after BMT, following confirmation of bone marrow engraftment by flow cytometry (Figure S1C), mice received an orthotopic injection of EO771 tumor cells (5×105). We compared the non-hematopoetic populations of bone marrow in WT and CD-/- mice (n=3 each group) to confirm that population percentage of potential stromal progenitors were statistically insignificant based on CD45neg/Ter119neg bone marrow derived cells (Figure S1D).
Tumors were excised 5 weeks after engraftment and subjected to multiplex immunofluorescent imaging to infer the origin of the tumor stromal participants: donor BM (RFP+ WT or β-gal+ CD44-/-) or recipient host (GFP+ WT). The activated fibroblasts in this model were defined by FAP and FSP while the activated myofibroblasts were defined by αSMA and NG2. BM derived incorporation of activated fibroblasts into tumors showed significant discrepancy between transplant groups whereas there was no significant difference in the activated myofibroblast derived from the BM or host tissue (Figure S2A-C). A small percentage (0.05%+/-0.018%) of the activated fibroblasts were CD44-/- BM derived whereas 0.27%+/-0.044% of the activated fibroblasts were WT (RFP+) BM derived (p<0.001; Figure 2A). Furthermore, the lack of CD44-/-BM derived incorporation of activated fibroblasts was compensated by a significant increase (p<0.05) in WT host derived (GFP+) activated fibroblasts; 2.5%+/-0.47% in the CD44-/-BMT group versus 1.4%+/-0.25% in the WT BMT group (Figure 2B, 2C).
Figure 2. Tumors engrafted in mice with CD44-/- bone marrow transplants are deficient in activated fibroblast incorporation.
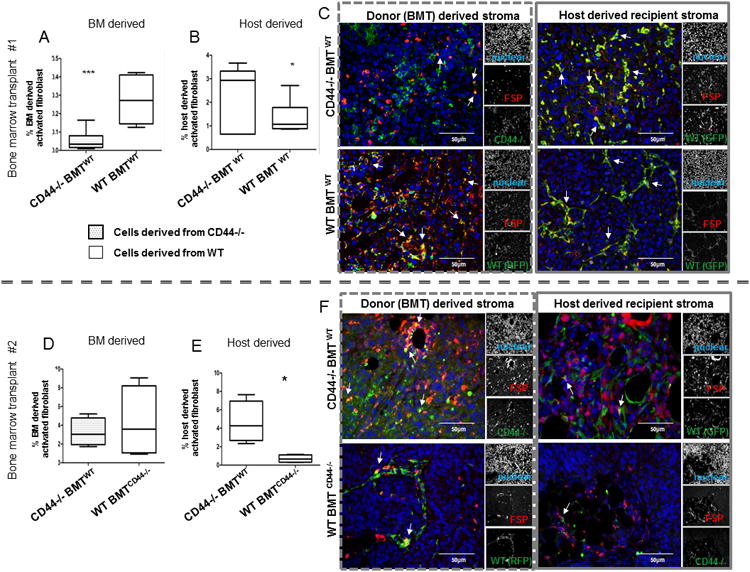
Percentage of double positive (DP) stromal cells expressing activated fibroblasts (FAP or FSP) with (A) donor (β-gal or RFP) BM derived cells in EO771 tumors (n=20 total) is significantly less (p<0.005) in CD44-/-BMTWT mice (0.05%+/-0.018%) compared to WT BMTWT mice (0.27%+/-0.044%); and (B) FAP/FSP expressing host-derived (GFP) cells within tumors is significantly increased (p<0.05) in CD44-/-BMTWT mice (2.5%+/-0.47%) compared to WT BMTWT mice (1.4%+/-0.25%). (C) Fluorescent images of FSP+ stromal cells in EO771 tumors in CD44-/-BMTWT mice compared to WT BMTWT mice co-stained with BM or host derived markers. Arrowheads emphasize the co-localized expression in each image. (D) Percentages of the DP activated fibroblasts incorporated into ID8 tumors (n= 10 total) are smaller in BM derived fraction in CD44-/-BMTWT mice (3.25%+/-0.75%) compared to WT BMTCD44-/- mice (4.28%+/-1.92%); and (E) there is a significantly higher activated fibroblast population derived from host cells in the CD44-/-BMTWT mice (4.63%+/-1.13%) compared to WT BMTCD44-/- mice (0.69%+/-0.21%). (F) Fluorescent images of FSP+ stromal cells in ID8 tumors in CD44-/-BMTWT mice compared to WT BMTCD44-/- mice co-stained with BM or host derived markers. Arrowheads emphasize the co-localized expression in each image.
Endogenous tumor incorporation of local tissue derived stromal components
This model was recapitulated in an inverse transplant series to assess the derivation of tumor stroma in a CD44-/- recipient/host background with WT BMT (WT BMTCD44-/-) compared to WT recipient/host with CD44-/- BMT (CD44-/- BMTWT). Using the orthotopic C57BL/6 ID8 ovarian tumor model (29) that mimics human progression of ovarian cancer and recruits more stroma than the C57BL/6 EO771 breast cancer tumor used previously, we compared the incorporation of the activated fibroblast and myofibroblast populations derived from host tissue or donor bone marrow. The BM derived activated fibroblasts from WT BMTCD44-/- mice were more abundant in engrafted tumors compared to those from CD44-/-BMTWT mice which followed the trend from the first transplant series without statistical significance (Figure 2D). But, we show that the host derived activated fibroblast was significantly higher (p<0.01) in the tumors engrafted in the CD44-/-BMTWT mice (4.63%+/-1.13) compared to those in the WT BMTCD44-/- mice (0.69%+/-0.21; Figure 2E, 2F) suggesting a compensation of the decreased activated fibroblast population derived from the CD44-/-BM. In this model, we also observed a significant increase in the BM derived myofibroblast populations from the CD44-/-BMTWT versus the WT BMTCD44-/- (0.34%+/-0.15% vs 6.3%+/-1.05%; Figure S2D-F).
CD44-/- murine MSC produce fewer TAF in vivo
The bone marrow transplant studies showed limited tumor stroma participation among the activated fibroblast populations derived from CD44-/- compared to WT. The MSC is a stromal precursor; therefore, we next wanted to investigate whether the loss of CD44 expression on MSC could hinder the capacity to form FAP+/FSP+ activated fibroblasts. To address this question, we increased the physiological ratio of stromal cells to tumor cells to emphasize and clearly visualize the stromal contribution within the tumor microenvironment. MSC derived from the CD44-/- mice were phenotypically similar to WT controls and could differentiate into chondrocyte, osteoblast (Figure S3B-D) and adipocyte lineages (not shown). To generate tumors, we used the 4T1 murine breast carcinoma line which produces a solid orthotopic tumor mass as opposed to the hemolytic EO771 breast cancer tumor model and is more easily accessible in the mammary fat pad as opposed to the IP cavity. Briefly, 4T1 tumor cells were engrafted into one mammary fat pad of SCID mice, and a 1:1 admixture of 4T1 cells with WT MSC or CD44-/- MSC was injected into the contralateral mammary fat pad of these mice.
We measured the FAP and FSP expression in the admixed tumors to confirm the presence of the activated fibroblasts as in the previous BMT models. We also looked at CD31 incorporation within the tumors to assess the potential to promote angiogenesis. FAP (p<0.001), FSP (p<0.001), and CD31 (p<0.005) were expressed at significantly higher levels in the WT MSC admixed tumors compared to the 4T1-only tumors. FAP (p<0.001; Figure 3A), FSP (p<0.001; Figure 3B) and CD31 (p<0.05; Figure 3C) were expressed significantly lower in CD44-/- MSC admixed tumors than in WT MSC admixed tumors. These data confirm that CD44-/- MSC are defective in producing FAP+/FSP+ activated fibroblasts, and the lack of stromal support is associated with a decrease in CD31+ cells within the tumor.
Figure 3. Tumor associated fibroblast marker expression is significantly decreased in tumors admixed with CD44-/- derived murine MSC.
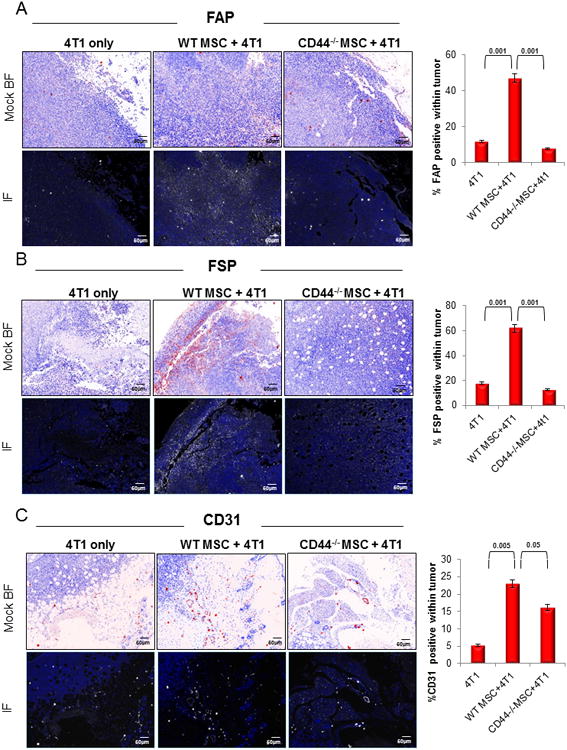
(A) Expression of FAP was significantly lower (p<0.001) in CD44-/-MSC admixed 4T1 tumors than in WT MSC admixed 4T1 tumors (n=20 total). (B) Similarly, FSP expression was significantly reduced (p<0.001) in CD44-/- MSC admixed 4T1 tumors than in WT MSC admixed 4T1 tumors. (C) CD31 expression, used as evidence of vascularization, was also significantly reduced (p<0.05) in CD44-/-MSC admixed 4T1 tumors compared to WT MSC admixed 4T1 tumors. Mock brighfield images were digitally generated from the fluorescent images and are shown in this figure to emphasize the staining and the tissue architecture on a white background.
Tumor conditioned medium activates differentiation and signaling cascades in MSC through CD44
Our preliminary studies suggest that binding of CD44 to its ligands (i.e.: HA and osteopontin) can trigger signaling cascades in MSC that can lead to TAF differentiation and increased migration potential. To analyze the effects of long term tumor stimulation on MSC in vitro, we first needed to identify a model tumor cell line that secreted a known CD44 ligand. We screened several pancreatic, breast, and ovarian human cancer cell lines for expression of CD44 ligands. More specifically, we looked at the potential for the tumor cell lines to produce osteopontin, as measured by ELISA, and HA by assessing the HA synthetase 2 (HAS2) expression levels. Most of the analyzed tumor cells lines -expressed low/negligible levels of HAS2 and osteopontin, however the ovarian carcinoma cell line Skov-3 expressed and secreted high quantities of osteopontin (Figure S4A-B). More importantly, we found that MSC cultured with Skov-3-tumor conditioned medium (TCM) expressed high levels of HAS2 suggesting the MSC use CD44 for autocrine and paracrine signals (Figure S4A). Skov-3 was thus chosen as our human tumor model for all subsequent experiments.
Human CD44 knockdown MSC are TAF deficient in vitro
Next, we wanted to confirm the role of CD44 deficiency in primary human MSC. We constructed a stable knockdown of CD44 in MSC using four shRNA hairpins (MSCsh44). By flow cytometry, MSCsh44 expressed surface markers common to the non-specific shRNA knockdown MSC (MSCshNS) including positive expression of CD105, CD90, CD73, CD146, CD166 and CD140b, and negative expression of CD45, CD31 and CD34 but the MSCsh44 were slightly hindered in their adipocyte differentiation capacity (Figure S3G) compared to the MSCshNS.
We then conditioned MSCsh44 and MSCshNS with tumor (conditioned) media (TCM) for 21 days in vitro in order to induce TAF marker expression. After long term tumor conditioning, the MSCshNS expressed FAP, FSP and α-SMA—three markers that we use as hallmarks of TAF. Importantly, the MSCsh44 expressed nearly negligible levels of FAP and FSP, similar to the results observed by IF in our mouse models. The MSCsh44 also expressed less α-SMA, vimentin and PDGFRβ than WT MSC, and expressed negligible levels of TSP1 and Twist1 (Figure 4A).
Figure 4. CD44-knockdown of human MSC decreases efficacy as tumor associated fibroblasts.
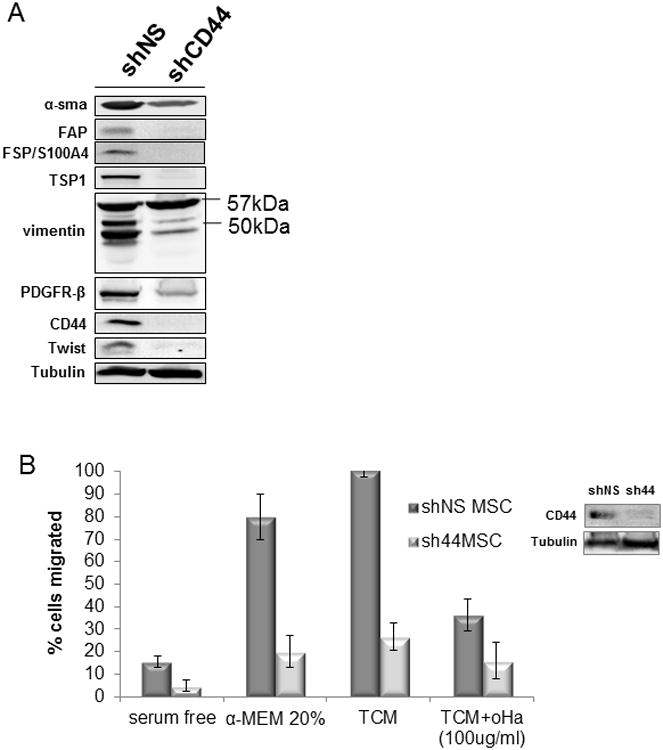
(A) Long-term (21 day) tumor-conditioned shCD44 MSC did not express FAP, FSP or CD44 and expressed diminished levels of α-SMA, PDGFRβ, TSP and Twist and were deficient in vimentin cleavage products when compared to the conditioned shNS MSC. (B) shCD44 MSC were deficient in migration toward TCM compared to shNS MSC. Hyaluronan oligomer (oHa) was used as a CD44 inhibition control on shNS MSC.
After we confirmed a deficiency in TAF marker expression in MSCsh44 compared to MSCshNS by western blot, we asked whether the knockdown of CD44 contributed to a migratory deficient phenotype in the MSCsh44 compared to MSCshNS. A transwell migration assay confirmed the defective migration of the MSCsh44 compared to MSCshNS towards normal serum media (Figure 4B). We then used a small HA oligomer (oHA) (28) to competitively bind to CD44 and block activity to show an alternative means of migratory inhibition through CD44 (Figure 4B). We next performed an in vivo migration study using a soluble CD44 (s44) which acts as a CD44 antagonist by competing for the available ligand (30). We engrafted human Skov-3 tumors subcutaneously in Nod/SCID mice to analyze the homing of intravenously injected ffLuc-labeled human MSC. One group was given an intratumoral injection of s44 and the other was given an intratumoral injection of PBS. Forty-eight hours after ffLuc-MSC injection, we assessed the capacity of these MSC to migrate to the tumor site by bioluminescent activity measured within the tumors. Bioluminescent signal was observed at significant lower levels (p<0.05) in the tumor injected with s44 (Figure S5A).
TCM induces CD44 activation on MSC
By flow cytometry, the surface expression of CD44 following 30min exposure to TCM was slightly lower than that of serum-cultured MSC and of MSC exposed to 24hr TCM (Figure S5B). Since activation and signaling via CD44 is known to be induced by a series of extracellular and intracellular cleavages (31), we sought to test whether TCM was activating CD44 by cleaving it. Because matrix metalloproteinase (MMP) 14 has been reported to cleave the extracellular portion of CD44 (32), we treated MSC with an MMP 14 inhibitor and showed a decrease in MSC transwell migration similar to the migration inhibition observed when cells were treated with soluble CD44 s44 or oHA antagonist (Figure S5C). Furthermore, the migration inhibition at 1μM MMP inhibitor coincided with the abrogation of CD44 cleavage products seen by western blot (Figure S5D).
CD44 activation by TCM stimulates Twist expression in MSC
One of the consequences of intracellular cleavage of CD44 is nuclear localization and transcriptional activation of target genes through association with Stat3 (33). By screening a number of Stat3-activated genes following TCM by PCR (data not shown), we only observed one gene, Twist1, that had a correlated increase in protein expression by immunofluorescence (Figure 5A). This observation was confirmed by western blot in the MSCshNS cells following TCM; furthermore, the MSCsh44 did not show increases in Twist1 expression comparable to MSCshNS by western (Figure 5B). We then confirmed by RT-PCR that Twist1 RNA expression was lower in the MSCsh44 than in naïve MSC or MSCshNS; this decrease was similar to the decrease in expression observed by s44 and MMP inhibition (Figure 5C) suggesting that the regulation of Twist1 by CD44 is transcriptional.
Figure 5. MSC-expressed CD44 acts as a transcriptional regulator under tumor conditioned mediain vitro.
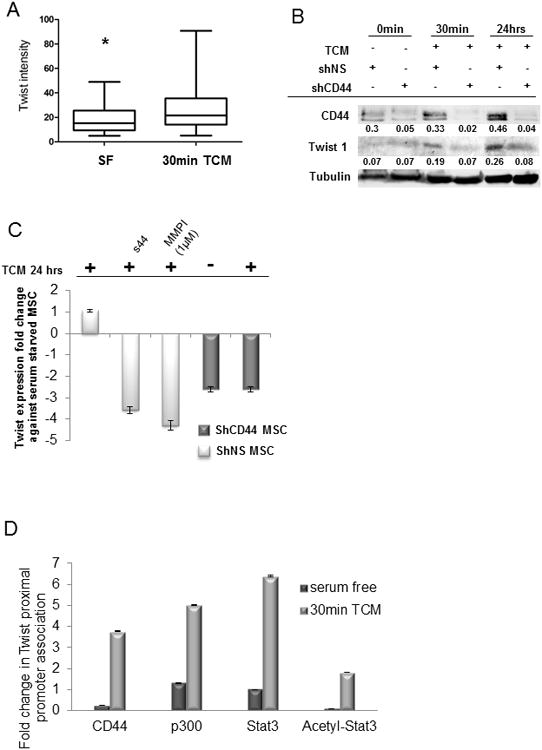
(A) Protein expression of Twist is increased (p<0.05) in MSC exposed to TCM compared to serum free medium by confocal microscopy. (B) By western blot, sh44MSC showed a decrease in Twist protein expression compared to shNS MSC after 30 min or 24 h of TCM. (C) By real-time PCR, shCD44MSC showed a decrease in Twist expression similar to shNS MSC treated with s44 antagonist or MMP inhibitor. Y axis is expression fold change. (D) After 30 min TCM, CD44 association with the Twist promoter increased by 3.5-fold. Furthermore, Stat3, acetyl-Stat3 and p300 association with the Twist promoter also increased.
To confirm the direct transcriptional regulation of Twist1 by CD44 in tumor-conditioned MSC we performed quantitative chromatin immunoprecipitation analysis. Results revealed that after 30 min exposure to TCM, CD44 association at the Twist1 promoter increased by 3.5-fold (Figure 5D). Furthermore, association of acetylated Stat3, Stat3, and p300 with the Twist1 promoter in MSC following TCM exposure increased by 2-fold, 6.5-fold and 5-fold, respectively (Figure 5D).
Modulation of tumor conditioned MSCsh44 with Twist re-expression
Next, we exogenously expressed Twist1 in MSCsh44 (Figures 6A-B) to see if we could regain function of MSC under TCM exposure. Expression of Twist in MSCsh44 increased the migration capacity under TCM by 25%, similar to the increase in migration capacity of MSCshNS (Figure 6C). We also examined expression of proangiogenic genes with and without exogenous Twist1 expression. After 24 h exposure to TCM, MSCsh44 ectopically expressing Twist1 had increases in Il8, VegfC, and EGF expression (Figure 6D). While protein secretion was not measured, we did analyze the functional effects of the proangiogenic factors produced by MSCsh44 ectopically expressing Twist1 compared to MSCsh44 and MSCshNS. Ectopic expression of Twist1 partially restored the ability of MSCsh44 conditioned medium to promote HUVEC tube formation (Figure 6E) suggesting the re-establishment of proangiogenic function of the MSCsh44.
Discussion
It is well established that MSC contribute to the tumor microenvironment in support of the tumor parenchyma. The interactions between tumor cells and stromal cells dictate tumorigenesis; stroma-rich breast tumors have a higher risk of relapse and confer shorter overall survival (34). In this current study, we showed that CD44 expression on tumor stromal precursors is necessary for their migration, incorporation and functionality within the tumor microenvironment as TAF—or more specifically as FAP/FSP-expressing activated fibroblasts.
Previous studies have shown the significance of CD44 in migration of MSC (20). Okamoto et al. proposed that CD44 transcription can be self-regulated and mediated by CBP/p300 in a glioma cell model (35). CD44, when appropriately stimulated, undergoes an extracellular cleavage that is followed by an intracellular cleavage leading to the translocation of the intracellular domain of CD44 into the nucleus where is has been shown to associate with Stat3 to promote transcription in a lung carcinoma model (33). We sought to identify the genes transcriptionally regulated by CD44 in MSC under exposure to TCM. Assessment of Stat3 regulated genes lead us to Twist, which was up-regulated in MSC under tumor conditioning, but significantly down-regulated under CD44 inhibition with s44. The mesoderm regulator Twist has been associated with migration, metastases, drug resistance, angiogenesis and EMT. (36-38) Decreased survival has been correlated to stromal fibroblast Twist expression in gastric cancer patients. (39) Our data support these findings and provide evidence that MSC whose CD44 expression is blocked results disruption of mesenchyme markers including Twist, FAP or FSP expression thereby producing dysfunctional activated fibroblasts.
TAF are known to have an inflammatory gene expression profile compared to normal fibroblasts (40), particularly in the breast tumor microenvironment (41). Pro-inflammatory gene expression leads to recruitment of macrophages, myofibroblasts, MSC, pericytes, and endothelial precursors that aid in tumor progression and angiogenesis. We showed a global decrease in the expression of proangiogenic factors from MSCsh44 under TCM in vitro. We suggest that the observed decrease in endothelial CD31 presence within the CD44-/-MSC admixed 4T1 tumors in vivo was due to lack of proangiogenic expression potential. All tumor models used in these studies were highly aggressive and therefore did not demonstrate a strong dependence on stromal support. This suggests that the differences observed between the CD44-/- and WT stromal microenvironments may be more pronounced in a less aggressive, or earlier stage tumor model.
Differences in tumor stroma recruitment have previously been described in the context of circulating bone marrow-derived cells and the “potency” of the tumor (42). The complex interface between the tumor parenchyma and the stroma collectively influences tumor progression. Our model shows a division of stroma participants based on marker specificity: the activated myofibroblast TAF and the activated fibroblast TAF. Individual expression of activated fibroblast markers FAP and FSP have been associated with disease and metastases (43-45). Our studies suggest that decrease in CD44 expression leads to decreases in both FAP and FSP expression, but further studies are needed to determine the complete mechanism of depletion of TAF gene expression as a result of CD44 knockdown. We did not observe a significant difference in size between tumors engrafted in the transgenic BMT models, nor did we observe metastatic lesions at the time the mice were sacrificed.
While the most profound difference in the transgenic BMT models was the activated fibroblast populations, we did see a significantly higher population of BM derived activated myofibroblasts (p<0.001) in the tumors engrafted in the CD44-/-BMTWT mice (6.32%+/-1.05) compared to those in the WT BMTCD44-/- mice (0.34%+/-0.15; Figure S2). This increase in myofibroblast from the CD44-/- BM cells could help explain the negligible tumor size difference between the BMT models because the BM derived cells that typically provide the activated fibroblast population (5) was able to supplement the lack of activated fibroblasts with an increase in activated myofibroblast population. In our exaggerated tumor stroma model where MSC were admixed with tumor cells at a 1:1 ratio, we did observe a significant difference in size between the 4T1 tumors contralateral to the CD44-/- admixed tumors compared to the 4T1 tumors contralateral to the WT admixed tumors suggesting that the CD44-/- MSC were not able to generate the factors necessary to elicit a proinflammatory signature that recruits additional tumor-supportive stromal participants including macrophages and endothelial cells (data not shown).
Anti-CD44 targeting methods have shown promising anti-tumor efficacy in animal model systems including the use of a soluble CD44 antagonist (27, 30). We used an intratumoral injection of adenoviral expressed soluble CD44 to reduce MSC incorporation into the tumor microenvironment indicating therapeutic avenues to suppress stroma incorporation within the tumor are possible. However, further studies are required to assess the global effects of CD44 inhibition on the normal cells and not just the tumor associated cell populations. One potential therapeutic avenue is to distinguish between CD44 variant forms among normal cells and tumor associated cells.
Our findings regarding the formation activated fibroblasts evokes broader implications on tumor biology as CD44 expression has been used as one of the definitive markers of cancer stem cell populations, particularly in breast cancer (46). We provide evidence in MSC that CD44 can be activated by tumor paracrine factors to induce and directly regulate Twist expression. This suggests that CD44 expression in mesenchymal transitioned (EMT) cancer cells may also have the ability to propagate their mesenchymal phenotype (10) and thus their own microenvironment through CD44 regulation of Twist. In fact, CD44 stimulation in breast cancer cells has been shown to activate Twist expression through lysyl oxidase activation to regulate an EMT phenotype.(47) Recently, Shangguan et al. reported that inhibition of TGF beta signaling by transducing MSC with BMP and Activin Membrane Bound Inhibitor blocked expression of FAP, FSP and α-SMA in tumor conditioned MSC (48). Furthermore, the TGFβ receptor contains a CD44 binding site through which CD44 stimulation has previously been shown to activate downstream TGFβ signaling (49). Our findings suggesting the importance of CD44 signaling in MSC role within the tumor microenvironment supports the potential for CD44 activations to stimulate multiple downstream pathways including TGFβ signaling.
In summary, we show that CD44-deficient stromal mesenchyme is incapable of participating in the tumor microenvironment as fully functional TAF. In particular, they are unable to form activated fibroblasts, but retain the ability to express activated myofibroblast markers. They display deficiencies in (1) tumor-tropic migratory capacity, (2) ability to induce angiogenesis, and (3) expression of activated fibroblast markers FAP and FSP. We show that tumor paracrine stimulation on the MSC activates CD44, which is able to directly regulate expression of Twist through interaction with Stat3. These results implicate CD44 on the tumor stromal population as a potential therapeutic target as tumor progression is associated with an increased presence of stromal cells. Furthermore, because of the breadth of potential cells from which stromal cells can be derived including cancer cells themselves, CD44 provides a potential anti-tumor target on the supportive stromal population as well as cancer cell population. Ongoing studies utilizing a pharmacological anti-CD44 antibody to inhibit tumor stroma formation are currently underway.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Cancer Institute (CA-1094551 and Ca-116199 for FCM, CA-55164, CA-16672, and CA-49639 for MA, CA073839 and CA082867 to BPT) and by the Paul and Mary Haas Chair in Genetics (MA). FCM is also supported in part by grants from the Susan G Komen Breast Cancer Foundation. ELS is also supported by the Army Department of Defense (BC083397). We gratefully acknowledge Dr Shannon Kidd for thoughtful discussions during the progress of this work. We would also like to thank Wendy Schober, Amy Hazen and Jared Burks of the MD Anderson Flow Cytometry and Cellular Imaging Core for technical assistance.
Footnotes
Authors have no conflicts of interest.
References
- 1.Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, Lamarca HL, et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3806–11. doi: 10.1073/pnas.0900244106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K, et al. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010 doi: 10.3109/14653241003631815. [DOI] [PubMed] [Google Scholar]
- 3.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct Evidence of Mesenchymal Stem Cell Tropism for Tumor and Wounding Microenvironments Using In Vivo Bioluminescence Imaging. Stem cells (Dayton, Ohio) 2009 doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the Tumor Microenvironment: Quantitative Assessment of Adipose-Derived and Bone Marrow–Derived Stroma. PLoS ONE. 2012;7:e30563. doi: 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20009–14. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviewsCancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 8.Silzle T, Randolph GJ, Kreutz M, Kunz-Schughart LA. The fibroblast: sentinel cell and local immune modulator in tumor tissue. International journal of cancerJournal international du cancer. 2004;108:173–80. doi: 10.1002/ijc.11542. [DOI] [PubMed] [Google Scholar]
- 9.Udagawa T, Puder M, Wood M, Schaefer BC, D'Amato RJ. Analysis of tumor-associated stromal cells using SCID GFP transgenic mice: contribution of local and bone marrow-derived host cells. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:95–102. doi: 10.1096/fj.04-3669com. [DOI] [PubMed] [Google Scholar]
- 10.Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem cells. 2010;28:1435–45. doi: 10.1002/stem.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? Journal of Cellular Biochemistry. 2007;101:830–9. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF-C Mediates the Angiogenic and Tumorigenic Properties of Fibroblasts Associated with Tumors Refractory to Anti-VEGF Treatment. Cancer Cell. 2009;15:21–34. doi: 10.1016/j.ccr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Sasser AK, Mundy BL, Smith KM, Studebaker AW, Axel AE, Haidet AM, et al. Human bone marrow stromal cells enhance breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3D tumor environments. Cancer Letters. 2007;254:255–64. doi: 10.1016/j.canlet.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 15.Bauer M, Su G, Casper C, He R, Rehrauer W, Friedl A. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene. 2010;29:1732–40. doi: 10.1038/onc.2009.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer research. 2008;68:918–26. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Zhang X, Wang S, Qian H, Zhu W, Cao H, et al. Isolation and comparison of mesenchymal stem-like cells from human gastric cancer and adjacent non-cancerous tissues. Journal of cancer research and clinical oncology. 2010:1–10. doi: 10.1007/s00432-010-0908-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinoshita K, Nakagawa K, Hamada JI, Hida Y, Tada M, Kondo S, et al. Imatinib mesylate inhibits the proliferation-stimulating effect of human lung cancer-associated stromal fibroblasts on lung cancer cells. International journal of oncology. 2010;37:869–77. doi: 10.3892/ijo_00000738. [DOI] [PubMed] [Google Scholar]
- 19.Li L, Heldin CH, Heldin P. Inhibition of platelet-derived growth factor-BB-induced receptor activation and fibroblast migration by hyaluronan activation of CD44. Journal of Biological Chemistry. 2006;281:26512–9. doi: 10.1074/jbc.M605607200. [DOI] [PubMed] [Google Scholar]
- 20.Zhu H, Mitsuhashi N, Klein A, Barsky LW, Weinberg K, Barr ML, et al. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem cells (Dayton, Ohio) 2006;24:928–35. doi: 10.1634/stemcells.2005-0186. [DOI] [PubMed] [Google Scholar]
- 21.Teramoto H, Castellone MD, Malek RL, Letwin N, Frank B, Gutkind JS, et al. Autocrine activation of an osteopontin-CD44-Rac pathway enhances invasion and transformation by H-RasV12. Oncogene. 2005;24:489–501. doi: 10.1038/sj.onc.1208209. [DOI] [PubMed] [Google Scholar]
- 22.Bao LH, Sakaguchi H, Fujimoto J, Tamaya T. Osteopontin in metastatic lesions as a prognostic marker in ovarian cancers. Journal of Biomedical Science. 2007;14:373–81. doi: 10.1007/s11373-006-9143-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Chao L, Ma G, Chen L, Tian B, Zang Y, et al. Increased expression of osteopontin in patients with triple-negative breast cancer. European journal of clinical investigation. 2008;38:438–46. doi: 10.1111/j.1365-2362.2008.01956.x. [DOI] [PubMed] [Google Scholar]
- 24.Weber GF, Lett GS, Haubein NC. Osteopontin is a marker for cancer aggressiveness and patient survival. British journal of cancer. 2010;103:861–9. doi: 10.1038/sj.bjc.6605834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koyama H, Kobayashi N, Harada M, Takeoka M, Kawai Y, Sano K, et al. Significance of Tumor-Associated Stroma in Promotion of Intratumoral Lymphangiogenesis: Pivotal Role of a Hyaluronan-Rich Tumor Microenvironment. American Journal of Pathology. 2008;172:179–93. doi: 10.2353/ajpath.2008.070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wernicke M, Piñeiro LC, Caramutti D, Dorn VG, Raffo MML, Guixa HG, et al. Breast cancer stromal myxoid changes are associated with tumor invasion and metastasis: A central role for hyaluronan. Modern Pathology. 2003;16:99–107. doi: 10.1097/01.MP.0000051582.75890.2D. [DOI] [PubMed] [Google Scholar]
- 27.Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells invivo by disruption of tumor cell surface CD44 function. Journal of Experimental Medicine. 1997;186:1985–96. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng C, Toole BP, Kinney SD, Kuo JW, Stamenkovic I. Inhibition of tumor growth in vivo by hyaluronan oligomers. International Journal of Cancer. 1998;77:396–401. doi: 10.1002/(sici)1097-0215(19980729)77:3<396::aid-ijc15>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–91. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 30.Peterson RM, Yu Q, Stamenkovic I, Toole BP. Perturbation of hyaluronan interactions by soluble CD44 inhibits growth of murine mammary carcinoma cells in ascites. The American journal of pathology. 2000;156:2159–67. doi: 10.1016/S0002-9440(10)65086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagano O, Saya H. Mechanism and biological significance of CD44 cleavage. Cancer Sci. 2004;95:930–5. doi: 10.1111/j.1349-7006.2004.tb03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura H, Suenaga N, Taniwaki K, Matsuki H, Yonezawa K, Fujii M, et al. Constitutive and Induced CD44 Shedding by ADAM-Like Proteases and Membrane-Type 1 Matrix Metalloproteinase. Cancer research. 2004;64:876–82. doi: 10.1158/0008-5472.can-03-3502. [DOI] [PubMed] [Google Scholar]
- 33.Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. Journal of Cell Biology. 2009;185:949–57. doi: 10.1083/jcb.200812060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Kruijf EM, Van Nes JGH, Van De Velde CJH, Putter H, Smit VTHBM, Liefers GJ, et al. Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast cancer research and treatment. 2011;125:687–96. doi: 10.1007/s10549-010-0855-6. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, et al. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. JCell Biol. 2001;155:755–62. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer research. 2007;67:1979–87. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- 37.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu L, Roth JM, Brooks P, Ibrahim S, Karpatkin S. Twist is required for thrombin-induced tumor angiogenesis and growth. Cancer research. 2008;68:4296–302. doi: 10.1158/0008-5472.CAN-08-0067. [DOI] [PubMed] [Google Scholar]
- 39.Sung CO, Lee KW, Han S, Kim SH. Twist1 is up-regulated in gastric cancer-associated fibroblasts with poor clinical outcomes. Am J Pathol. 2011;179:1827–38. doi: 10.1016/j.ajpath.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erez N, Truitt M, Olson P, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell. 2010;17:135–47. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 41.Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 42.McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic Endocrine Instigation of Indolent Tumor Growth Requires Osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henry LR, Lee HO, Lee JS, Klein-Szanto A, Watts P, Ross EA, et al. Clinical implications of fibroblast activation protein in patients with colon cancer. Clinical Cancer Research. 2007;13:1736–41. doi: 10.1158/1078-0432.CCR-06-1746. [DOI] [PubMed] [Google Scholar]
- 44.Forst B, Hansen MT, Klingelhöfer J, Möller HD, Nielsen GH, Grum-Schwensen B, et al. Metastasis-inducing S100A4 and RANTES cooperate in promoting tumor progression in mice. PLoS ONE. 2010;5:1–11. doi: 10.1371/journal.pone.0010374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Helfman DM, Kim EJ, Lukanidin E, Grigorian M. The metastasis associated protein S100A4: role in tumour progression and metastasis. British journal of cancer. 2005;92:1955–8. doi: 10.1038/sj.bjc.6602613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips TM, McBride WH, Pajonk F. The response of CD24-/low/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute. 2006;98:1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 47.El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A. 2012;109:17460–5. doi: 10.1073/pnas.1206653109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, et al. Inhibition of TGF-beta/Smad Signaling by BAMBI Blocks Differentiation of Human Mesenchymal Stem Cells to Carcinoma-Associated Fibroblasts and Abolishes Their Pro-Tumor Effects. Stem Cells. 2012 doi: 10.1002/stem.1251. [DOI] [PubMed] [Google Scholar]
- 49.Bourguignon LY, Singleton PA, Zhu H, Zhou B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor beta receptor I in metastatic breast tumor cells. J Biol Chem. 2002;277:39703–12. doi: 10.1074/jbc.M204320200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.