

Abstract
The mechanisms by which natural rewards such as sugar affect synaptic transmission and behavior are largely unexplored. Here, we investigate regulation of nucleus accumbens synapses by sucrose intake. Previous studies have shown that AMPA receptor (AMPAR) trafficking is a major mechanism for regulating synaptic strength, and that in vitro, trafficking of AMPARs containing the GluA1 subunit takes place by a two-step mechanism involving extrasynaptic and then synaptic receptor transport. We report that in rat, repeated daily ingestion of a 25% sucrose solution transiently elevated spontaneous locomotion and potentiated accumbens core synapses through incorporation of Ca2+-permeable AMPA receptors (CPARs), which are GluA1-containing, GluA2-lacking AMPARs. Electrophysiological, biochemical, and quantitative electron microscopy studies revealed that sucrose training (7 d) induced a stable (>24 h) intraspinous GluA1 population, and that in these rats a single sucrose stimulus rapidly (5 min) but transiently (<24 h) elevated GluA1 at extrasynaptic sites. CPARs and dopamine D1 receptors were required in vivo for elevated locomotion after sucrose ingestion. Significantly, a 7 d protocol of daily ingestion of a 3% solution of saccharin, a noncaloric sweetener, induced synaptic GluA1 similarly to 25% sucrose ingestion. These findings identify multistep GluA1 trafficking, previously described in vitro, as a mechanism for acute regulation of synaptic transmission in vivo by a natural orosensory reward. Trafficking is stimulated by a chemosensory pathway that is not dependent on the caloric value of sucrose.
Introduction
Sucrose overconsumption is a significant public health problem (Hu and Malik, 2010), but the mechanisms by which natural, orosensory rewards such as sucrose regulate synaptic transmission to influence behavior are not known. Synaptic plasticity in the nucleus accumbens, an integral component of brain reward circuitry (Sesack and Grace, 2010), contributes to many forms of motivated behavior, including reward learning (Day and Carelli, 2007), responses to social stress (LaPlant et al., 2010), and addiction pathologies (Lüscher and Malenka, 2011). Repeated cocaine exposure causes synaptic plasticity in neurons of the accumbens and the ventral tegmental area (VTA) (Thomas et al., 2001; Ungless et al., 2001; Brebner et al., 2005; Mameli et al., 2009; Grueter et al., 2010; Pascoli et al., 2012). Upon extended access cocaine self-administration followed by prolonged withdrawal, synapses are potentiated through the incorporation of Ca2+-permeable, GluA2-lacking AMPA-type glutamate receptors (CPARs), whose signaling mediates incubation of cocaine craving (Conrad et al., 2008; McCutcheon et al., 2011a). Similar to cocaine, orosensory rewards such as sucrose robustly elevate accumbens dopamine (Smith, 2004), but orosensory reward induction of accumbens plasticity has not been investigated.
AMPA receptors (AMPARs) are primary mediators of CNS excitatory transmission, and their trafficking contributes to diverse neural processes, including learning and memory (Rumpel et al., 2005; Whitlock et al., 2006; Nedelescu et al., 2010). AMPARs are composed of four different subunits, GluA1–4. GluA2-containing AMPARs are Ca2+-impermeable and traffic constitutively to synapses, while GluA2-lacking receptors (CPARs), which are predominately GluA1 homomers, conduct Ca2+ and exhibit inward rectification. GluA1 undergoes activity-dependent synaptic trafficking by a two-step pathway in which Ser 845 phosphorylation by cAMP-dependent protein kinase A and cGMP-dependent protein kinase II (cGKII) promotes receptor accumulation at extrasynaptic sites in the plasma membrane (Esteban et al., 2003; Sun et al., 2005, 2008; Serulle et al., 2007). After lateral diffusion to the synapse, phosphorylation of Ser 818 by protein kinase C stabilizes AMPARs within the synapse (Boehm et al., 2006), anchored to the postsynaptic density (PSD; Oh et al., 2006; Ehlers et al., 2007; Serulle et al., 2007). Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of Ser 567 and Ser 831 also contributes to synaptic incorporation and extrasynaptic targeting (Roche et al., 1996; Lu et al., 2010), respectively. However, it is not known whether in vivo incorporation of CPARs employs these rapid, multistep mechanisms described in vitro.
To investigate the mechanisms by which orosensory rewards such as sucrose regulate accumbens excitatory synapses, we have used a paradigm of brief sucrose ingestion and measured changes in synaptic transmission in accumbens neurons. We observe that repeated sucrose ingestion potentiates accumbens synapses through incorporation of CPARs, and that in a sucrose-trained animal, a single sucrose stimulus is sufficient to induce rapid GluA1 trafficking to extrasynaptic sites. Because saccharin, a noncaloric sweetener, induced synaptic trafficking similarly to sucrose, trafficking is a response to orosensory rather than caloric pathways. Furthermore, CPAR blockade prevented sucrose-induced elevations of spontaneous locomotor activity in vivo, further identifying accumbens CPARs as important regulators of responses to natural rewards.
Materials and Methods
Subjects and surgical procedures.
The subjects were male Sprague Dawley rats (Taconic; behavioral experiments) weighing 150–300 g on arrival and female E18 pregnant Sprague Dawley rats (Taconic; cell culture experiments). The rats were housed two per cage for behavioral experiments on a 12 h light/dark cycle (lights out at 18:00) and had access to food and water ad libitum at all times. All experimental procedures were approved by the New York University School of Medicine Institutional Animal Care and Use Committee and were performed in accordance with the “Principles of Laboratory Animal Care” (National Institutes of Health publication number 85-23).
Sucrose training and locomotor measurements.
Rats were transported to the test room 3 consecutive days for 2 h/d in their home cages. On the fourth day bottles containing water or 25% sucrose were introduced through the cage lid for 5 min. Bottles were then weighed. For all experiments, rats were required to drink at least 1 g of sucrose during the 5 min access within 3 d of training commencement to be included in the study; in fact, all rats met this criterion. After bottle removal, rats remained in the test room for 30 min before transport back to the animal facility. On the day of kill, rats were rendered unconscious by CO2, decapitated by guillotine, and tissue samples were collected on ice. For locomotor experiments, rats were placed in locomotor measurement chambers (AccuScan) for a total of 35 min. After 15 min in the chamber, a bottle with a bead stopper was introduced through the top of the chamber and stabilized. The bottle was removed from the top of the chamber after 5 min, and the rats remained in the chamber for an additional 15 min after bottle removal. This procedure was repeated identically for 7 consecutive days. Distance traveled was measured using the VersaMax System (AccuScan), which monitored animal activity via a grid of 16 × 16 infrared light beams that traverse the animal cage (42 × 42 × 30 cm) front to back and left to right. Information about beam status, scanned at a rate of 100 times per second, was stored to disk. Activity was expressed as ambulatory distance measured in cm during 12 different 3 min bins in a 35 min session (the final bin was 2 min).
Saccharin training.
To compare sucrose-induced trafficking of GluA1 with the effects of saccharin ingestion, 12 adult male rats (250 g) were housed in the animal facility on a 12 h light/dark cycle. All rats were then habituated to the test room by being transported to the test room, left for 2 h, and transported back to the animal facility. One the fourth day (after 3 d of habituation), rats were given access bottles containing, water, sucrose, or saccharin. Four rats were given access to a bottle containing water rested on the top of the cage with the spout protruding into the cage through the lid. Access time was 5 min, then the bottle was removed, and after 15 additional minutes rats were transported back to the animal facility. Four rats were given access to 25% sucrose solution and four rats were given access to 3% saccharin solution (Sweet'N Low). The volume of fluid consumed was measured. This procedure was repeated for 7 d. On the seventh day of drinking, immediately following bottle removal, rats were killed and accumbens core was harvested and GluA1 levels were probed by Western blot.
Electrophysiology.
Rats were sucrose trained as described above in clear plastic cages and, after bottle removal on day 7, were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and transcardially perfused with cold saline (miniature EPSC (mEPSC) experiments) or decapitated immediately (rectification experiments). Brains were quickly removed into artificial CSF (ACSF) that consisted of the following (in mm): for mEPSC experiments: 118 NaCl, 2.5 KCl, 3 CaCl2, 1 MgCl2, 26 NaHCO3, 1 NaH2PO4, and 10 d-glucose, osmolarity adjusted to 325 mOsm and aerated by 95% O2/5% CO2, pH 7.4; for rectification experiments: 75 sucrose, 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 6 H2O, 25 NaHCO3, and 10 dextrose, bubbled with 95% O2/5% CO2, pH 7.4. Coronal slices (300 μm thick) containing the nucleus accumbens were cut in ice-cold ACSF using a vibratome (Leica VT1200S) and kept submerged in ACSF containing the following (in mm[scap]): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.5 CaCl2, 1.5 MgSO4, 7 H2O, 26 NaHCO3, and 10 dextrose for <30 min; then kept in a slice pre-incubator at room temperature for at least 1 h to allow for recovery. For mEPSC experiments a single slice was then transferred to a recording chamber in which it was held submerged by a nylon net at 32°C with a TC324B in-line solution heater and controller (Warner Instruments). The chamber was continuously perfused by ACSF at a constant rate of 2 ml/min. Medium spiny neurons (MSNs) from the nucleus accumbens core region were identified under visual guidance using infrared-differential interference contrast (IR-DIC) video microscopy (Hamamatsu C5405) with an Olympus BX50WI upright microscope fitted with 40× long working distance water-immersion objective. Patch electrodes (4–6 MΩ) filled with an intracellular pipette solution consisting of the following (in mm): 145 CsCl, 10 HEPES, 0.5 EGTA, and 5 MgATP. Osmolarity was adjusted to 290 mOsm with sucrose, and pH was adjusted to 7.4 with CsOH. mEPSCs were recorded in the presence of bicuculline (10 μm) and tetrodotoxin (1 μm) using Axopatch 200B amplifier (Molecular Devices) and digitized by Digidata 1322A (Molecular Devices). For rectification experiments slices were transferred to the recording chamber and perfused (2.0–2.5 ml min−1) with oxygenated ACSF at 33−35°C containing 50 μm picrotoxin to isolate EPSCs. Somatic whole-cell recordings were made from core region MSNs in voltage clamp with a Multiclamp 700B amplifier (Molecular Devices) using IR-DIC video microscopy. Patch pipettes (4–6 MΩ) were filled with intracellular solution consisting of the following (in mm[scap]): 125 Cs-gluconate, 2 CsCl, 5 TEA-Cl, 4 Mg-ATP, 0.3 GTP, 10 phosphocreatine, 10 HEPES, 0.5 EGTA, and 3.5 QX-314. Data were filtered at 2 kHz, digitized at 10 kHz, and analyzed with Clampfit 10 (Molecular Devices). Extracellular stimulation (0.01–1 ms, 5–150 μA, 0.2 Hz) was applied with a small glass bipolar electrode 0.05–0.5 mm from the recording electrode. After ∼10 min of baseline recording, a solution containing Naspm (200 μm) was perfused into the bath for 10 min. Changes in EPSC amplitude were measured before and after drug application at holding potentials of −70, −50, −30, 0, +20, +40, and +60 mV. The rectification index (ir) was calculated by correcting any potential shifts in reversal potential and computed from the following equation: ir = (I−70/70)/(I+40/40), where I−70 and I+40 are the EPSC amplitudes recorded at −70 mV and +40 mV, respectively.
Subcellular fractionation and Western blotting.
Accumbens were collected on ice as described above. When core and shell were separately dissected, separation was confirmed by probing synaptosome fractions for dopamine β-hydroxylase, an enzyme found in the terminals of axons to the shell but not the core (Sesack and Grace, 2010). Whole-cell, synaptosome, and postsynaptic density (PSD) fractions were prepared as described previously (Jordan et al., 2004). Synaptosomal pellets were resuspended in 200 μl of 25 mm Tris with 1% Triton X-100, rocked at 4°C for 30 min, and centrifuged at 13,800 × g for 15 min in a microcentrifuge to pellet PSDs. The pellet containing crude PSDs was resuspended in 25 mm Tris with 2% SDS. Fractions were analyzed by Western blot by SDS-PAGE gels as described previously (Jordan et al., 2004). The following antibodies were used: dopamine β-hydroxylase (1:1000; Abcam), GluA1 (1:1000; Millipore), phosphor-Ser 845 GluA1 (1:1000; Millipore), GluA2 (1:1000; Millipore), and tubulin (1:10,000; Sigma).
Electron microscopy.
On the day of tissue harvesting (day 7 of sucrose training), rats from three test groups (Water, Sucrose/Water, Sucrose; three rats/test group) were placed in locomotor measurement chambers for 15 min; at 15 min a bottle was introduced through the chamber top. Rats in the Water group received water, rats in the Sucrose group received 25% sucrose, rats in the Sucrose/Water group, which had consumed 25% sucrose for 6 d, received water. Rats were deeply anesthetized with Nembutal (50 mg/kg, i.p.) and transcardially perfused with 0.1 m phosphate buffer, pH 7.4, containing 4% paraformaldehyde and 0.1% glutaraldehyde at a rate of 50 ml/min during the first 3 min, then at a rate of 20 ml/min for the succeeding 7 min. Tissue was prepared for postembed immunogold (PEG) labeling and images were captured as described previously (Nedelescu et al., 2010). Immunolabels were categorized according to their position relative to the PSD at asymmetric synaptic junctions as “cleft,” “near PSD” (within 1 PSD width from the PSD), “at PSD,” “intraspinous,” or “extrasynaptic membrane.” From each animal, 93 synapses were sampled from the core of the accumbens. Random sampling was ensured by analyzing all of the first 93 randomly encountered synapses as we swept systematically across the grid, then pooling the same number of synapses from each of the three animals that received identical antemortem treatments. Two types of quantification were performed. One was to evaluate GluR1-immunoreactivity level by tallying the number of PEG particles that occurred at discrete functional domains of the spine. The other was to assess the proportion of synapses labeled at the PSD by any number of PEG particles. Even synapses labeled by just 1 PEG particle were considered labeled, based on earlier work demonstrating specificity of the GluR1-PEG procedure (Nedelescu et al., 2010). Treatment effects on the proportion and level of GluR1 immunolabeling were analyzed by one-way ANOVA with planned post hoc comparisons (Fisher's LSD). To eliminate experimental bias, the data were triple blinded: one experimenter performed sucrose training and kept records of the animals in the three test groups, a second experimenter created the electron micrographs and assigned a new alphanumeric code to each micrograph and kept the code sealed, and three additional experimenters scanned the micrographs and quantified PEG particles. After PEG quantification was complete, the experimenters convened to reveal the identities of each micrograph.
Cannula implantation and intracranial injections.
Intracranial injection was used to deliver Naspm and aminophosphonovalerate (APV) to the accumbens core. For cannula implantation, as described previously (Carr et al., 2010), rats were deeply anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and injected postoperatively with the analgesic benamine (1 mg/kg, s.c.). Rats were stereotaxically implanted with two 26 gauge guide cannulae (PlasticsOne) bilaterally in the accumbens core with coordinates: 1.6 mm anterior to bregma; 2.9 mm lateral to the sagittal suture, tips angled 8° toward the midline, 5.6 mm ventral to skull surface. Cannulae were held in place by dental acrylic and patency was maintained with an occlusion stylet. For intracranial injections, Naspm and APV solutions were loaded into two 30 cm lengths of PE-50 tubing attached at one end to 25 μl Hamilton syringes filled with distilled water and at the other end to 31 gauge injector cannulae, which extended 2.0 mm beyond the implanted guides. The syringes were mounted on the twin holders of a Harvard 2272 microliter syringe pump that delivered the 0.5 μl injection volumes over a period of 100 s. One minute following completion of injections, injector cannulae were removed from guides, stylets were replaced, and animals were placed into locomotor testing chambers for sucrose training. After killing the animal, cryogenic brain sections were analyzed for cannula localization; 2 of 15 animals were excluded from the study because of improper cannula placement.
Statistical analysis.
One-way ANOVA followed by Fisher post hoc tests was used for electron microscopy, immunocytochemistry, and biotinylation experiments. Two-tailed Student's t tests were used for electrophysiology. For sucrose hyperactivity experiments, two-way ANOVA was used, followed by Fisher post hoc tests.
Results
Characterization of a sucrose ingestion paradigm
We used a sucrose ingestion paradigm to investigate the effects of a natural, orosensory reward on synaptic transmission (Fig. 1A). Adult male rats were transported to a test room on 3 consecutive days. On the fourth day (first day of training), the rats were placed in a locomotor measurement chamber. After 15 min of locomotor activity measurement in the chamber, bottles containing either water (for Water animals) or 25% sucrose solution (for Sucrose animals) were introduced into the measurement chambers through holes in the lid of the chamber. The bottles were removed after 5 min and locomotor activity was measured for an additional 15 min before animals were returned to home cages. We repeated this procedure for 7 consecutive days. In some experiments, sucrose training was extended to an eighth day. This brief, noncontingent access to a highly palatable solution allowed us to investigate both acute and cumulative effects of sucrose intake, as animals reliably imbibed sucrose vigorously during the access window within 3 d of training (Fig. 1B). These experimental conditions enabled the comparison of test groups immediately following cessation of ingestion. Our criterion for inclusion in the study was that rats begin to consume at least 1 g of sucrose during the access period within 3 d of training commencement; no animals were excluded from the study on the basis of this criterion.
Figure 1.

Repeated sucrose ingestion causes transient elevations of spontaneous locomotion. A, Adult male rats (n = 8, water; n = 10, sucrose) were habituated to a test room for 3 d before being placed in a locomotor measurement chamber for 7 consecutive days; spontaneous locomotor activity was measured 15 min before and 15 min after 5 min access to a bottle containing water or 25% sucrose. B, Sucrose rats consumed significantly more liquid than Water rats starting on day 3. C–E, Total distance traveled (centimeters) was significantly greater in Sucrose than Water rats in the 3 min time bin immediately after Sucrose on days 7 and 8 while pre-sucrose activity did not change. F, Distance traveled in the 3 min time bin immediately after bottle removal was positively correlated with the amount of sucrose consumed. B–E, Data are expressed as mean + SEM and were analyzed using one-way ANOVA on each training day followed by Fisher's post hoc tests. F, Data were analyzed using a Kendall tau rank correlation test (τ = 0.285, Z = 3.156).
We observed that within 3 d of training, Sucrose animals consumed significantly more sucrose solution than Water animals consumed water (Fig. 1B). Additionally, while no significant differences in spontaneous locomotion were observed on training days 1–6 (data not shown), we observed significant elevation of total distance traveled in Sucrose animals compared with Water animals in the 3 min after bottle removal on day 7 (Fig. 1D), and this difference was also present on day 8 (Fig. 1E). No differences in total distance traveled were observed between Water and Sucrose animals in the 3 min before bottle introduction on any of the test days (Fig. 1C), suggesting elevated locomotion was an acute response to sucrose ingestion specific to the sucrose-trained rat, rather than a conditioned response to the locomotor chamber. In keeping with this possibility, there was a significant positive correlation between the amount of sucrose consumed and the total distance traveled (Fig. 1F). There was no difference in animal weights between the Sucrose and Water groups before or after the 7 d of training (data not shown).
Sucrose ingestion induces CPAR incorporation
Sucrose training led to a transient elevation of locomotion on the final training day. To determine whether this consequence of sucrose ingestion was accompanied by electrophysiological changes in the nucleus accumbens, a region that regulates reward behavior, we prepared nucleus accumbens slices immediately following bottle removal on day 7 and recorded from neurons of the accumbens core (Fig. 2A). The core subregion has been implicated in locomotor responses to rewarding stimuli (Sesack and Grace, 2010). We found that both the amplitude and the frequency of spontaneous mEPSCs were significantly greater in the accumbens core of Sucrose animals compared with Water animals (Fig. 2B). This demonstrated that repeated sucrose consumption could positively regulate synaptic transmission in the nucleus accumbens core. To determine whether CPAR incorporation played a role in potentiation after sucrose, we determined rectification indices for accumbens core neurons by measuring EPSCs at different membrane potentials (Fig. 2C–E). CPARs are inwardly rectifying at depolarized potentials because of endogenous polyamine blockade. We observed significant rectification in recordings from neurons of Sucrose animals, as indicated by nonlinearity in the I/V relationship, compared with Water animals (Fig. 2E), in addition to a significant increase in rectification index (Fig. 2F).
Figure 2.
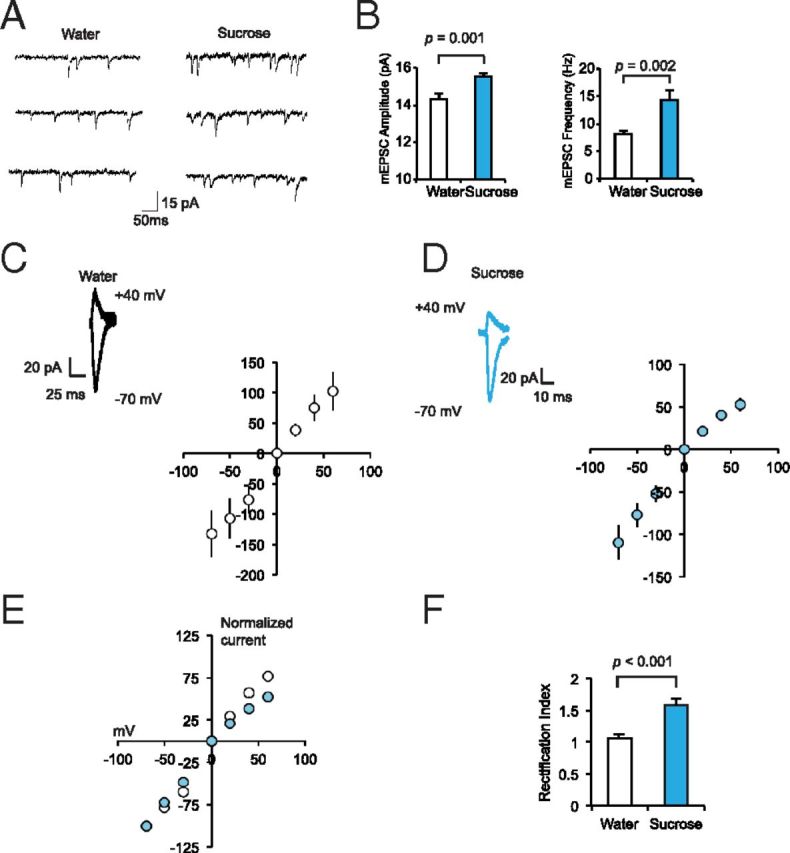
Accumbens core synapses are potentiated after repeated sucrose ingestion. A–C, Spontaneous EPSCs in accumbens core neurons (n = 10 cells from 3 rats, Water; n = 10 cells from 3 rats, Sucrose) after 7 d sucrose or water training. Slices were prepared immediately after bottle removal. D, E, Evoked EPSCs in accumbens core neurons (n = 10 cells from 4 rats, Water; n = 11 cells from 5 rats, Sucrose) measured at −70, −50, −30, 0, +20, +40, and +60 mV. In F, evoked EPSCs from individual records were normalized to the current at −70 mV and averaged for water and sucrose animals as indicated. Rectification index (F) calculated as EPSC−70/EPSC+40. B–G, Data are represented as mean ± SEM and analyzed using unpaired, two-tailed t tests.
To confirm elevation of CPAR levels by another method, we recorded from accumbens core neurons after inclusion of the specific CPAR blocker, Naspm in the bath. We found that Naspm significantly decreased EPSC amplitude in recordings from neurons from Sucrose but not Water animals (Fig. 3A–C). Additionally, after Naspm treatment, the I/V relationship in neurons from Sucrose animals became linear, reflecting the inhibition of CPARs in Sucrose animal synapses, while no significant effect on the I/V relationship was observed after Naspm treatment in neurons from Water animals (Fig. 3D). These results demonstrate that repeated sucrose ingestion induces the incorporation of CPARs in synapses of the accumbens core.
Figure 3.

Sucrose ingestion causes incorporation of Ca2+-permeable AMPARs. A, B, Evoked EPSCs before and after bath Naspm (200 μm) wash-in. B, Mean evoked EPSCs before and after bath Naspm wash-in (n = 9 cells from 4 rats, water; n = 10 cells from 5 rats, sucrose). C, Mean EPSCsNaspm/baseline. D, I/V relationship before and after wash-in of Naspm into the bath. C, D, Data are represented as mean ± SEM and analyzed with an unpaired, two-tailed t test (C).
Sucrose ingestion induces GluA1 trafficking
CPARs are AMPARs that lack the GluA2 AMPAR subunit. Thus, synaptic incorporation of CPARs most often involves synaptic activity-dependent trafficking of the GluA1 subunit (Plant et al., 2006; Isaac et al., 2007; Liu and Zukin, 2007; He et al., 2009). To confirm CPAR synaptic incorporation following sucrose training, we investigated whether sucrose ingestion increased synaptic expression of GluA1. Rats were given access to sucrose as described above for 7 consecutive days. On days 1, 3, 5, and 7, we isolated the whole-cell, synaptosome and PSD fractions from three brain regions: accumbens core (core), accumbens shell (shell), and somatosensory cortex (cortex). We analyzed the whole-cell and PSD fractions by Western blot for expression of GluA1 and GluA2.
We found no changes in GluA1 or GluA2 in the whole-cell fractions of accumbens lysates on the test days examined, suggesting repeated sucrose consumption does not regulate the overall levels of these proteins (Fig. 4A–C). In the accumbens PSD fractions, however, GluA1 increased significantly on day 7 in the core but not in the shell while GluA2 did not change significantly in either fraction (Fig. 4D–F; data not shown). We observed no significant increase in GluA1 in the accumbens core PSD fractions on the preceding test days (Fig. 4D–F) and GluA1 or GluA2 did not change in the cortex PSD fractions on any of the test days (data not shown). Increased GluA1, especially relative to GluA2, in accumbens core PSDs after repeated sucrose ingestion is in keeping with the increased rectification observed in accumbens core neurons, as described above.
Figure 4.

PSD GluA1, but not GluA2, is increased in nucleus accumbens core after sucrose ingestion. A, Western blots of accumbens core whole-cell lysates harvested from Water or Sucrose rats on days 1, 3, 5, and 7 of training (n = 3 rats for Water and Sucrose on each training day). B, C, GluA1:tubulin or GluA2:tubulin integrated density of Sucrose rats normalized to Water rats. D, Western blots of accumbens core PSD fractions harvested from Water or Sucrose rats on days 1, 3, 5, and 7 of training (n = 3 rats for Water and Sucrose on each training day). E, F, GluA1 or GluA2 integrated density of Sucrose rats normalized to Water rats. B, C, E, F, Data are represented as mean + SEM and were analyzed using one-way ANOVA followed by Fisher's post hoc tests on each test day.
Activity-dependent GluA1 trafficking has been shown to contribute synaptic plasticity in vitro and also in vivo (Lu and Roche, 2012). A rapid, multistep mechanism for GluA1 trafficking has been demonstrated in vitro (Sun et al., 2005, 2008,Serulle et al., 2007). Thus far, however, the contribution of this multistep mechanism to GluA1 synaptic accumulation in vivo has not been tested. To determine whether sucrose training induces GluA1 trafficking acutely by the multistep mechanism, we localized GluA1 at nucleus accumbens synapses of sucrose- and water-trained animals by quantitative electron microscopy. Accumbens core tissue was harvested on the seventh day of sucrose training from three test groups of rats. These were rats that consumed (1) water for 7 d (Water), (2) sucrose for 7 d (Sucrose), and (3) sucrose for 6 d and water on day 7 (Sucrose/Water). Rats were killed on the seventh day, 5 min following consumption of sucrose or water. Thus, comparison of two of the test groups, Sucrose/Water and Sucrose animals, to each other and to Water animals revealed the timescale of postsynaptic changes induced by sucrose consumption in sucrose-trained rats. We measured PEG-labeled GluA1 in five different postsynaptic compartments: dendritic spine cytosol (intraspinous), extrasynaptic plasma membrane (membrane), PSD, near PSD, and synaptic cleft, with the final three compartments being grouped together as “PSD” (Fig. 5A). To eliminate experimenter bias, electron micrograph test group identities were triple blinded.
Figure 5.

Electron microscopy reveals induction of multistep GluA1 trafficking by sucrose ingestion. A, GluA1 was PEG labeled and particles were classified into five postsynaptic regions: (1) intraspinous, (2) extrasynaptic membrane, or (3) PSD (cleft + at PSD + near PSD). B, Electron micrographs were prepared from Water, Sucrose/Water, and Sucrose animals (n = 3 animals/test group) and the number of GluA1 PEG particles was quantified for each individual synapse (n = 279 synapses/condition). Scale bar, 500 nm. C–F, Repeated sucrose ingestion elevates intraspinous and PSD GluA1 while acute sucrose ingestion induces rapid trafficking of GluA1 to the extrasynaptic membrane. Data are represented as mean + SEM and were analyzed using one-way ANOVA followed by Fisher's post hoc tests. C–E, Data are presented as averages of the number of particles per spine.
Both Sucrose and Sucrose/Water animals exhibited significantly elevated intraspinous GluA1 relative to Water animals (Fig. 5B,C). This suggests that chronic sucrose consumption increases an intracellular pool of GluA1-containing AMPARs adjacent to synaptic sites, receptors that may be readily available for synaptic trafficking, and, importantly, that this intracellular pool can persist for 24 h following the final consumption of sucrose. We then explored the significant question of whether an acute sucrose stimulus can induce rapid GluA1 trafficking. We observed that extrasynaptic plasma membrane GluA1 was significantly increased in Sucrose animals compared with both Sucrose/Water and Water animals (Fig. 5B,D). This observation indicates that a natural, orosensory reward provided by a single sucrose stimulus can rapidly (<5 min) but transiently (decay time <24 h) elevate the extrasynaptic population of GluA1-containing AMPARs, creating a labile pool from which the receptors may traffic to the synapse.
Significantly, in vitro studies have suggested that synaptic incorporation of AMPARs takes place in two steps. In the first, glutamate- or dopamine-dependent Ser 845 phosphorylation elevates the levels of receptors at extrasynaptic sites in the plasma membrane (Esteban et al., 2003; Sun et al., 2005, 2008, Serulle et al., 2007), while in the second, Ser 818 phosphorylation promotes synaptic incorporation (Boehm et al., 2006). Our electron microscopy comparison of Sucrose animals with Sucrose/Water and Water animals demonstrates that the first step of GluA1 trafficking observed in vitro (Makino and Malinow, 2009), rapid trafficking to the extrasynaptic membrane, also takes place in vivo following provision of an orosensory reward.
In keeping with electrophysiology and biochemical results described above, PEG electron microscopy demonstrated that sucrose intake also induced the second step in GluA1 trafficking—GluA1 receptor entry to the synapse—since the level of GluA1-immunoreactivity at the PSD was significantly greater for Sucrose compared with Water rats, and there was a trend toward increased GluA1 in Sucrose/Water compared with Water rats (Fig. 5B,E). The increase in Sucrose/Water animals is consistent with either rapid incorporation of GluA1, which decays with a synaptic half-life of ∼24 h, or rapid incorporation of GluA1 and replacement by synaptic GluA1/2 over a similar time period. The percentage of synapses expressing GluA1 in the PSD was also significantly greater in Sucrose rats compared with Water rats (Fig. 5F), suggesting that GluA1 trafficked to synapses that previously lacked GluA1. This also suggests that the increase in mEPSC amplitude observed in Sucrose rats results from an increase in synaptic GluA1, and that the increase in mEPSC frequency may result from recruitment of GluA1 to previously silent accumbens synapses, although potentiation of glutamate release cannot be dismissed. We also measured the number of synapses in each of the three test groups to determine whether sucrose ingestion induced synaptogenesis; there were no differences among the three test groups (data not shown). We conclude that repeated sucrose ingestion elevates a stable (>24 h) intraspinous pool of GluA1, and a single sucrose stimulus to a sucrose-trained (6 d) rat is sufficient to rapidly (5 min) elevate GluA1 in the extrasynaptic plasma membrane, potentially drawing receptors from the intraspinous pool. We suggest that a portion of these extrasynaptic receptors is stably incorporated into the PSD, leading to the observed rectification index and PSD GluA1 changes, before the extrasynaptic pool returns to baseline at 24 h following stimulation. These results reveal that a natural reward can acutely induce rapid GluA1 trafficking in a trained animal.
CPAR activity is required for elevated locomotion after sucrose ingestion
MSNs receive both dopaminergic and glutamatergic inputs (Calabresi et al., 1992). To assess the involvement of glutamate signaling in the elevated spontaneous locomotion that we observed after sucrose ingestion in sucrose-trained rats, we implanted cannulae into the accumbens core of rats, and trained animals in locomotor measurement chambers as described above. On day 8 of sucrose training, we microinjected Naspm into the core before placement in the locomotor test chamber. The injection decreased the total distance traveled of the Sucrose animals and eliminated the difference between Sucrose and Water animals seen immediately following bottle removal (Fig. 6A). To verify that stress caused by handling of the animals had not affected the sucrose response, we injected saline into the core on the following day (day 9 of sucrose training); again significant hyperactivity was observed in the Sucrose animals immediately after bottle removal (Fig. 6B). This demonstrates that Naspm had specifically inhibited sucrose-induced elevation of locomotion. Injection of the NMDAR antagonist, APV, into the core on the following days also eliminated the difference between Sucrose and Water animals (Fig. 6C), demonstrating that NMDARs are also required for elevation of spontaneous locomotion after sucrose ingestion. To determine whether a conditioned response to the test chamber played a role in hyperactivity induction, animals were placed in the chamber for 35 min without bottle introduction; no differences in distance traveled were observed between Sucrose and Water animals (Fig. 6D). Naspm and APV did not affect sucrose consumption (Fig. 6E), demonstrating that core CPARs and NMDARs are not required for vigorous intake of sucrose. Animals in which cannulae were not placed in the accumbens core (2 of 15 animals), as evaluated post kill (Fig. 6F), were not included in the study. In conclusion, these data together demonstrate that consumption of sucrose by a sucrose-trained rat induces synaptic trafficking of GluA1 in 5 min, and that blockade of signaling mechanisms that control this trafficking prevents the elevation of spontaneous locomotor activity after sucrose.
Figure 6.

Elevated spontaneous locomotion after sucrose ingestion requires CPARs and NMDARs. A–D, Total distance traveled in 3 min time bin after sucrose or water bottle removal (n = 6 Water rats, 7 Sucrose rats). Bilateral microinjections of Naspm (40 μg/side) or APV (4 μg/side) were performed before placement of rats in locomotor measurement chambers. E, Sucrose or water consumed after microinjections. F, Placement of cannulae in the accumbens core. Data are represented as mean + SEM and were analyzed using one-way ANOVA followed by Fisher post hoc tests.
Two pathways for sucrose signaling may be envisaged. One, a strictly chemosensory or orosensory pathway, is initiated by sucrose binding to the sweet taste receptor, which corresponds to the heteromeric G-protein-coupled receptor complex, T1R2/T2R3 (Kitagawa et al., 2001; Max et al., 2001; Nelson et al., 2001; Sainz et al., 2001). Calorie-rich nutrients can also regulate brain function by metabolic pathways independent of taste, although the mechanisms are not well understood (de Araujo et al., 2008). To distinguish between these two alternatives for the pathway of GluA1-trafficking induced by sucrose, we repeated the training protocol with three groups of rats (four rats/group) that were given access for 5 min to bottles containing, water, 25% sucrose solution, or 3% saccharin (Sweet'N Low). The bottles were removed and the rats remained for 15 min longer in the training cage. Training was repeated for 7 d. The volumes of liquid consumed by the sucrose and saccharin test groups were not significantly different from one another and both were greater than consumption by the water group, consistent with reward by both sweet substances (Fig. 7A). On the seventh day of drinking, immediately following bottle removal, rats were killed, the accumbens core tissue was harvested and pooled for each test group, the PSD fraction isolated, and GluA1 levels probed by Western blot (Fig. 7B). As before, animals that consumed sucrose showed elevated GluA1 in the PSD fraction relative to the water group (Fig. 7C). Significantly, GluA1 was also elevated in the PSD fraction of animals that consumed saccharin. There was no significant difference in the levels of GluA1 in the whole-cell fractions from accumbens core of water, sucrose, and saccharin animals, suggesting that the GluA1 increase was specific to the synaptic fraction (Fig. 7D). Because saccharin stimulates the same heteromeric G-protein-coupled taste receptor complex as sucrose (Nelson et al., 2001; Masuda et al., 2012), but lacks caloric value, we conclude that stimulation of the sweet taste receptor is sufficient to initiate signaling that elevates GluA1 levels at nucleus accumbens core synapses.
Figure 7.

Saccharin training induces an increase in synaptic GluA1 similar to sucrose training. Rats were given access to water, 3% saccharin, or 25% sucrose daily (4 rats per group), in a training cage for 5 min per day, whereupon the feeding bottle was removed and the rats were maintained in the cage for 15 additional minutes. This training was performed for 7 consecutive days. A, The mass of Saccharin and Sucrose solution consumed were similar, and both were greater than the mass of Water consumed. B, Western blot analysis of GluA1 and actin (control) in the PSD and whole-cell fractions of the nucleus accumbens core isolated from Water, Saccharin, and Sucrose rats. Relative to Water training, Saccharin and Sucrose training significantly elevated GluA1 levels similarly in the PSD (Fc = 10.6; *p = 0.009, **p < 0.001) (C) but not the whole-cell fractions (D) of the accumbens core. Western blot quantitation of GluA1 was normalized to actin levels.
Discussion
We have shown that an orosensory reward, repeated sucrose consumption, can acutely induce GluA1 synaptic incorporation through a multistep trafficking mechanism previously described in vitro. Repeated sucrose consumption over 6–7 d potentiated accumbens core synapses electrophysiologically through the insertion of CPARs. This effect was accompanied by accumulation of GluA1 but not GluA2 in the PSD of the core, and was regionally and temporally specific, as no changes were observed before training day 7 in the core, and no change was observed in the accumbens shell or in somatosensory cortex. Electron microscopic analysis revealed that repeated sucrose ingestion elevated a relatively stable (t1/2 > 24 h) population of intraspinous GluA1-containing receptors. Sucrose also rapidly (5 min) and transiently (t1/2 <24 h) elevated levels of GluA1-containing receptors at extrasynaptic sites in sucrose-trained animals, increasing the population of AMPARs capable of diffusing laterally into the synapse. Synaptic GluA1, both as represented by the PSD fraction and as detected by PEG-EM, was significantly increased in Sucrose compared with Water animals. From these results we propose that the two-step mechanism of extrasynaptic exocytosis followed by synaptic trafficking for AMPAR synaptic insertion previously described in vitro (Sun et al., 2005; Boehm et al., 2006; Oh et al., 2006; Serulle et al., 2007; Makino and Malinow, 2009) can be initiated rapidly in vivo by a natural, orosensory reward.
Changes in synaptic GluA1 levels were only observed after seven training sessions, suggesting that a several day-long process is required for potentiation. In biochemical experiments in vivo, we did not observe significant increases in accumbens core PSD GluA1 levels on days 1, 3, and 5 of sucrose training; only after 7 d of sucrose training was GluA1 in the PSD significantly elevated. In electron microscopy experiments, we observed that Sucrose/Water animals, which had been sucrose trained for 6 d but had not received a sucrose stimulus in 24 h, exhibited a trend toward increased PSD GluA1. These animals also exhibited elevated intraspinous GluA1 compared with Water animals, but no change in extrasynaptic membrane GluA1 was observed. From these results we draw three conclusions. First, GluA1-containing AMPARs accumulate intraspinously with successive sucrose stimulations. Given that previous studies have demonstrated that sucrose ingestion induces dopamine release in the accumbens (Rada et al., 2005; Cacciapaglia et al., 2012; McCutcheon et al., 2012), and that D1 receptors (D1Rs) can drive local GluA1 translation in dendrites (Smith et al., 2005), dopamine release after sucrose ingestion may trigger local GluA1 synthesis that leads to intraspinous GluA1 accumulation. Alternatively the intraspinous increase may reflect trafficking of GluA1 from distal sites. It is likely that exocytotic trafficking from this elevated intraspinous pool contributes to the extrasynaptic pool in the plasma membrane. Second, the observation of an increase in extrasynaptic membrane GluA1 in Sucrose, but not in Sucrose/Water or Water animals, suggests that the extrasynaptic receptors either move through a second step into the synapse or are endocytosed within the 24 h following sucrose consumption, making the extrasynaptic pool transient. Third, the elevation of Sucrose animal PSD GluA1 compared with Water animals, but not Sucrose/Water animals, also suggests that after each sucrose stimulus, receptors move laterally into the synapse from the pool of receptors trafficked rapidly to the extrasynaptic plasma membrane. We cannot rule out that GluA1 trafficks directly from the intraspinous pool to the synapse. Such a path, however, seems unlikely given the studies that show GluA1 is inserted extrasynaptically (Sun et al., 2005; Boehm et al., 2006; Oh et al., 2006; Serulle et al., 2007; Makino and Malinow, 2009). These findings represent the first demonstration that the time course for GluA1 trafficking (<5 min) and pathway observed in vitro are also observed in vivo. In addition, our results suggest that repeated rewarding stimuli modify the capacity for the potentiation of a synapse by elevating the pool of intraspinous receptors capable of being trafficked.
Because saccharin induced GluA1 trafficking similarly to sucrose, the caloric content of sucrose is not required. Saccharin stimulates the same sweet taste receptor, T1R2/T2R3, as sucrose (Nelson et al., 2001; Masuda et al., 2012), suggesting activation of this receptor likely initiates the incorporation of GluA1 into MSN synapses. Sucrose elevates dopamine release in the accumbens from VTA neurons (Rada et al., 2005; Cacciapaglia et al., 2012; McCutcheon et al., 2012) leading to GluA1 surface trafficking. Thus the pathway that links the sweet taste receptor to the VTA is likely to be central to the plasticity studied here.
It is likely that rapid GluA1 trafficking after sucrose ingestion plays a role in regulating spontaneous locomotion. Indeed, in sucrose-trained animals, inhibition of CPARs prevented elevation of spontaneous locomotor activity immediately after sucrose ingestion. Total distance traveled by rats after sucrose consumption measured on consecutive days was significantly elevated only for the period 3 min immediately after sucrose consumption on the seventh day of training. Increased activity immediately after sucrose was observed starting in day 3 of training, but did not become significantly different until day 7. This time course of activity correlates with the time course of accumulation of GluA1 in accumbens core dendrites. The increased locomotion was a functional consequence of CPAR trafficking to MSN synapses in the accumbens core since Naspm injection into the core inhibited the increases in activity. Prevention of elevated locomotion by an NMDAR inhibitor demonstrated that glutamate signaling via NMDARs as well as CPARs was necessary to elevate locomotor activity. Sucrose ingestion, however, was not affected by blockade of glutamate signaling, in keeping with previous studies demonstrating that the accumbens core is involved in the orchestration of motor responses related to orosensory reward but not consumption itself (Smith, 2004). A similar time course for the development of hyperlocomotion has been reported for development of conditioned hyperactivity in animals fed their daily meal in a distinctive environment (Matthews et al., 1996). If the present response were a conditioned hyperactivity arising from pairing of context and sucrose, however, it would have preceded sucrose delivery, which was not observed. It is possible that the subjects are displaying an exploratory arousal. Further experiments would be necessary to distinguish whether elevated locomotion after sucrose ingestion was arousal of exploration as opposed to a form of motor sensitization or other behavior. In any case, the elevation of spontaneous locomotion required glutamate signaling, and resulted, at least in part, from the incorporation of CPARs in the accumbens core.
The increased locomotor activity following sucrose ingestion may result directly from the observed potentiation of accumbens core synapses, as increased output from the direct basal ganglia pathway promotes locomotion through disinhibition of motor thalamus (Sesack and Grace, 2010). The potentiated synapses most likely reside on direct pathway accumbens core neurons, which express D1Rs. Potentiation of direct pathway neurons synapses would result if D1R activity induced trafficking GluA1-containing AMPARs to synapses in these neurons following robust dopamine release. The resulting potentiation would increase activity in inhibitory projections of direct pathway neurons to basal ganglia output nuclei, thus disinhibiting motor thalamus and promoting motor cortex activity (Kravitz et al., 2010; Sesack and Grace, 2010; Gerfen and Surmeier, 2011). The synaptic potentiation observed after repeated sucrose ingestion likely takes place specifically in direct pathway neurons because dopamine acting through the D1R can induce GluA1 S845 phosphorylation, leading to surface trafficking.
A number of studies have examined the effects of repeated stimulation with cocaine followed by withdrawal, a treatment that exerts profound effects on reward system function and eventually leads to cocaine sensitization, which is characterized by elevated motor response to cocaine, drug craving, and relapse (Kalivas et al., 1998). Repeated intraperitoneal injection with cocaine for 5–10 d followed by withdrawal resulted in a gradual increase over 14 d in surface GluA2-containing AMPARs (Boudreau et al., 2007; Kourrich et al., 2007). However, at 45 d of withdrawal following 10 d of self-administration, a large increase in rectification index was observed in rat MSNs (McCutcheon et al., 2011b) indicating an increase in CPARs. Thus, CPAR trafficking has been observed following both sucrose ingestion, in the current work, and cocaine self-administration, albeit under very different treatment conditions. Because the immediate consequences of cocaine self-administration or injection (e.g., at 5 min post) are not known, cocaine action cannot be compared directly to the current sucrose work. Likewise, it is not known if CPARs persist in MSN synapses of sucrose-trained animals following cessation of sucrose training or if such animals display sucrose sensitization following lengthy withdrawal.
Understanding how rewarding stimuli regulate accumbens plasticity and behavior are critical to addressing addiction, hyperphagia, pathological gambling, and other behavioral disorders (Berridge, 2009; Basar et al., 2010; Lüscher and Malenka, 2011). Sugar overconsumption contributes to the obesity epidemic (Hu and Malik, 2010), and although potentially similar to drug abuse (Avena et al., 2008), its mechanism has not been extensively explored. The current findings establish basic elements of reward-induced plasticity from which future studies can address regulation of complex behavior, potentially providing novel avenues to confront reward-related pathologies.
Footnotes
This work was supported by National Institute of Mental Health Predoctoral Fellowship F31MH76617-01 and National Institutes of Health Training Grant 5T32DC000063 to the New York University Training Program in the Neurosciences (D.S.T.), R01NS061920 from the National Institute of Neurological Disorders and Stroke (E.B.Z.), 1R21MH091445-01 from the National Institute of Mental Health and Office of Research on Women's Health, Klarman Family Foundation Grants Program in Eating Disorders Research, NYU's Research Challenge Fund and P30EY13079 (C.A.), National Institute on Drug Abuse Grant DA003956 and an Independent Investigator Award from NARSAD (K.D.C.), National Institute on Deafness and other Communications Disorders Grant DC009635 to R.C.F., and by a seed grant in the Center of Excellence on Addiction from the New York University Langone Medical Center. We thank members of the Ziff Laboratory, past and present, for technical assistance and helpful discussions, including H. Girma, L. Lee, and Drs. B. Fernholz, B. Jordan, W. Lu, G. Rameau, S. Restituito, and Y. Serulle.
The authors declare no competing financial interests.
References
- Avena NM, Rada P, Hoebel BG. Evidence for sugar addiction: behavioral and neurochemical effects of intermittent, excessive sugar intake. Neurosci Biobehav Rev. 2008;32:20–39. doi: 10.1016/j.neubiorev.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basar K, Sesia T, Groenewegen H, Steinbusch HW, Visser-Vandewalle V, Temel Y. Nucleus accumbens and impulsivity. Prog Neurobiol. 2010;92:533–557. doi: 10.1016/j.pneurobio.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Berridge KC. ‘Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiol Behav. 2009;97:537–550. doi: 10.1016/j.physbeh.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Cacciapaglia F, Saddoris MP, Wightman RM, Carelli RM. Differential dopamine release dynamics in the nucleus accumbens core and shell track distinct aspects of goal-directed behavior for sucrose. Neuropharmacology. 2012;62:2050–2056. doi: 10.1016/j.neuropharm.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr KD, Chau LS, Cabeza de Vaca S, Gustafson K, Stouffer M, Tukey DS, Restituito S, Ziff EB. AMPA receptor subunit GluR1 downstream of D-1 dopamine receptor stimulation in nucleus accumbens shell mediates increased drug reward magnitude in food-restricted rats. Neuroscience. 2010;165:1074–1086. doi: 10.1016/j.neuroscience.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Carelli RM. The nucleus accumbens and Pavlovian reward learning. Neuroscientist. 2007;13:148–159. doi: 10.1177/1073858406295854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo IE, Oliveira-Maia AJ, Sotnikova TD, Gainetdinov RR, Caron MG, Nicolelis MA, Simon SA. Food reward in the absence of taste receptor signaling. Neuron. 2008;57:930–941. doi: 10.1016/j.neuron.2008.01.032. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Heine M, Groc L, Lee MC, Choquet D. Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron. 2007;54:447–460. doi: 10.1016/j.neuron.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–1525. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1–S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu FB, Malik VS. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: epidemiologic evidence. Physiol Behav. 2010;100:47–54. doi: 10.1016/j.physbeh.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Pierce RC, Cornish J, Sorg BA. A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol. 1998;12:49–53. doi: 10.1177/026988119801200107. [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Kusakabe Y, Miura H, Ninomiya Y, Hino A. Molecular genetic identification of a candidate receptor gene for sweet taste. Biochem Biophys Res Commun. 2001;283:236–242. doi: 10.1006/bbrc.2001.4760. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lu W, Isozaki K, Roche KW, Nicoll RA. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc Natl Acad Sci U S A. 2010;107:22266–22271. doi: 10.1073/pnas.1016289107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Roche KW. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol. 2012;22:470–479. doi: 10.1016/j.conb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Masuda K, Koizumi A, Nakajima K, Tanaka T, Abe K, Misaka T, Ishiguro M. Characterization of the modes of binding between human sweet taste receptor and low-molecular-weight sweet compounds. PloS One. 2012;7:e35380. doi: 10.1371/journal.pone.0035380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews K, Wilkinson LS, Robbins TW. Repeated maternal separation of preweanling rats attenuates behavioral responses to primary and conditioned incentives in adulthood. Physiol Behav. 1996;59:99–107. doi: 10.1016/0031-9384(95)02069-1. [DOI] [PubMed] [Google Scholar]
- Max M, Shanker YG, Huang L, Rong M, Liu Z, Campagne F, Weinstein H, Damak S, Margolskee RF. Tas1r3, encoding a new candidate taste receptor, is allelic to the sweet responsiveness locus Sac. Nat Genet. 2001;28:58–63. doi: 10.1038/ng0501-58. [DOI] [PubMed] [Google Scholar]
- McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011a;31:5737–5743. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011b;31:14536–14541. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Beeler JA, Roitman MF. Sucrose-predictive cues evoke greater phasic dopamine release than saccharin-predictive cues. Synapse. 2012;66:346–351. doi: 10.1002/syn.21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelescu H, Kelso CM, Lázaro-Muñoz G, Purpura M, Cain CK, Ledoux JE, Aoki C. Endogenous GluR1-containing AMPA receptors translocate to asymmetric synapses in the lateral amygdala during the early phase of fear memory formation: an electron microscopic immunocytochemical study. J Comp Neurol. 2010;518:4723–4739. doi: 10.1002/cne.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJ, Zuker CS. Mammalian sweet taste receptors. Cell. 2001;106:381–390. doi: 10.1016/s0092-8674(01)00451-2. [DOI] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Rada P, Avena NM, Hoebel BG. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience. 2005;134:737–744. doi: 10.1016/j.neuroscience.2005.04.043. [DOI] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sainz E, Korley JN, Battey JF, Sullivan SL. Identification of a novel member of the T1R family of putative taste receptors. J Neurochem. 2001;77:896–903. doi: 10.1046/j.1471-4159.2001.00292.x. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP. Accumbens dopamine mediates the rewarding effect of orosensory stimulation by sucrose. Appetite. 2004;43:11–13. doi: 10.1016/j.appet.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron. 2005;45:765–779. doi: 10.1016/j.neuron.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Milovanovic M, Zhao Y, Wolf ME. Acute and chronic dopamine receptor stimulation modulates AMPA receptor trafficking in nucleus accumbens neurons cocultured with prefrontal cortex neurons. J Neurosci. 2008;28:4216–4230. doi: 10.1523/JNEUROSCI.0258-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]