

Abstract
We have developed a method for the detection of genetic markers associated with high pathogenicity in influenza. The assay consists of an array of 5′-thiolated ssDNA oligonucleotides immobilized on the surface of a Ag nanorod substrate that serve as capture probes for the detection of synthetic RNA sequences coding for a genetic mutation in the influenza PB1-F2 protein. Hybridization of the DNA probes to their complementary RNA sequences was detected using surface-enhanced Raman spectroscopy (SERS). Multivariate statistical analysis was used to differentiate the spectra of the complementary DNA probe-RNA target hybrids from those of the non-complementary DNA probes containing a single base pair polymorphism. Hierarchical cluster analysis (HCA) was able to distinguish with 100% accuracy the spectra of the complementary DNA probe – RNA target from the spectra of the immobilized DNA probes alone, or the DNA probes incubated with non-complementary RNA sequences. Linearity of response and limits of sensitivity of the SERS-based assays were determined using a partial least squares (PLS) regression model; detection limits computed by PLS was determined to be ~10 nM. The binding affinity of the DNA probes to their complementary RNA sequences was confirmed using enzyme-linked immunosorbent assay (ELISA); however, the detection limits observed using ELISA were approximately 10× higher (~100 nM) than those determined by PLS analysis of the SERS spectra.
Introduction
Influenza A is a segmented, negative-strand RNA virus that circulates worldwide and infects a wide range of species.1 Global, annual cases of seasonal influenza cause between 3–5 million severe illness and 250,000 to 500,000 deaths.2, 3 However, in certain years, highly virulent influenza pandemics occur that have been responsible for approximately 50 million deaths in 1918–1919, and 1 million deaths in 1956–1957.4 The most recent influenza pandemic scare was caused in 1997 by a H5N1 highly pathogenic avian influenza virus (HPAIV) originated in Hong Kong.5–7
Current work on HPAIV viruses aims to identify the factors responsible for the virulent pathogenicity of these influenza virus infections.8–13 Recent studies suggest that specific mutated proteins, including the proapoptotic PB1-F2 protein that is conserved among H5N1 HPAIV strains, may offer a mechanism for the observed increase in virulence, and as such are considered as potential precursors for future influenza pandemics.14–18 PB1-F2 is an 87-amino acid protein that has previously been correlated with increased pathogenicity in-vivo.19 A single amino acid mutation at position 66 in the PB1-F2 sequence (N66S) is consistent among HPAIV strains, including the H5N1 (HK/97) and 1918 influenza virus strains, and is a likely factor indicating increased virulence.14
Current routine laboratory diagnosis of influenza virulence factors relies entirely on genomic sequencing and alignment.20, 21 Other common immunological approaches, such as the conventional microarray techniques, have not yet been developed for analysis of HPAIV virulence. As a result, there is a critical need for new, rapid approaches for the detection of virulence factors in influenza.
This current report uses surface-enhanced Raman scattering (SERS) for the detection of virulence factors common to highly pathogenic influenza. SERS is a signal amplification technique based on electromagnetic enhancement resulting from localized surface plasmons.22, 23 The advantage of sensing using the intrinsic SERS spectra without further labeling is that Raman provides a unique chemically-specific molecular fingerprint, unlike other label-free methods, which rely on a general signal response for the captured analyte.
Studies have shown that SERS possesses high sensitivity and specificity, reliable quantification, and multiplexing ability for detection of oligonucleotide sequences.24–31 A number of reports have demonstrated direct, label-free detection of DNA/RNA sequences using SERS.32–37 In these studies, the intrinsic SERS spectra were demonstrated to be statistically unique and sensitive to the hybridization of matched and mismatched target sequences, including single nucleotide polymorphisms, molecular orientation and packing, and conformational changes upon ligand binding.32, 36, 38–40
Our research laboratories have previously demonstrated the use of Ag nanorod arrays to rapidly, sensitively, and quantitatively detect gene sequences using SERS.41–46 The current work expands on our previous studies on influenza detection via SERS44, 46 and shows that oligonucleotide-modified Ag nanorod arrays can be used for SERS-based profiling of RNA expression, allowing identification of the genetic mutations in the PB1-F2 protein related to influenza virulence and pandemic potential.
Experimental Methods
Material and Reagents
6-mercapto-1-hexanol (MCH) was purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of analytical grade and used without any further purification.
Preparation of Ag Nanorod SERS Substrates and Microwell Array Fabrication
Ag nanorod SERS-active substrates were fabricated using a custom-designed electron-beam/sputtering evaporation system by an oblique-angle vapor deposition (OAD) technique according to previously published procedures.47, 48 Full details on the nanofabrication, cleaning, and patterning of the Ag nanorod arrays are provided in Supporting Information.
DNA Probes and Synthetic RNA Targets
All DNA probes and synthetic RNA target sequences were purchased from Integrated DNA Technologies (IDT, Coralville, IA). From the analysis of the PB1-F2 protein sequence, DNA probes were designed corresponding to the minimal mitochondrial targeting sequence for both the high and the low virulence strains.14 The gene sequence targeted is comprised of 11 amino acids in the position 60 to 70 of the PB1-F2 protein sequence. Tables 1 and 2 present the amino acid, RNA codon, complementary DNA and reverse DNA probe sequences used in these experiments for both the low and high virulence genes, respectively. Five thymine (T) bases were added at the beginning of each DNA probe sequence to provide more flexibility to the probe and promote hybridization of the complementary RNA target sequences on the surface of the Ag nanorods.
Table 1.
Amino acid, RNA codon, complementary DNA and reverse DNA probe sequences used to construct samples for low virulence influenza.
| Low Virulence | PB1-F2 Sequence (Position 60–70) |
|---|---|
| Amino Acid | Q W L S L K S P T Q D |
| RNA Codon | 5′-CAA UGG CUU UCC UUG AAG A G U CCC ACC CAG GAC-3′ |
| Complementary DNA | 3′-GTT ACC GAA AGG AAC TTC T C A GGG TGG GTC CTG-5′ |
| Reverse DNA Probe | 5′-GTC CTG GGT GGG A C T CTT CAA GAA AAG CCA TTG-3′ |
| Thiolated DNA Probe | 5′-/5ThioMC6-D/ TTTTT GTC CTG GGT GGG A C T CTT CAA GAA AAG CCA TTG-3′ |
| Complementary RNA Target | 3′-CAG GAC CCA CCC U G A GAA GUU CCU UUC GGU AAC-5′ |
Table 2.
Amino acid, RNA codon, complementary DNA and reverse DNA probe sequences used to construct samples for high virulence influenza.
| High Virulence | PB1-F2 Sequence (Position 60–70) |
|---|---|
| Amino Acid | Q W L S L K N P T Q D |
| RNA Codon | 5′-CAA UGG CUU UCC UUG AAG A A U CCC ACC CAG GAC-3′ |
| Complementary DNA | 3′-GTT ACC GAA AGG AAC TTC T T A GGG TGG GTC CTG-5′ |
| Reverse DNA Probe | 5′-GTC CTG GGT GGG A T T CTT CAA GAA AAG CCA TTG-3′ |
| Thiolated DNA Probe | 5′-/5ThioMC6-D/ TTTTT GTC CTG GGT GGG A T T CTT CAA GAA AAG CCA TTG-3′ |
| Complementary RNA Target | 3′-CAG GAC CCA CCC U A A GAA GUU CCU UUC GGU AAC-5′ |
The complementary RNA targets used for the ELISA experiments were modified with the fluorescent label TAMRA™ (Azide) at the 5′ end of the RNA sequence. TAMRA™ has an excitation wavelength of 546 nm and an emission wavelength of 579 nm.
Binding Buffer
The binding buffer used in the hybridization experiments was 20 mM Tris HCl, 15 mM NaCl, 4 mM KCl, 1 mM MgCl2 and 1 mM CaCl2 in molecular biology grade water at pH 7.3 and stored at 4 °C. The buffer and working tools were DNase free.
Raman Spectroscopy
Raman measurements were performed using a confocal Raman microscope (InVia, Renishaw, Inc., Gloucestershire, United Kingdom). Laser excitation was provided by a 785 nm near-infrared diode laser. The incident laser beam was delivered to the sample by epi-illumination through a 20× (NA = 0.40) objective onto an automated sample stage. The laser illumination spot produced by this system through this objective creates a rectangular pattern of dimensions 4.8 × 27.8 μm. The laser power used was 0.1% (~0.42 mW), as measured at the sample. SERS spectra were collected from five different spots within a given microwell using a 30 s acquisition time with one accumulation. Spectra were collected between 2000 and 500 cm−1.
Data Analysis
Off-line spectral manipulation and analysis, i.e., baseline correction, band height, and peak frequency determination, were performed using GRAMS 32/AI Version 6.0 (Galactic Industries Corporation, Nashua, NH). Multivariate statistical analysis of the samples, including Hierarchical Cluster Analysis (HCA) and Partial Least Squares (PLS) regression analysis, was performed with PLS Toolbox version 6.2 (Eigenvector Research Inc., Wenatchee, WA), operating in a MATLAB environment (R2012a, The Mathworks Inc., Natick, MA).
ELISA 96-Well Plate Functionalization and Measurements
Maleimide activated black 96-well plates were purchased from Thermo Scientific (Waltham, MA). Prior to use, the plate wells were washed three times with the binding buffer. 100 μL of thiolated DNA probes were then added to each well at a concentration of 10 μg/mL (~1000 nM) in the binding buffer (pH 7.3) and incubated overnight at 4° C; the DNA solution was then removed and the wells rinsed three times with binding buffer. 100 μL of cysteine solution was added to each well at a concentration of 10 μg/mL in the binding buffer and incubated for one hour at room temperature to inactivate excess maleimide groups. After one hour, the cysteine solution was removed and the wells were rinsed three times with the binding buffer. The fluorescent RNA target sequences were then added to each well at varying concentrations (1000, 800, 600, 400, 200, 100, 50, 10, 1, and 0.1 nM) in the binding buffer at 37°C for 2 hours. The RNA solution was removed and each well was rinsed three times with the binding buffer prior to measurements.
Fluorescent measurements were performed using a POLARstar OPTIMA multidetection microplate reader (BMG Labtech, Ortenberg, Germany). Excitation was accomplished at 544 nm and detection of the emitted fluorescent signal was recorded at 590 nm. The instrument gain was set to 95% for the wells containing the highest concentration of complementary RNA (1000 nM) and 5% for the blank wells, i.e. DNA probes incubated with the binding buffer only. Each plate was read twice by the instrument; the results presented in this report are averages of 4 plates for a total of 8 measurements.
Results and Discussion
Experimental Design
The thiolated DNA probes were immobilized on the surface of the Ag nanorod microwells for 6 hours by self-assembly to form oligonucleotide monolayers.49 This immobilization step was followed by addition of the spacer molecule, 6-mercapto-1-hexanol (MCH) molecule for 6 hours at a concentration of 100 nM.50 The oligonucleotide-modified microwells were then incubated with the RNA target sequences. Full details of these experimental procedures used for immobilization and hybridization are provided in Supporting Information, as well as Figure S.1 outlining the various steps of the experimental design.
SERS Spectra of the DNA Probes
Figures 1A (top left) and 1B (top right) show raw, unprocessed SERS spectra of the high and low virulence DNA probe-spacer complexes, respectively. In order to ensure reproducibility of results, 10 spectra were collected in two different wells (5 spectra in each well) and on two different Ag nanorod substrates for a total of 20 spectra for each DNA sequence. Figures 1A and 1B show a high degree of spectral reproducibility and present similar spectral features. The number and position of the bands in Figs. 1A and 1B are in agreement with those of previous studies of thiolated DNA bound to Au surfaces.51, 52 These spectra are also in close agreement with previous studies from our laboratory of DNA aptamers bound to Ag nanorod surfaces.44, 46
Figure 1.

SERS spectra of: (A) high virulence DNA probe (1000 nM) with 6-mercapto-1-hexanol spacer (100 nM) on a Ag nanorod substrate, and (B) low virulence DNA probe-spacer complex. (C) Dendrogram produced by hierarchical cluster analysis (HCA) of the SERS spectra shown in panels A and B and label-coded as follows: (A) high virulence DNA probe calculated from spectra in panel A, and (B) low virulence DNA probe calculated from spectra in panel B. Panels A and B show 20 individual SERS spectra collected using multiple wells and multiple substrates; spectra are presented without any processing.
The dominant features present in the spectra shown in Figs. 1A and 1B are the bands assigned to the backbone phosphate stretching mode of nucleic acids (1089 cm−1) and the mixed in-plane ring skeleton stretching vibration (1332 cm−1).33, 53 Prominent nucleic acid vibrations include the bands at 731 and 1454 cm−1 (ring stretching modes of A),52, 54 1275 cm−1 (ring stretching and C-H bending of T),55 1496 cm−1 (C8=N7 vibration of the G ring),53 1023 cm−1 (the amino group vibration of C),54 and 1045 cm−1 (asymmetric out-of-plane deformation of NH2 in C).52 Additional prominent vibrations of nucleic acid bases appear in these spectra, for example, the 793 cm−1 combined ring breathing mode of C and T,51 and the band at 623 cm−1 attributed to the ring bending mode of A and the ring breathing mode of G.55 A complete table of the observed SERS bands in the spectra of the high and low virulence DNA probe-spacer complexes shown in Figs. 1A and 1B, with their literature assignments, is provided in Table S.1 in Supporting Information.
Cluster Analysis of the DNA Probe Spectra
Multivariate statistical analysis was used to establish statistically significant differences between the SERS spectra shown in Fig. 1. Figure 1C (bottom) illustrates the hierarchical cluster analysis (HCA) dendrogram based on the 40 SERS spectra shown in Figs. 1A and 1B. The oligonucleotide spectra were clustered based on Ward’s linkage method for minimizing variance.56 The vertical bars in the dendrogram specify which samples and classes are linked, while the horizontal bars represent the distance between the linked classes. The dendrogram shown in Fig. 1C reveals two distinct clusters. The green cluster (A) corresponds to the 20 SERS spectra of the high virulence probe complex shown in Fig. 1A, while the red cluster (B) corresponds to the 20 SERS spectra of the low virulence DNA probe complex shown in Fig. 1B. The results from this dendrogram provide evidence that HCA can differentiate with 100% accuracy the subtle differences in the SERS spectra of two DNA probes that differ only by a single base pair polymorphism.
SERS Spectra of the DNA Probes-RNA Target Hybrids
Figure 2A shows three panels of SERS spectra of the high virulence DNA. Figure 2A (i) is the spectra of the probe-spacer complex alone; Fig. 2A (ii) is the spectra of the probe-spacer complex incubated with the complementary high virulence synthetic RNA target sequence; Fig. 2A (iii) is the spectra of the probe-spacer complex incubated with a non-complementary low virulence synthetic RNA sequence. The SERS spectra of the DNA probes before hybridization, i.e. Fig. 2A (i), resemble those of the DNA probe-RNA target complexes after hybridization, i.e. Fig. 2A (ii) and (iii). Subtle spectral changes are seen in Fig. 2A (ii) upon full hybridization of the DNA probe sequences to the synthetic RNA target sequence due to sequence complementarity. In addition, incubation of the DNA probes with the non-complementary RNA sequence in Fig. 2A (iii) results in minor spectral changes due to mismatched hybridization. Overall, we observe a distinct SERS signature for DNA probe-RNA target hybrids when compared to the DNA probes alone, as we have previously observed with DNA aptamers.44, 46
Figure 2.
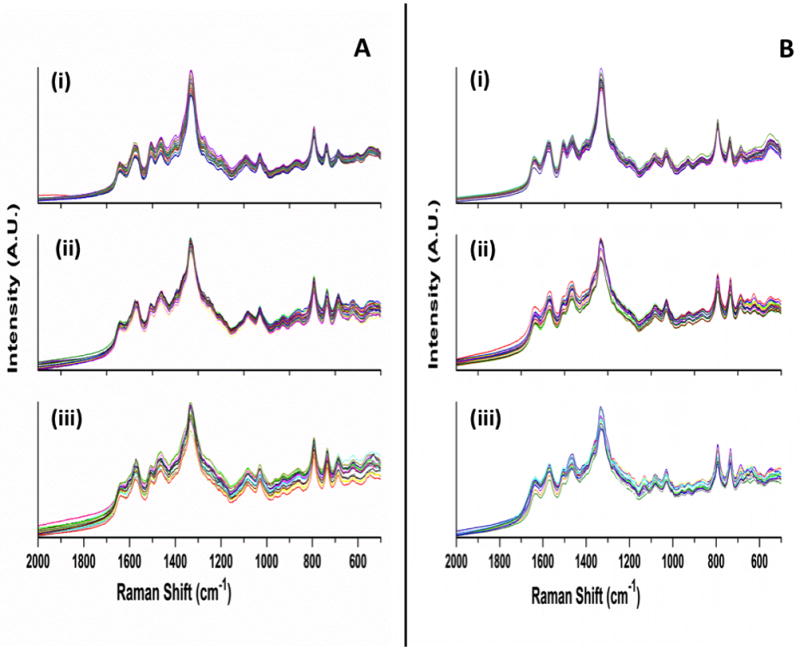
(A) SERS spectra of: (i) high virulence DNA probe, (ii) DNA probe incubated with the complementary high virulence synthetic RNA target sequence, and (iii) DNA probe incubated with non-complementary low virulence synthetic RNA sequence. (B) SERS spectra of: (i) low virulence DNA probe (ii) DNA probe incubated with complementary low virulence synthetic RNA target sequence, and (iii) DNA probe incubated with non-complementary high virulence synthetic RNA sequence. Each plot shows 20 individual SERS spectra collected using multiple wells and multiple substrates; spectra are presented without any processing.
Figure 2B presents three panels of the SERS spectra of the low virulence DNA. Figure 2B (i) is the spectra of the probe-spacer complex alone; Fig. 2B (ii) is the spectra of the probe-spacer complex incubated with the complementary low virulence synthetic RNA target sequence; Fig. 2B (iii) is the spectra of the probe-spacer complex incubated with a non-complementary low virulence synthetic RNA sequence. The SERS spectra of the low virulence DNA probes before hybridization, i.e. Fig. 2B (i), closely resemble those of the DNA probe-RNA target complexes after hybridization, i.e. Fig. 2B (ii) and (iii). The interpretation of the SERS spectra of the low virulence complexes shown in Fig. 2B follows directly from the discussion of the high virulence spectra presented in Fig. 2A. The high spectral similarity observed among the high and low virulence spectra in Figs. 2A and 2B requires the use of multivariate statistical methods to detect minor spectral differences among the SERS spectra and allow differentiation based on their intrinsic SERS signatures.
Cluster Analysis of DNA Probe-Synthetic RNA Target Complex Spectra
Figure 3 presents the HCA dendrogram generated using Ward’s algorithm and computed from the SERS spectra of the DNA probe-synthetic RNA target complexes shown in Fig. 2. One hundred and twenty spectra, corresponding to 20 spectra for each of the six sample categories described above, and seen in Fig. 2, were used to generate this dendrogram. Figure 3 illustrates that the SERS spectra of the low and high virulence DNA probes, along with their complementary and non-complementary targets, completely separate into six distinct clusters. The A and B clusters resulted from the spectra of the high (A) and low (B) virulence DNA probe complexes, respectively, i.e. Figs. 2A (i) and 2B (i). The C and D clusters represent the spectra of the high (C) and low (D) virulence DNA probes incubated with their complementary RNA target sequences, and correspond to the spectra presented in Fig. 2A (ii) and Fig. 2B (ii), respectively. The E and F clusters represent the spectra of the high (E) and low (F) virulence DNA probes incubated with their non-complementary RNA target sequences, and correspond to the spectra presented in Fig. 2A (iii) and Fig. 2B (iii), respectively.
Figure 3.
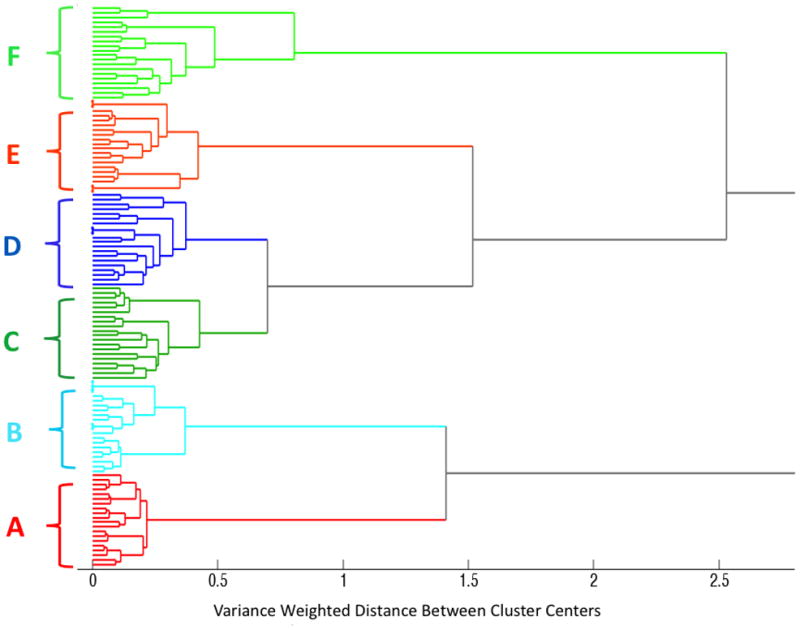
Dendrogram produced by hierarchical cluster analysis (HCA) of the SERS spectra shown in Figure 2, and label-coded as follows: (A) high virulence DNA probe, (B) low virulence DNA probe, (C) high virulence DNA probe with its complementary high virulence RNA target, (D) low virulence DNA probe with its complementary low virulence RNA target, (E) high virulence DNA probe with a non-complementary low virulence RNA target, and (F) low virulence DNA probe with a non-complementary high virulence RNA target. A total of 120 spectra were used to generate this dendrogram, corresponding to 20 spectra in each of the 6 DNA probe-RNA sample categories.
The results in Fig. 3 demonstrate that HCA classified each of the six separate groups of SERS spectra into its own cluster with 100% accuracy, showing the robustness of HCA classification in this case for differentiating highly similar SERS spectra. Interestingly, the six lowest level clusters combine to form two distinct higher order clusters. The DNA probe complexes alone (clusters A and B) form a single higher order cluster, while the DNA-RNA hybrids (complementary clusters C and D, and non-complementary clusters E and F) form their own higher order cluster. These results demonstrate that cluster analysis is able to distinguish the small but real spectral differences between the high and low virulence DNA probes alone and in comparison with their DNA-RNA probe-target complexes.
Sensitivity and Limit of Detection of Synthetic RNA Target Sequences Binding to Complementary DNA Probes
We investigated the sensitivity of the assay by incubating the oligonucleotide-modified SERS substrates with varying concentrations of complementary RNA target sequences. Following immobilization of the DNA probe-spacer complex on the surface of the Ag nanorods, incubation of the complementary RNA sequences was accomplished as described in the Experimental Methods section. Twenty SERS spectra were acquired from each of eight serial dilutions (1000 nM to 1 pM) of complementary RNA. These spectra were analyzed using a partial least squares (PLS) regression model to determine linearity of response and limits of sensitivity for the SERS-based hybridization assay.57
Figure 4 shows the PLS regression plots for both the high virulence (Fig. 4A) and the low virulence (Fig. 4B) assays, respectively. Each plot was generated using a total of 160 spectra, i.e., 20 for each the 8 target RNA concentrations. The PLS regression plots reveal the correlation between the predicted complimentary RNA concentration for cross-validated samples versus the true measured concentration based on the intrinsic SERS spectra of the DNA-RNA complexes. Figure 4 shows that the PLS model has a linear relationship for detection of synthetic RNA target sequences binding to their complementary DNA probes for concentrations ≥10 nM of complementary RNA in both the high and low virulence assays. The PLS model deviates from the predicted regression line at concentrations lower than 10 nM, meaning that the regression model can no longer quantify hybridization of the DNA probes for concentrations of complementary RNA target sequences lower than this value. This observation is consistent for both the high virulence (Fig. 4A) and the low virulence (Fig. 4B) assays.
Figure 4.
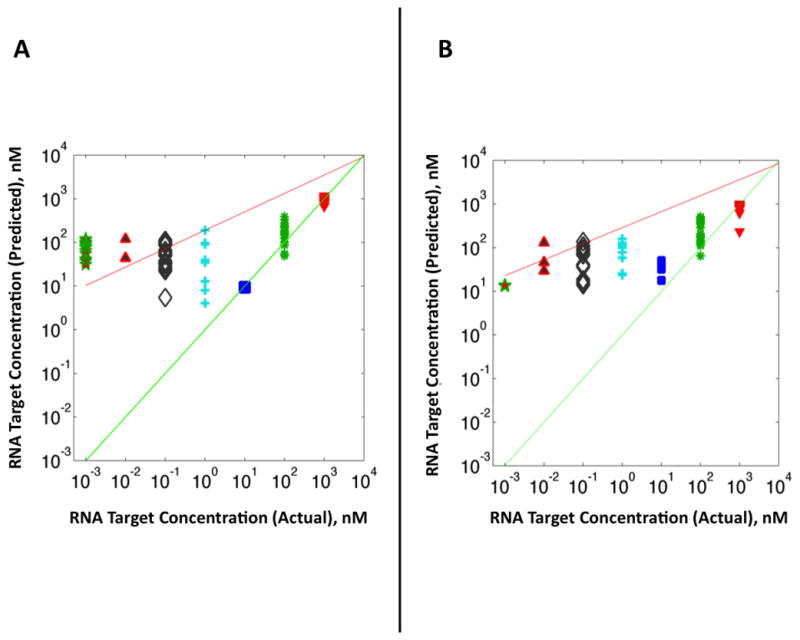
PLS regression analysis based on SERS spectra of serial dilutions of complementary RNA target sequence for (A) high and (B) low virulence assays, respectively. Measured values along abscissa axis correspond to serial 10-fold dilutions of complementary RNA target sequences from 1000 nM to 1 pM incubated with the DNA probe-spacer complex at 37°C for 2 hours. Green line indicates a theoretical perfect regression correlation index (R2 = 1) for all concentrations. Red line indicates a “best fit” line drawn through the actual measured RNA concentrations.
Detection of Fluorescently Tagged RNA Target Sequences Binding to DNA Probes by ELISA
Enzyme-linked immunosorbent assay (ELISA) was used to confirm the binding of the RNA target sequences to the immobilized DNA probes. ELISA has previously been used to determine the binding affinity and specificity of influenza virus targets.58 Both high and low virulence thiolated DNA probes were immobilized on the surface of the maleimide activated black 96-well plates via formation of stable thioether bonds. Following functionalization of the plate wells, the fluorescently labeled synthetic RNA target sequences corresponding to the minimal mitochondrial targeting sequence of the PB1-F2 protein with and without the mutation at position 66 were added to the oligonucleotide-modified wells at 37°C for 2 hours at various concentrations. The plate wells were then rinsed with copious amount of the binding buffer and read on a microplate reader. Full details on the DNA probes immobilization, RNA target sequences incubation, and fluorescent measurements are provided in Experimental Methods.
Figure 5 shows the plots of the fluorescent intensity for the high (Fig. 5A) and low (Fig. 5B) virulence assays recorded on the 96-well plates as a function of RNA concentration. Both high and low virulence DNA-RNA complexes display a steady, quasi-linear increase in fluorescent intensity over the 10–1000 nM concentration range of complementary RNA target sequences (black symbols and line). In contrast, the fluorescent intensity increases only incrementally when the oligonucleotide-modified wells are incubated with non-complementary RNA sequences that differ only by a single base pair polymorphism (red symbols and line). The fluorescent intensity for non-complementary RNA sequences remains at a baseline level even for high concentration of non-complementary RNA, suggesting that binding of DNA probes to RNA sequences has a high affinity only for complementary sequences.
Figure 5.
ELISA fluorescent intensity recorded from oligonucleotide-modified 96-well plates incubated with the complementary and non-complementary fluorescent RNA target sequences at 37°C for 2 hours for the high (A) and low (B) virulence assays. Each data point is an average of 8 measurements recorded on four different plates. Error bars represent the standard deviation.
These ELISA results confirm the SERS evidence that hybridization of DNA probes to complementary synthetically labeled RNA target sequences can be spectroscopically detected even with a single base pair mismatch. The detection limit of the ELISA assay can be determined by subtracting the baseline level of the non-complementary RNA signal from the fluorescent signal generated by each of the complementary RNA concentration. The lowest complementary RNA concentration for which the intensity value is positive after subtraction is 100 nM, meaning that binding of complementary RNA targets to DNA probes can no longer be detected by ELISA below this concentration. The plots shown in Figure 5 support this statement for both the high and low virulence assays. A comparison of Figs. 4 and 5 shows that the ELISA detection limit (100 nM) is approximately an order of magnitude greater than that obtained via PLS regression of the SERS spectra (10 nM).
Conclusion
The report describes a sensitive SERS-based assay consisting of an array of DNA oligonucleotides immobilized on the surface of a Ag nanorod substrate. These 5′-thiol-modified ssDNA sequences are employed as probes to capture complementary RNA target sequences corresponding to a segment of the PB1-F2 gene coding for the N66S mutation that has been linked to increased virulence influenza. The intrinsic SERS spectra of the immobilized DNA probes (Fig. 1) are reproducible and distinguishable from that of the spectra of the DNA-RNA hybrids (Fig. 2), demonstrating that SERS provides a non-amplified, label-free optical method to detect hybridization in this system. Multivariate statistical analysis using hierarchical cluster analysis (HCA) was used to differentiate: i) the spectra of the high and low virulence probes from one another (Fig. 1), ii) the spectra of the complementary high and low virulence DNA-RNA target hybrids from one another (Fig. 3), and iii) the spectra of the non-complementary high and low virulence DNA-RNA target hybrids from one another (Fig. 3).
In another set of experiments, both high and low virulence DNA probes were immobilized onto the Ag nanorod surface. A series of eight serial dilutions (1000 nM to 1 pM) were prepared of the complementary RNA for each DNA probe. A partial least squares (PLS) regression model was used for analysis of the resulting SERS spectra as a function of target RNA sequence concentration (Fig. 4). The PLS model predicted linearity of spectral response for RNA target sequence concentrations ≥10 nM of the synthetic to their immobilized DNA probes. The affinity of binding between the DNA probes to their complementary RNA sequences was confirmed using enzyme-linked immunosorbent assay (ELISA, Fig. 5). ELISA supported the PLS results and confirmed the evidence that DNA probes preferentially hybridize to their complementary RNA target sequences. The limit of detection for binding of these DNA-RNA hybrids, as determined using the ELISA assay, was found to be 100 nM. Therefore, the SERS-based assay described here is more sensitive than the ELISA standard by an order of magnitude.
The results of these studies indicate that SERS-based methods provide a new tool to accurately detect genetic markers related to virulence potential for early detection of influenza A viruses. This novel technology could potentially lead to the rapid identification of diagnostic indicators of disease severity and facilitate rapid implementation of disease intervention strategies. Future work will aim at extending the feasibility of this diagnostic strategy and directly test the validity of this assay for viral RNA samples extracted from clinical isolates of recombinant H5N1 influenza strains with and without the PB1-F2 gene mutation.
Supplementary Material
Acknowledgments
The work described here was supported by the U.S. Public Health Service through NIH Grant GM102546.
Footnotes
Supporting Information. 1) A schematic diagram illustrating the overall DNA immobilization, RNA binding, and detection strategy (Figure S.1). 2) Detailed experimental methods for SERS substrate fabrication. 3) Detailed experimental methods for preparation and immobilization of synthetic DNA probes and binding of RNA targets. 4) Tables of the observed SERS peaks, including band assignments to particular vibrational modes, in the spectrum of the DNA and RNA samples (Table S.1).
References
- 1.Easterday BC, Hinshaw VS. Influenza. Iowa State University Press; Ames: 1991. [Google Scholar]
- 2.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Influenza (Seasonal) http://www.who.int/mediacentre/factsheets/fs211/en/index.html.
- 4.Beveridge WIB. Hist Phil Life Sci. 1991;13:223–235. [PubMed] [Google Scholar]
- 5.Dybing JK, Schultz-Cherry S, Swayne DE, Suarez DL, Perdue ML. J Virol. 2000;74:1443–1450. doi: 10.1128/jvi.74.3.1443-1450.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuen KY, Chan PKS, Peiris M, Tsang DNC, Que TL, Shortridge KF, Cheung PT, To WK, Ho ETF, Sung R, Cheng AFB. Lancet. 1998;351:467–471. doi: 10.1016/s0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen-Van-Tam JS, Hampson AW. Vaccine. 2003;21:1762–1768. doi: 10.1016/s0264-410x(03)00069-0. [DOI] [PubMed] [Google Scholar]
- 8.de Jong MD, Simmons CP, Thanh TT, Hien VM, Smith GJD, Chau TNB, Hoang DM, Chau NVV, Khanh TH, Dong VC, Qui PT, Van Cam B, Ha DQ, Guan Y, Peiris JSM, Chinh NT, Hien TT, Farrar J. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li ZJ, Jiang YP, Jiao PR, Wang AQ, Zhao FJ, Tian GB, Wang XJ, Yu KZ, Bu ZG, Chen HL. J Virol. 2006;80:11115–11123. doi: 10.1128/JVI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mase M, Tanimura N, Imada T, Okamatsu M, Tsukamoto K, Yamaguchi S. J Gen Virol. 2006;87:3655–3659. doi: 10.1099/vir.0.81843-0. [DOI] [PubMed] [Google Scholar]
- 11.Muramoto Y, Le TQM, Phuong LS, Nguyen T, Nguyen TH, Sakai-Tagawa Y, Horimoto T, Kida H, Kawaoka Y. J Vet Med Sci. 2006;68:735–737. doi: 10.1292/jvms.68.735. [DOI] [PubMed] [Google Scholar]
- 12.Muramoto Y, Le TQM, Phuong LS, Nguyen T, Nguyen TH, Sakai-Tagawa Y, Iwatsuki-Horimoto K, Horimoto T, Kida H, Kawaoka Y. J Vet Med Sci. 2006;68:527–531. doi: 10.1292/jvms.68.527. [DOI] [PubMed] [Google Scholar]
- 13.Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 14.Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. PLoS Pathog. 2007;3:1414–1421. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamarin D, Garcia-Sastre A, Xiao XY, Wang R, Palese P. PLoS Pathog. 2005;1:40–54. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zamarin D, Ortigoza MB, Palese P. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munier S, Larcher T, Cormier-Aline F, Soubieux D, Su B, Guigand L, Labrosse B, Cherel Y, Quere P, Marc D, Naffakh N. J Virol. 2010;84:940–952. doi: 10.1128/JVI.01581-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou HB, Yu ZJ, Hu Y, Tu JG, Zou W, Peng YP, Zhu JP, Li YT, Zhang AD, Yu ZN, Ye ZP, Chen HC, Jin ML. PLoS One. 2009:4. doi: 10.1371/journal.pone.0006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen WS, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, Basta S, O’Neill R, Schickli J, Palese P, Henklein P, Bennink JR, Yewdell JW. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 20.Kash JC, Tumpey TM, Proll SC, Carter V, Perwitasari O, Thomas MJ, Basler CF, Palese P, Taubenberger JK, Garcia-Sastre A, Swayne DE, Katze MG. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salomon R, Franks J, Govorkova EA, Ilyushina NA, Yen HL, Hulse-Post DJ, Humberd J, Trichet M, Rehg JE, Webby RJ, Webster RG, Mann EH. J Exp Med. 2006;203:689–697. doi: 10.1084/jem.20051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kneipp K, Moskovits M, Kneipp H, editors. Surface-Enhanced Raman Scattering. Springer-Verlag; Berlin: 2006. [Google Scholar]
- 23.Stiles PL, Dieringer JA, Shah NC, Duyne RPV. Annu Rev Anal Chem. 2008;1:601–626. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 24.Park T, Lee S, Seong GH, Choo J, Lee EK, Kim YS, Ji WH, Hwang SY, Gweon DG. Lab on a Chip. 2005;5:437–442. doi: 10.1039/b414457k. [DOI] [PubMed] [Google Scholar]
- 25.Yea K, Lee S, Kyong JB, Choo J, Lee EK, Joo SW, Lee S. Analyst. 2005;130:1009–1011. doi: 10.1039/b501980j. [DOI] [PubMed] [Google Scholar]
- 26.Farquharson S, Shende C, Inscore FE, Maksymiuk P, Gift A. J Raman Spectrosc. 2005;36:208–212. [Google Scholar]
- 27.Grajcar L, El Amri C, Ghomi M, Fermandjian S, Huteau V, Mandel R, Lecomte S, Baron MN. Biopolymers. 2006;82:6–28. doi: 10.1002/bip.20455. [DOI] [PubMed] [Google Scholar]
- 28.Berdat D, Rodriguez ACM, Herrera F, Gijs MAM. Lab Chip. 2008;8:302–308. doi: 10.1039/b712609c. [DOI] [PubMed] [Google Scholar]
- 29.Huh YS, Chung AJ, Erickson D. Microfluid Nanofluid. 2009;6:285–297. [Google Scholar]
- 30.Kustner B, Gellner M, Schutz M, Schoppler F, Marx A, Strobel P, Adam P, Schmuck C, Schlucker S. Angew Chem Int Edit. 2009;48:1950–1953. doi: 10.1002/anie.200804518. [DOI] [PubMed] [Google Scholar]
- 31.Monaghan PB, McCarney KM, Ricketts A, Littleford RE, Docherty F, Smith WE, Graham D, Cooper JM. Anal Chem. 2007;79:2844–2849. doi: 10.1021/ac061769i. [DOI] [PubMed] [Google Scholar]
- 32.Green M, Liu FM, Cohen L, Kollensperger P, Cass T. Faraday Discuss. 2006;132:269–280. doi: 10.1039/b506636k. [DOI] [PubMed] [Google Scholar]
- 33.Barhoumi A, Zhang D, Tam F, Halas NJ. J Am Chem Soc. 2008;130:5523–5529. doi: 10.1021/ja800023j. [DOI] [PubMed] [Google Scholar]
- 34.Barhoumi A, Zhang D, Halas NJ. J Am Chem Soc. 2008;130:14040–14041. doi: 10.1021/ja804367c. [DOI] [PubMed] [Google Scholar]
- 35.Levin CS, Kundu J, Barhoumi A, Halas NJ. Analyst. 2009;134:1745–1750. doi: 10.1039/b909080k. [DOI] [PubMed] [Google Scholar]
- 36.Moody B, McCarty G. Anal Chem. 2009;81:2013–2016. doi: 10.1021/ac802247y. [DOI] [PubMed] [Google Scholar]
- 37.Papadopoulou E, Bell SEJ. Chem-Eur J. 2012;18:5394–5400. doi: 10.1002/chem.201103520. [DOI] [PubMed] [Google Scholar]
- 38.Dong LQ, Zhou JZ, Wu LL, Dong P, Lin ZH. Chem Phys Lett. 2002;354:458–465. [Google Scholar]
- 39.Barhoumi A, Halas NJ. J Am Chem Soc. 2010;132:12792–12793. doi: 10.1021/ja105678z. [DOI] [PubMed] [Google Scholar]
- 40.Prado E, Daugey N, Plumet S, Servant L, Lecomte S. Chem Commun. 2011;47:7425–7427. doi: 10.1039/c1cc11925g. [DOI] [PubMed] [Google Scholar]
- 41.Driskell JD, Seto AG, Jones LP, Jokela S, Dluhy RA, Zhao YP, Tripp RA. Biosens Bioelec. 2008;24:917–922. doi: 10.1016/j.bios.2008.07.060. [DOI] [PubMed] [Google Scholar]
- 42.Driskell JD, Primera-Pedrozo OM, Dluhy RA, Zhao YP, Tripp RA. Appl Spectrosc. 2009;63:1107–1114. doi: 10.1366/000370209789553183. [DOI] [PubMed] [Google Scholar]
- 43.Driskell JD, Tripp RA. Chem Commun. 2010;46:3298–3300. doi: 10.1039/c002059a. [DOI] [PubMed] [Google Scholar]
- 44.Negri P, Kage A, Nitsche A, Naumann D, Dluhy RA. Chem Commun. 2011;47:8635–8637. doi: 10.1039/c0cc05433j. [DOI] [PubMed] [Google Scholar]
- 45.Abell JL, Garren JM, Driskell JD, Tripp RA, Zhao Y. J Am Chem Soc. 2012;134:12889–12892. doi: 10.1021/ja3043432. [DOI] [PubMed] [Google Scholar]
- 46.Negri P, Chen G, Kage A, Nitsche A, Naumann D, Xu B, Dluhy RA. Anal Chem. 2012;84:5501–5508. doi: 10.1021/ac202427e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaney SB, Shanmukh S, Zhao YP, Dluhy RA. Appl Phys Lett. 2005;87:31908–31910. [Google Scholar]
- 48.Driskell JD, Shanmukh S, Liu Y, Chaney SB, Tang XJ, Zhao YP, Dluhy RA. J Phys Chem C. 2008;112:895–901. [Google Scholar]
- 49.Basu S, Jana S, Pande S, Pal T. J Coll Inter Sci. 2008;321:288–293. doi: 10.1016/j.jcis.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 50.Herne TM, Tarlov MJ. J Am Chem Soc. 1997;119:8916–8920. [Google Scholar]
- 51.Kneipp K, Flemming J. J Mol Struct. 1986;145:173–179. [Google Scholar]
- 52.Suh JS, Moskovits M. J Am Chem Soc. 1986;108:4711–4718. [Google Scholar]
- 53.Pagba CV, Lane SM, Wachsmann-Hogiu S. J Raman Spectrosc. 2010;41:241–247. [Google Scholar]
- 54.Kundu J, Neumann O, Janesko BG, Zhang D, Lal S, Barhoumi A, Scuseria GE, Halas NJ. J Phys Chem C. 2009;113:14390–14397. [Google Scholar]
- 55.Otto C, Vandentweel TJJ, Demul FFM, Greve J. J Raman Spectrosc. 1986;17:289–298. [Google Scholar]
- 56.Beebe KR, Pell RJ, Seasholtz MB. Chemometrics: A Practical Guide. J. Wiley & Sons, Inc; New York: 1998. [Google Scholar]
- 57.Barker M, Rayens W. J Chemometr. 2003;17:166–173. [Google Scholar]
- 58.Song DS, Lee YJ, Jeong OM, Kim YJ, Park CH, Yoo JE, Jeon WJ, Kwon JH, Ha GW, Kang BK, Lee CS, Kim HK, Jung BY, Kim JH, Oh JS. J Vet Sci. 2009;10:323–329. doi: 10.4142/jvs.2009.10.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.