

Abstract
With the advent of improved molecular biology techniques, the genetic basis of an increasing number of reproductive disorders has been elucidated. Mutations in at least 20 genes cause hypogonadotropic hypogonadism including Kallmann syndrome in about 35–40% of patients. The two most commonly involved genes are FGFR1 and CHD7. When combined pituitary hormone deficiency includes hypogonadotropic hypogonadism as a feature, PROP1 mutations are the most common of the six genes involved. For hypergonadotropic hypogonadism, mutations in 14 genes cause gonadal failure in 15% of affected females, most commonly in FMR1. In eugonadal disorders, activating FSHR mutations have been identified for spontaneous ovarian hyperstimulation syndrome; and WNT4 mutations have been described in mullerian aplasia. For other eugonadal disorders, such as endometriosis, polycystic ovary syndrome, and leiomyomata, specific germline gene mutations have not been identified, but some chromosomal regions are associated with the corresponding phenotype. Practical genetic testing is possible to perform in both hypogonadotropic and hypergonadotropic hypogonadism and spontaneous ovarian hyperstimulation syndrome. However, clinical testing for endometriosis, polycystic ovary syndrome, and leiomyomata is not currently practical for the clinician.
Keywords: Genetics of hypogonadism, Mullerian aplasia, Endometriosis, Polycystic ovary syndrome, Spontaneous ovarian hyperstimulation, syndrome, Leiomyomata
1. Introduction
In humans, sexual development and reproductive function occur by the actions of the hypothalamic–pituitary–gonadal axis instigated by gonadotropin releasing hormone (GnRH). GnRH neurons originate outside the brain in the nasal region, but migrate alongside olfactory nerves across the cribriform plate into the hypothalamus during embryological development (Tobet and Schwarting, 2006). Once reaching the hypothalamus, these neurons send projections from their location in the arcuate nucleus to the median eminence where nerve terminals contact hypophy-seal-portal vessels. Pulsatile GnRH is released into these capillaries where it is delivered and binds to its cell surface receptor on pituitary gonadotropes, which are then induced to synthesize and secrete follicle stimulating hormone (FSH) and luteinizing hormone (LH). The gonadotropins in turn bind their cognate G-protein coupled membrane receptors in the gonads, which then elaborate sex steroids and gametes. The sex steroids then exert negative feedback at both the hypothalamus and pituitary to control the gonadotropin stimulus (Plant, 2008). A number of other peptide factors, including GnIH (gonadotropin inhibitory hormone), inhibins, and antimullerian hormone also play important roles in reproductive function (Plant, 2008; Bentley et al., 2009).
In the past decade, much has been learned about the genetic etiology of reproductive dysfunction in women. Reproductive disorders in humans can somewhat arbitrarily be divided into hypogonadal and eugonadal dysfunction (Layman, 2002). Manifesting low levels of estrogen, hypogonadal states represent a more severe perturbation in reproductive function than eugonadal states. However, the molecular basis of hypogonadism has been better understood, and increasing numbers of disorders now are known to be caused by mutations in specific genes, which have functional consequences. Similarly, for some eugonadal disorders, such as spontaneous ovarian hyperstimulation syndrome and mullerian aplasia, some genes have been elucidated. In contrast, the genetic basis of eugonadal disorders such as polycystic ovary syndrome (PCOS), endometriosis, and leiomyomata is largely unknown, although some genomic loci are associated with their phenotypes. In this review, the genetic basis of hypogonadal disorders and five selected eugonadal disorders will be summarized with an emphasis, not upon all individual genes involved, but rather on the overriding concepts and practical diagnostic recommendations for clinicians.
2. Hypogonadism
2.1. Hypogonadotropic hypogonadism
2.1.1. Phenotype
When females manifest symptoms of estrogen deficiency, such as absent breast development or hypoestrogenic amenorrhea, there is a lack of negative feedback to the hypothalamus and pituitary gland. Serum gonadotropin levels in these patients are low (or inappropriately normal), indicating that the defect is in the hypothalamus or pituitary. These patients usually have GnRH deficiency, which is often termed idiopathic (or isolated) hypogonadotropic hypogonadism at age ≥17 (age ≥18 in males) once eating disorders, strenuous exercise, or extreme stress are excluded (Bhagavath et al., 2006). The remainder of pituitary function is typically normal and MRI does not reveal a pituitary tumor. If the sense of smell is normal, this disorder is termed normosmic hypogonadotropic hypogonadism (nHH); when an impaired sense of smell accompanies hypogonadotropic hypogonadism, Kallmann syndrome (KS) is present. Although traditionally thought to be an irreversible disorder, this is no longer true for a small percentage of patients who regain reproductive function when treatment is discontinued (Pitteloud et al., 2005). In addition, it is now appreciated that certain somatic anomalies may be present in IHH/KS patients, including midline facial defects (cleft lip/palate and dental agenesis), neurologic disorders (ataxia, synkinesia, visual impairment, and deafness), unilateral renal agenesis, and cardiac defects (Kim et al., 2008). The presence of these congenital anomalies complicates their medical care, and must be considered when nHH/KS patients pursue pregnancy.
2.1.2. The first genes for Kallmann syndrome and normosmic hypogonadotropic hypogonadism
Although difficult to ascertain, the prevalence of nHH/KS is uncommon, but has been found to be higher in males than females. It would be expected that a KS gene would direct the migration of GnRH and olfactory neurons, such that a mutation should impair these functions and cause anosmia (or hyposmia) and GnRH deficiency. This is exactly what was found. The molecular basis of nHH/KS for 35–40% of all patients has been uncovered during the past 20 years beginning with the discovery of the KAL1 gene. In 1991, two different groups of investigators identified the KAL1 gene by positional cloning utilizing very valuable patients with partial X chromosome deletions and translocations (Franco et al., 1991; Legouis et al., 1991). Since KAL1 is localized to the X chromosome and inherited in an X-linked recessive fashion, only males are affected. Interestingly, the rodent ortholog has not been identified, but expression patterns in humans and other organisms, such as chick, have correlated very nicely with the phenotypic features seen in humans (Table 1) (Rugarli and Ballabio, 1993; Lutz et al., 1994). This has clinical relevance in affected patients as up to 50% of males with KAL1 mutations may have unilateral renal agenesis (Hardelin et al., 1993).
Table 1.
KAL1 gene expression in human and chick correlates with the phenotype in patients with Kallmann syndrome (Rugarli and Ballabio, 1993; Lutz et al., 1994).
| Expression | Phenotype |
|---|---|
| Olfactory bulb | Anosmia/hyposmia |
| Cerebellum | Nystagmus Ataxia |
| Spinal cord (corticospinal tract) | Synkinesia |
| Oculomotor nucleus | Eye movement abnormalities |
| Retina | Visual defects |
| Facial mesenchyme | Cleft palate |
| Mesonephros and metanephros | Renal agenesis |
| Limb bud | Pes cavus, limb anomalies |
Initially, it was thought that most IHH/KS would be inherited as an X-linked disorder. However, the most obvious candidate for mutations was GnRH (GNRH1), an autosomal gene, particularly since the naturally occurring hypogonadal mouse had a phenotype of hypogonadotropic hypogonadism due to a partial Gnrh1 deletion (Mason et al., 1986). Amazingly, no human mutations were identified until more than 20 years later (Bouligand et al., 2009; Chan et al., 2009).
In late 1997/early 1998, two groups independently identified compound heterozygous mutations in the GnRH receptor (GNRHR) gene in nHH (de Roux et al., 1997; Layman et al., 1998). The pedigree of one of the families is shown in Fig. 1. The proband was 30 years of age and completely lacked breast development, as did her two younger sisters (ages 21 and 17). The affected brother at age 29 had never shaved and had a very low testosterone. All four affected nHH patients had either undetectable or low but measurable FSH and LH (Layman et al., 1998). The proband had been treated with very high doses of GnRH, but failed to respond, suggesting GnRH resistance. Utilizing denaturing gradient gel electrophoresis and PCR-based DNA sequencing, compound heterozygous GNRHR mutations were identified in all four siblings, each of which markedly impaired GnRH signaling in vitro in this G-protein coupled receptor (Layman et al., 1998).
Fig. 1.
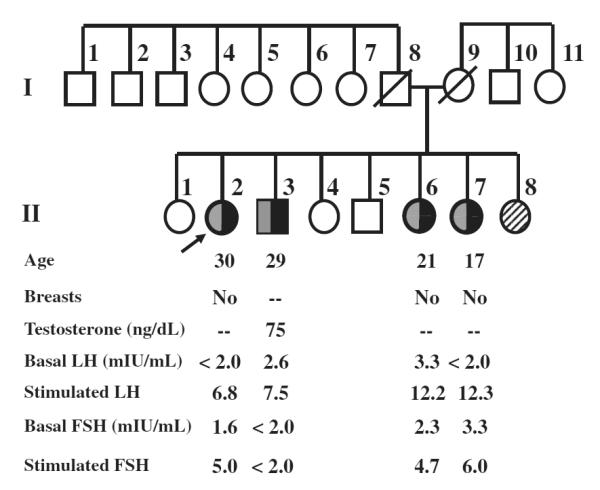
Pedigree of the family with four affected nHH patients with compound heterozygous GNRHR mutations denoted by shaded squares or circles. Female patient II-8 has delayed puberty, but the genotype is unknown. Modified from Layman et al. (1998).
GNRHR mutations were the first mutations identified in nHH patients and represented the first autosomal recessively inherited form of IHH (de Roux et al., 1997; Layman et al., 1998). Therefore, the GNRHR gene was the first gene involved in nHH/KS in women. Most of the mutations subsequently identified were compound heterozygous missense mutations that impaired GnRH binding and/or signaling in vitro. Of clinical interest was the observation that the phenotypes could range from very severe (a complete lack of puberty) to partial pubertal development or constitutional delay of puberty (Kim et al., 2010). To date, GNRHR mutations cause nHH without other additional somatic anomalies, but only account for ~4% of nHH patients (Bhagavath et al., 2005). It was clear that some females manifest KS, which suggested the presence of additional autosomal disease-producing genes (Bhagavath et al., 2006).
2.1.3. Additional nHH/KS genes affecting the hypothalamus
The discovery of KAL1 and GNRHR gene mutations paved the way for additional gene discovery which has resulted in the identification of 24 more genes that have nHH/KS as a phenotypic feature (Table 2). Most of these genes affect the hypothalamus, but at least one—NR0B1, which encodes DAX1 (Muscatelli et al., 1994; Habiby et al., 1996), has both hypothalamic and pituitary effects, while others principally affect hypothalamic or pituitary function. Not surprisingly, at least six ligand/receptor partners have been identified, typically with mutations of the receptor being more common than those of the ligand. These ligand/receptor combinations include KAL1 (Franco et al., 1991; Legouis et al., 1991) and FGF8 (Falardeau et al., 2008)/FGFR1 (Dode et al., 2003) since both FGF8 and anosmin-1, the protein product of KAL1, bind FGFR1; and HS6ST1 encodes a heparan sulfotransferase important in FGF signaling) (Tornberg et al., 2011). Other ligand/receptors include LEP (Strobel et al., 1998)/LEPR (Clement et al., 1998), GNRH1 (Bouligand et al., 2009; Chan et al., 2009)/GNRHR (de Roux et al., 1997; Layman et al., 1998), PROK2/PROKR2 (Dode et al., 2006), KISS1 (Topaloglu et al., 2012)/KISS1R (de Roux et al., 2003; Seminara et al., 2003), and TAC3 (Topaloglu et al., 2009)/TACR3 (Topaloglu et al., 2009). Two of these ligand/receptor complexes—KAL1/FGF8/FGFR1/HS6ST1 and PROK2/PROKR2 predominantly affect GnRH neuron migration since patients with mutations in these genes most commonly present with KS (see below). In contrast, mutations in GNRH1/GNRHR, KISS1/KISS1R, LEP/LEPR, and TAC3/TACR3 result in nHH, suggesting that they more likely affect GnRH regulation. In fact, within the hypothalamus, some neurons secrete kisspeptin, neurokinin B, and dynorphin leading some to term them as KNDy neurons, which have a positive stimulatory effect upon GnRH neurons (Cheng et al., 2010).
Table 2.
Gene mutations causing hypogonadotropic hypogonadism in females. The presumed inheritance patterns are shown as supported by literature. The first description of each gene mutation along with the prevalence of mutations is shown. In publications where digenic mutations are included, only the prevalence of monogenic mutations is shown (it is recognized that for older publications, this will not be apparent, so the stated prevalence could be exaggerated). The six ligand/receptor combinations are shown first and grouped together (1–4, 5–6, 7–8, 9–10, 11–12, 13–14), followed by additional genes (Chan et al., 2009; de Roux et al., 1997; Layman et al., 1998; Kim et al., 2010; Bhagavath et al., 2005; Muscatelli et al., 1994). Genes 21 and 22 are involved in isolated gonadotropin deficiency; genes 23–28 have mutations in combined pituitary hormone deficiency (CPHD) including hypogonadotropic hypogonadism. XLR = X-linked recessive, AD = autosomal dominant; AR = autosomal recessive; SOD = septo-optic dysplasia; IGHD = isolated growth hormone deficiency.
| Gene | Reproductive phenotype |
Nonreproductive phenotype | Inheritance | Prevalence in nHH/KS |
|---|---|---|---|---|
| 1 KAL1(Franco, 1991
#293;Legouis, 1991
#569;Bhagavath, 2007 #1114) |
KS | See Table 1 | XLR | 5–10%; 30–70% in X- linked families |
| 2 FGFR1(KAL2)(Dode, 2004 #243;Pitteloud, 2006 #744) |
nHH and KS | Synkinesia, dental agenesis, bony abnormalities; cleft palate/lip, unilateral absence of nasal cartilage, iris coloboma; clinodactyly |
AD | 10% |
| 3 FGF8(Falardeau, 2008 #265) | nHH and KS | Cleft lip/palate, hearing loss, camplodactyly, | AD? | 1.3% |
| 4 HS6ST1(Tornberg, 2011 #1450) | nHH and KS | Cleft palate; genu valgus | AD? | 0.9% |
| 5 GNRH1(Bouligand, 2009 #110;Chan, 2009 #161) |
nHH | AR | 0.3–0.8% | |
| 6 GNRHR(Layman, 1998 #1125;Bhagavath, 2005 #1117;de Roux, 1997 #224) |
nHH | AR | 4% | |
| 7 KISS1(Topaloglu, 2012 #2505) | nHH | AR | 1% | |
| 8 KISS1R(Seminara, 2003 #858;de Roux, 2004 #220) | nHH | AR | 1.2% | |
| 9 LEP(Strobel, 1998 #906) | nHH | Morbid obesity | AR | ?/rare |
| 10 LEPR(Farooqi, 2007 #272;Clement, 1998 #181) | nHH | Morbid obesity | AR | ?/rare; 3% early-onset obesity |
| 11 PROKR2 (KAL3)(Abreu, 2008 #3;Dode, 2006 #246;Sarfati, 2010 #2701) |
nHH and KS | AR | 2% | |
| 12 PROK2 (KAL4)(Abreu, 2008 #3;Dode, 2006 #246;Sarfati, 2010 #2701) |
KS and nHH | AR | 0–0.9% | |
| 13 TACR3(Gianetti, 2010 #1326;Topaloglu, 2009 #952) |
nHH | AR | 5.5% | |
| 14 TAC3(Gianetti, 2010 #1326;Topaloglu, 2009 #952) |
nHH | AR | 0.2% | |
| 15 NR0B1(Muscatelli, 1994 #677;Zanaria, 1994 #1063) |
nHH | Adrenal hypoplasia congenita; pituitary effects upon gonadotropins | XLR (affected females reported) |
0 in nHH/KS; 82.4%with AHC/IHH |
| 16 PCSK1(Jackson, 1997 #413) | nHH | Obesity | AR | Rare (1 case) |
| 17 CHD7 (KAL5)(Kim, 2008 #1110) | nHH and KS | CHARGE features | AD or sporadic |
6% |
| 18 NELF(Xu, 2011 #1321) | nHH and KS | AR | 0.6% | |
| 19 WDR11(Kim, 2010 #1269) | nHH and KS | AD? | 1.5% | |
| 20 SEMA3A(Hanchate, 2012 #2543;Young, 2012 #2544) |
nHH and KS | AD? | 1–2% | |
| 21 LHB(Lofrano-Porto, 2007 #592) | Isolated LH deficiency |
AR | Rare | |
| 22 FSHB(Layman, 1997 #1127;Matthews, 1993 #629) |
Isolated FSH deficiency |
No hirsutism despite elevated LH | AR | Rare |
| 23 PROP1(CPHD2)(Wu, 1998 #1042;Kelberman, 2009 #2643;Park, 2004 #1119) |
nHH | CPHD (Deficiency of GH, TSH, LH, FSH, prolactin, and ACTH) | AR | 0% nHH; 12% sporadic and 50% familial CPHD |
| 24 HESX1(Dattani, 1998 #212;Newbern, Submitted #2700;Kelberman, 2009 #2643) |
KS | CPHD, SOD, IGHD | AR or AD | 1.4% of KS; 1% of SOD |
| 25 LHX3(CPHD3)(Kelberman, 2009 #2643;Netchine, 2000 #686) |
HH | CPHD (GH, TSH, LH, FSH, prolactin), short cervical spine with limited neck rotation; pituitary small, normal or large |
AR | 1.3% of CPHD |
| 26 LHX4(CPHD4)(Kelberman, 2009 #2643;Machinis, 2001 #1108) |
HH | CPHD (GH, TSH, ACTH, LH, FSH) with cerebellar abnormalities, IGHD, ectopic pituitary gland, small pituitary gland |
AD | 1.2% of CPHD |
| 27 SOX2(Kelberman, 2009 #2643;Kelberman, 2006 #452) |
HH | CPHD, anophthalmia/microphthalmia, other eye defects, anterior pituitary hypoplasia, SOD, hypothalamic hamartomas, sensorineural hearing loss, esophageal atresia, intellectual disability |
AD | 3.4–10% of CPHD, hypothalamic– pituitary disorders |
| 28 SOX3(Kelberman, 2009 #2643;Woods, 2005 #1036) |
HH | CPHD, IGHD, intellectual disability | XLR | 6–7% (males) |
The additional genes that result in nHH/KS are shown in Table 2, along with their frequency, phenotype, and mode of inheritance. It can be seen that KAL1 mutations cause anosmia/hyposmia (KS), while GNRH1/GNRHR, KISS1/KISS1R, TAC3/TACR3, and LEP/LEPR result in nHH. However, the other genes such as FGF8/FGFR1/HS6ST1, PROK2/PROKR2, CHD7 (Kim et al., 2008), NELF (Xu et al., 2011), WDR11 Kim et al., 2010, and SEMA3A (Hanchate et al., 2012; Young et al., 2012) may cause either KS or nHH. These interesting findings blur the lines between the hypothesis that some genes just affect GnRH neuron migration, while others only affect GnRH secretion. In addition, associated non-reproductive phenotypic findings may occur with mutations in half the involved genes (Table 2). For example, FGFR1 mutations may result in cleft lip/palate, dental agenesis, and synkinesia. This gene is particularly interesting since activating mutations have been described in craniosynostosis disorders such as Pfeiffer syndrome (Muenke et al., 1994), Jackson–Weiss syndrome (Roscioli et al., 2000), and other disorders (Kress et al., 2000; Hurley et al., 2004), while inactivating mutations result in KS (and this locus has in fact been designated as KAL2).
Another gene worth considering is CHD7, which was initially found to cause CHARGE syndrome, consisting of a constellation of anomalies that include eye Coloboma, Heart defects, Atresia of the choanae (obstruction of the posterior nasopharynx), Retardation of growth/development, Genital abnormalities, and Ear (both auditory and vestibular dysfunction) anomalies (Vissers et al., 2004). Subsequently, CHD7 mutations were found in nHH/KS patients without CHARGE syndrome, indicating that nHH/KS is a mild allelic disease variant of CHARGE syndrome (Kim et al., 2008). The identification of this important syndrome has very important clinical implications since although it is usually sporadic, it may be autosomal dominant (Vissers et al., 2004), thereby portending a 50% recurrence to offspring.
When the inheritance patterns of known IHH/KS genes are evaluated, it can clearly be seen that the original hypothesis that X-linked recessive disease was the most common has not been substantiated. In fact of the 20 genes identified, only two (KAL1 and NR0B1) are X-linked recessive (Franco et al., 1991; Legouis et al., 1991; Muscatelli et al., 1994), while 12 are autosomal recessive, and 6 are autosomal dominant. However, there has been some disagreement in the literature regarding inheritance of some gene mutations. This is understandable when new genes are first identified, as the inheritance is not known a priori and cannot be adequately determined without sufficient numbers of families for study. This has been particularly true for PROK2 and PROKR2 since in the initial description, either heterozygous or biallelic mutations were identified (Dode et al., 2006). This led to confusion that the inheritance was either autosomal dominant or autosomal recessive. It was not until some informative families were studied supporting autosomal recessive inheritance for both the ligand and its receptor that the issue was clarified (Abreu et al., 2008). Similarly, only six heterozygous mutations in WDR11 have been identified, but no family members were available so although autosomal dominant inheritance seems likely, it is not currently well established (Kim et al., 2010). Further studies with affected families should reveal the mode of inheritance.
The recent identification of digenic, and even oligogenic, gene mutations has complicated genetic counseling. First described in IHH/KS in 2006 with NELF/FGFR1 and FGFR1/GNRHR (Pitteloud et al., 2007), other descriptions have followed. The prevalence of digenic disease is difficult to ascertain since it will depend upon the number of genes studied, and this number keeps increasing. The largest study involved the analysis of eight genes in 397 nHH/KS patients—10/88 (11%) of patients with a known mutation in one gene had a mutation in a second gene, while 2.5% of all patients had digenic disease (Sykiotis et al., 2010). In a study of 13 genes in 48 patients, 6/24 (25%) IHH/KS patients with a known mutation had a mutation in a second gene, while 0/24 without a known mutation had digenic disease (Quaynor et al., 2011).
Five digenic patterns were reported in 2011 and the other 13 reported cases at the time of publication were reviewed (Quaynor et al., 2011). What is interesting about the digenic disease is that of the 18 cases described, a single gene could be sufficient to cause the disease (without a mutation in the second gene) in 16 of 18 cases (Table 3) Quaynor et al., 2011. The presence of a second gene mutation could certainly modify the disease phenotype. In the case of digenic heterozygous mutations in two different genes (each of which normally results disease only when biallelic) could cause IHH/KS by synergistic heterozygosity (Quaynor et al., 2011). There is precedence for this in other genetic diseases; and implicates the two genes as being involved in the same pathway (Phillips et al., 2008; Schuler et al., 2005; Vockley et al., 2000).
Table 3.
Digenic nHH/KS patients as reviewed in Quaynor et al (2011). M = male; F = female. The genotypes are shown with the number of alleles affected in parentheses. Genes outlined in bold indicate those that would be sufficient to cause disease without a mutation in a second gene.
| Sex | Phenotype | Genotype | |
|---|---|---|---|
| 1 | M | KS | KAL1 (1)/PROKR2 (1) |
| 2 | M | KS | KAL1 (1)/PROKR2 (1) |
| 3 | M | KS | FGFR1 (1)/NELF (1) |
| 4 | F | nIHH | FGFR1 (1)/GNRHR (2) |
| 5 | F | KS | PROK2 (1)/PROKR2 (1) |
| 6 | M | nIHH | FGFR1 (2)/FGF8 (2) |
| 7 | M | nIHH | FGFR1 (1)/FGF8 (1) |
| 8 | F | nIHH | FGFR1 (1)/GNRHR (2) |
| 9 | F | nIHH | FGFR1 (1)/PROKR2 (1) |
| 10 | M | KS | NELF (1)/TACR3 (1) |
| 11 | F | nIHH | FGFR1 (1)/KAL1 (1) |
| 12 | M | KS | FGFR1 (1)/FGF8 (1) |
| 13 | M | KS | FGR1 (1)/NELF (1) |
| 14 | M | KS | KAL1 (1)/NELF/PROKR2 (1/1) |
| 15 | M | KS | KAL1 (1)/TACR3 (1) |
| 16 | M | KS | WDR11 (1)/KAL1 (1) |
| 17 | M | nIHH | WDR11 (1)/KAL1 (1) |
| 18 | M | nIHH | WDR11 (1)/GNRHR (1) |
2.1.4. Clinical considerations
How does the clinician seeing a patient with nHH/KS decide if genetic testing will be beneficial in the management of the patient? Fortunately, FGFR1 (10%) and CHD7 (6%) are the two most commonly involved genes in either nHH or KS. For the nHH patient, DNA sequencing of these two genes could be performed, and if GNRHR (5%) and TACR3 (6%) are added, it would be estimated that ~25% of nHH patients would have a mutation in one of these genes. These findings could guide the genetic counseling. The risk for transmission if the nHH patient has an autosomal recessive disorder with biallelic GNRHR or TACR3 mutations should be low since it is unlikely the male partner will be a carrier (he could be tested if there are concerns). However, mutations in FGFR1 and CHD7 are autosomal dominant, which carry a 50% chance risk for each child. For the female with KS, FGFR1 (10%) and CHD7 (6%) are the two most common genes, but the detection rate for mutations will be less (16%) for KS than for nHH since mutations in other KS genes are less frequent. KAL1 mutations should be considered in males, but not in females since it is X-linked recessive and the KAL1 gene escapes X-inactivation. There is not convincing evidence that KAL1 mutations cause KS in females.
2.1.5. nHH/KS Genes affecting the pituitary
Mutations in an increasing number of genes cause combined pituitary hormone deficiency (CPHD), which consists of growth hormone deficiency and deficiency in at least one more pituitary hormone. At least six genes have been found to cause CPDH in which there is also deficiency in gonadotropins. These disorders should be kept in mind when seeing patients with nHH/KS because of associated deficiencies, particularly for adrenal failure and/or hypothyroidism. In patients with a family history of hypogonadotropic hypogonadism, it is important to ascertain if pituitary failure is present, which could be manifested as short stature due to deficiencies of TSH and growth hormone. The inheritance patterns should also be considered since CPHD can be autosomal dominant, autosomal recessive, or X-linked recessive. HESX1 mutations may cause either autosomal dominant or autosomal recessive disease (Table 2). For patients with CPHD who have gonadotropin deficiency as one of its components, mutation in the PROP1 gene are by far the most common for this autosomal recessive disease (Kelberman et al., 2009).
Mutations have rarely been reported in gonadotropin-beta subunit genes—LHB, first in a male (Weiss et al., 1992) and later in a female (Lofrano-Porto et al., 2007), as well as in FSHB (Matthews et al., 1993; Layman et al., 1997), but not in the gene encoding the alpha-subunit (CGA). Sometimes an unusual, but enlightening disease entity may shed light upon normal physiologic function, as in the case of isolated FSH deficiency. This disorder is due to an autosomal recessive mutation in FSHB, which has been reported in at least six females (references in Siegel et al., 2012). All presented with primary amenorrhea with or without breast development with very low estradiol and FSH levels, but elevated LH levels. This pattern of serum gonadotropins is quite consistent with the negative feedback mechanisms known for the HPG axis. Since estradiol is low, GnRH increases, which then increases LH, but FSH cannot rise since there is a mutation in FSHB. The mutations have all resulted in unmeasurable immuno- and bioactive FSH in vitro, which is probably because of impaired dimer formation (Siegel et al., 2012). One FSH-deficient female underwent overnight 10-min sampling of LH and demonstrated an elevated baseline LH (35–50 mIU/mL) with 9 pulses overnight (Barnes et al., 2000, 2002). It is interesting that this LH secretion pattern is very similar to a woman with PCOS, but unlike PCOS women, the FSH-deficient patient had no hirsutism. These studies, along with other literature, support the concept that FSH has some role in androgen production in women, even though LH is the principle gonadotropin important in androgen production (Siegel et al., 2012; Barnes et al., 2000, 2002).
2.2. Hypergonadotropic hypogonadism
2.2.1. Phenotype
As indicated above, when females manifest symptoms of estrogen deficiency, such as absent breast development or hypoestrogenic amenorrhea, there is a lack of negative feedback to the hypothalamus and pituitary gland. If serum FSH and LH remain elevated on several occasions, hypergonadotropic hypogonadism is present and the defect is in the ovary (Layman, 1999). When a patient has hypergonadotropic hypogonadism, it is always important to think about Turner syndrome (pure 45,X or mosaic forms). As ascertained from two very large series of amenorrhea, (Reindollar et al. (1981) found that approximately 25% of women with primary amenorrhea and 0.5–1% of women with secondary amenorrhea (Reindollar et al., 1986) had a 45,X cell line. These patients are uniformly short, and although about 10% of patients with only a 45,X cell line and up to 50% of women with 45,X/X chromosome mosaicism may have pubertal development and menstruation, menstrual and ovarian function are usually very short-lived in those who are followed clinically (Zhong and Layman, 2012). Associated anomalies include cardiac in ~50% (coarctation, dilated aortic root, and bicuspid aortic valve), renal in one-third, and dysgenetic gonads in those with a 46,XY cell line. An initial screen with a karyotype, analyzing 30 cells or 60 cells will exclude 10% and 5% mosaicism, respectively with 95% confidence (Zhong and Layman, 2012). In patients with primary amenorrhea without breast development, but who have a vagina and uterus, it is also possible that the karyotype will be a pure 46,XY (Swyer syndrome). These phenotypic females have mutations in the SRY gene in about 15% of cases (Jager et al., 1990; Sim et al., 2008).
In patients with a 46,XX karyotype who have premature ovarian failure (POF), also known as premature ovarian insufficiency, at least 14 genes are known, but they only comprise 15% of all women (Table 4). The most common and clinically important gene is FMR1, the gene involved in fragile X syndrome. Fragile X syndrome is an X-linked dominant disorder with reduced penetrance that causes variable severity of intellectual disability in males, who may also have facial dysmorphism and macroorchidism. This gene normally has 5–50 repeats of the trinucleotide CGG in the 5′ untranslated region. If there is an expansion of this triplet repeat to 50–200 copies, it is termed a premutation. Females with premutation alleles are at increased risk of further expansion during meiosis and if >200 repeats are present in a male child, fragile X syndrome will be present. Diagnosis is confirmed by DNA analysis (PCR and Southern blot)—not cytogenetics. The mechanism for the full mutation is methylation and inactivation of the FMR1 promoter, which results in a null, inactivating mutation. In contrast, women with POF and older men with tremor/ataxia with a premutation allele, the disease mechanism results from RNA overexpression. If women are ascertained from well-characterized fragile X families, ~16% with the premutation allele will develop POF, but not with the full mutation (Allingham-Hawkins et al., 1999). On the other hand, when females are ascertained by the phenotype of POF, the risk of having a premutation allele is 3–4% when she is the only affected individual in the family, but 12–15% if a second female in the pedigree is affected with POF (Conway et al., 1998).
Table 4.
Gene regions and gene mutations causing hypergonadotropic hypogonadism in females. Only genes with mutations that have functional effects are shown (GDF9 and INHA are not included for this reason). LHR mutations are not included because they do not cause hypogonadism in 46,XX females, although menstrual abnormalities and anovulation occur.
| Gene | Reproductive phenotype |
Nonreproductive phenotype | Inheritance | Prevalence in POF |
|---|---|---|---|---|
| SRY(Jager, 1990
#415;Sim, 2008 #1385) |
PA and POF | Swyer syndrome in 46,XY males | Sporadic, Y linked | 10% in 46,XY |
| POF1 | Xq26-q28 | ? | ? | |
| POF2 | X13.3-q21.1 | ? | ? | |
| DIAPH2(Bione, 1998 #93) | Gene within POF2 |
Disruption in one X-autosome translocation |
1 Case; no point mutations in the gene |
|
| 1 FMR1(Conway, 1998 #194;Uzielli, 1999 #967) | POF (gene within POF1) |
XLD | 3–5% Sporadic; 12–15% familial | |
| 2 FOXL2(Crisponi, 2001 #205;Laissue, 2009 #1081) | POF3 | Blepharophimosis–ptosis– epicanthus syndrome |
AD | Rare without BPES |
| 3 BMP15(Di Pasquale, 2004 #238;Rossetti, 2009 #1080) |
POF4 | XLD | 2% | |
| 4 NOBOX (Qin, 2007
#1084;Qin, 2009 #2697) |
POF5 | AD, sporadic | 0.3% | |
| 5 FIGLA(Zhao, 2008 #1085) | POF6 | AD, sporadic | 2% | |
| 6 NR5A1(Lourenco, 2009 #1089) | POF7 | Adrenal failure | AR | 8% (2/25) |
| 7 FSHR(Aittomaki, 1995 #2268;Jiang, 1998 #2269) | POF | AR | Rare outside of Finland | |
| 8 AIRE(Consortium, 1997 #193;Nagamine, 1997 #682) |
POF | APECED | AR | Rare |
| 9 GALT(Kaufman, 1979 #448) | POF | Galactosemia | AR | Rare |
| 10 EIFB2(Fogli, 2004 #286;Fogli, 2003 #287) |
POF | Ovarioleukodystrophy | AR | Rare unless white matter abnormalities of brain |
| 11 EIFB4(Fogli, 2004 #286;Fogli, 2003 #287) |
POF | Ovarioleukodystrophy | AR | Rare unless white matter abnormalities of brain |
| 12 EIFB5(Fogli, 2004 #286;Fogli, 2003 #287) |
POF | Ovarioleukodystrophy | AR | Rare unless white matter abnormalities of brain |
| 13 CYP17A1(Yanase, 1991 #1050) | POF | Adrenal failure | AR | Rare |
| 14 CYP19 A1(Ito, 1993 #2202) | POF | Sexual ambiguity | AR | Rare |
Most of the other genes that result in ovarian failure occur in less than 1% of women studied. FSHR mutations are common in Finland (Aittomaki et al., 1995), but are quite rare outside of this country (Siegel et al., 2012). As can be seen from Table 4, isolated POF occurs in women with mutations in FMR1 (Allingham-Hawkins et al., 1999), FSHR (Aittomaki et al., 1995), BMP15 (Di Pasquale et al., 2004), FIGLA (Zhao et al., 2008), and NOBOX (Qin et al., 2007), while associated anomalies occur with others. At least three genes affecting steroid function, CYP17A1 (17-hydroxylase/17-20 lyase), CYP19A1 (aromatase), and NR5A1 (steroidogenic factor-1) have additional phenotypic findings. Wo men with CYP17A1 (Yanase et al., 1991) or NR5A1 (Lourenco et al., 2009) mutations may have coexistent adrenal failure, while those with CYP19A1 (Ito et al., 1993) or NR5A1 (Lourenco et al., 2009) mutations may have disorders of sexual differentiation. Although rare, autoimmune polyglandular syndrome type 1, also known as autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED) is a systemic autoimmune disorder in which adrenal and parathyroid dysfunction may accompany gonadal failure. Mutations in the AIRE gene result in autosomal recessive APECED, the first systemic autoimmune disorder for which the molecular basis was identified (Consortium, 1997; Nagamine et al., 1997). Another rare disorder, which has greatly furthered our understanding of normal ovarian function is the coexistence of ovarian failure with interesting eyelid abnormalities in blepharophimosis–ptosis–epicanthus syndrome (BPES) type 1. Mutations in FOXL2 cause this rare autosomal dominant disorder (Crisponi et al., 2001; Laissue et al., 2009). However, it is now known that FOXL2 is one of the principle genes in ovarian differentiation.
The inheritance patterns of genes for hypergonadotropic hypogonadism are shown in Table 4. Two are X-linked dominant (FMR1 and BMP15) and two are autosomal dominant (FOXL2 and FIGLA), while the remainder are autosomal recessive. The POF1 and POF2 regions are thought to contain ovarian genes and DIAPH2 on X was disrupted in an X-autosome translocation (Bione et al., 1998). However, no mutations are known in DIAPH2 and no other known ovarian genes are in the POF2 region (FMR1 is within the POF1 region).
3. Eugonadism
3.1. Spontaneous ovarian hyperstimulation syndrome
Although inactivating mutations in FSHR result in hypergonadotropic hypogonadism (Aittomaki et al., 1995), activating mutations cause spontaneous ovarian hyperstimulation syndrome (sOHSS) (Smits et al., 2003; Vasseur et al., 2003). Normally, this is an iatrogenic disorder resulting from the administration of exogenous gonadotropins for infertility treatment. However, at least five different females with sOHSS, including two familial cases, have been found to have activating FSHR mutations (Siegel et al., 2012). Familial transmission is consistent with autosomal dominant inheritance. Of these five mutant FSHRs, all of which have been studied in vitro, three are constitutively active, indicating that receptor signaling is propagated without ligand being present. In all cases, hCG is capable of activating the FSHR, and in some cases, TSH may stimulate the FSHR (Siegel et al., 2012).
The ovarian hyperstimulation in patients with activating FSHR mutations only occurs during pregnancy. As exemplified in first reported family with activating FSHR mutations, the affected proband had five pregnancies, four of which were affected by sOHSS and one spontaneous abortion, in which she was not affected (Vasseur et al., 2003). In each case, the sOHSS began in the first trimester with enlarged cystic ovaries, sometimes with ascites, and resolved after pregnancy. She had two liveborn children, one spontaneous abortion, and two therapeutic abortions. Her two affected sisters had 4/7 and 2/4 pregnancies affected with sOHSS; and her unaffected sister did not have sOHSS. This family exemplifies the spectrum that may be present in women with activating FSHR mutations. The sOHSS may not occur in all pregnancies, but when present, it starts in the first trimester and resolves postpartum. The pregnancy outcome could be a spontaneous abortion or term live birth (Vasseur et al., 2003).
3.2. Mullerian aplasia
Mullerian aplasia, also known as Mayer–Rokitansky–Kuster–Hauser syndrome, consists of the congenital absence of the uterus and vagina. This most severe uterine anomaly occurs in about 1/5000 women and 10% of women with primary amenorrhea (Reindollar et al., 1981). It may be associated with accompanying unilateral renal agenesis in one-third, skeletal anomalies in 10–15%, and less commonly deafness or congenital heart defects (Sultan et al., 2009). The molecular basis is largely unknown. No mutations have been identified in a number of candidate genes (CFTR, GALT, HOXA7, HOXA13, PBX1, HOXA10, AMH, AMHR, RARG, or RXRA), but heterozygous WNT4 mutations were described in one patient (Biason-Lauber et al., 2004) and 1/28 mullerian aplasia patients in a follow up study (Philibert et al., 2008). HNF1B mutations cause maturity onset diabetes of the young (MODY) type 5, some of whom have no vagina (Lindner et al., 1999).
More recently, investigators have utilized comparative genomic hybridization (CGH) arrays to screen for deletions and duplications—termed copy number variants (CNVs)—in mullerian aplasia patients. This method permits the detection of deletions or duplications at greater resolution than a karyotype. CNVs that are reported in more than one individual may provide a clue to genomic regions that contain causative gene. These repetitive CNVs have been reported in 5% of patients with isolated mullerian aplasia and 8% of those with associated anomalies (Nik-Zainal et al., 2011).
Although a number of chromosomal loci have been reported, a 0.55 Mb deletion on 16p11.2 and a 1.4 Mb deletion on 17q12 represent two of the more common regions (Nik-Zainal et al., 2011). Currently, it is not clear if mullerian aplasia may be a genomic disorder due to the loss of contiguous genes, or perhaps these regions contain etiologic genes.
3.3. Endometriosis
Endometriosis is an inflammatory disorder affecting 5–10% of reproductive aged women that results in pelvic pain and infertility. The endometriotic lesions contain endometrial stromal or epithelial cells, chronic bleeding, and signs of inflammation. There is evidence that endometriosis may be a polygenic or multifactorial disorder, and the risk is increased 7-fold in family members. The exact molecular basis is unknown, but there is evidence that increased estrogen, prostaglandins, and cytokines (such as IL1β, IL6, and TNF), as well as progesterone resistance are involved in its genesis (Bulun, 2009).
Since endometriosis is a complex disease (not apparently Mendelian), different types of genetic approaches have been undertaken to study its molecular basis. Largely, association studies have been used to study complex disorders such as endometriosis and polycystic ovary syndrome. Association studies are case-control studies comparing the frequency of a polymorphism in the study group vs. a control group (Spielman and Ewens, 1996). The polymorphisms, which are benign changes in DNA sequence that do not result in a disease phenotype, may be single nucleotide polymorphisms (SNPs), nucleotide repeats (such as CA repeats), or CNVs. A test of significance is then performed. Association studies do not rely upon other family members (which is necessary in linkage), but internal controls from parent-trios, as in the transmission disequilibrium test (TDT), improve power of association (Spielman and Ewens, 1996). Many of the first association studies utilized a candidate gene approach, but more recently many are genome wide association studies (GWAS).
When association is identified, there are several potential explanations (Ott et al., 2011). First, it is possible that the polymorphism causes the disease, but this is very rare. It is possible that the polymorphism does not cause the disease, but is in linkage disequilibrium with the disease-producing gene. This evidence could lead to the causative gene identification. However, it also possible that the polymorphism is more frequent in cases than controls because of artifacts of population admixture (population stratification) (Ott et al., 2011). For example, if the polymorphism is found more frequently in a certain race, its association with the disease could simply be related to the fact that more cases than controls are of that particular race (Ott et al., 2011). Lastly, association could occur by chance alone, which is why many journals require a replication study in which new samples of cases and controls are studied for genes that were associated in the initial study.
Logical candidate genes for endometriosis have been studied in genomic DNA for association with endometriosis (ESR1, COMT, IL6, IL10, CYP17A1, CYP19A1, CYP1A1, MMP1, and MMP9), but association was not demonstrated (Ewens et al., 2010). However, three chromosomal regions were identified to contain CNVs in endometriotic lesions themselves—deletions in 1p36, 7p22.1, and 22q12 (Gogusev, 1999). Although very challenging in endometriosis because it has characteristics of a complex disease, linkage analysis has been done. In more than 1000 families from Australia and the UK, each of which had two or more members with surgically documented endometriosis, significant linkage to 10q26 was demonstrated (Treloar et al., 2005). In addition, there was suggestive linkage to 20p13. Linkage requires the presence of family members; and in this study 10q26 was the most likely locus to contain a causative gene. However, no causative gene was identified (Treloar et al., 2005).
Recently, two large GWAS studies in endometriosis patients were performed (Painter et al., 2011; Uno et al., 2010). In both cases, a group of affected patients and controls were studied. Markers found to be associated were then studied for association in a new sample of cases and controls (replication study). In a Japanese population, significant association was demonstrated to 1p36, while none of the prior SNPs thought to be involved in endometriosis pathogenesis (estrogen action, cytokines, etc.) was associated (Uno et al., 2010). In another GWAS in Australia and the UK (Painter et al., 2011), significant association was found at 7p15.2 (near two genes NFE2L3 and HOXA10) and 1p36 (containing WNT4). It is remarkable that none of the loci were similar except for 1p36, which has demonstrated consistency among GWAS and CGH array studies.
3.4. Polycystic ovary syndrome
Polycystic ovary syndrome (PCOS) is usually defined as hyperandrogenic anovulation with or without polycystic appearing ovaries (Azziz et al., 2006). PCOS is the most common cause of anovulation and may affect 5–8% of reproductive aged women. The pathogenesis is not well understood, but it is clear that increased free estrogens (and reduced estradiol to estrone ratio) contribute to the increased risk of endometrial cancer and accompanying hyperinsulinemia portends an increased risk for type 2 diabetes (Azziz et al., 2006). Hyperandrogenemia results in hirsutism; and infrequent LH surges result in anovulatory infertility (Azziz et al., 2006).
The genetic basis of PCOS is not well understood, but familial cases have been recognized. It should be kept in mind that Mendelian forms of insulin resistance, for which the genetic basis is known, have been described. Mutations in the insulin receptor (INSR) gene may result in a broad range of phenotypes such as the severe autosomal recessive Donohue syndrome (Leprechaunism), which combines somatic anomalies with hyperinsulinemia, hypertrichosis, acanthosis nigricans, and hypoglycemia (Kadowaki et al., 1988). INSR mutations may also result in diabetes, insulin resistance, and acanthosis without other associated anomalies, as well. Another group of disorders to be considered are the lipodystrophies, which consist of a loss of body fat and insulin resistance (Garg, 2011). At least 11 genes with mutations have been reported. Two of the more common disorders include congenital generalized lipodystrophy, an autosomal recessive disorder with at least four genes (AGPAT2, BSCL2, CAV1, PTRF) identified, and familial partial lipodystrophy, an autosomal dominant disorder with at least four genes (LMNA, PPARG, AKT3, PLIN1) identified (Garg, 2011).
PCOS is thought to be a complex disease, and a number of association studies have been performed. Initially candidate genes were studied either individually or collectively, but association was rarely confirmed upon follow up studies. In a study of 14 genes (ACVR2A, CYP11A1, CYP17A1, CYP19A1, HSD17B2, IGF1R, INHBA, INHBB, INSL3, INSR, SMAD4, LEP, POMC, and SHBG) that had been previously reported to be associated with PCOS (Urbanek et al., 1999) and 12 other genes found to be associated in Caucasians (ADIPOQ, CAPN5, ENPP1, EPHX1, FEM1A, FEM1B, H6PD, HSD17B6, IL1A, SGTA, SRD5A1, and SRD5A2), Ewens et al. (2010) used family based association (transmission disequilibrium test), which tests for association and linkage, and found association with five genes. The most highly associated gene was FBN3. The insulin receptor (INSR) on 19p13 was previously found to be associated with marker D19S884, but in the current study, FBN3 was more significantly associated with this marker (Ewens et al., 2010). Importantly, PCOS was defined in this study as hyperandrogenemia and ≤6 menses/year.
Two GWAS in Han Chinese were recently reported (Shi et al., 2012), in which PCOS was defined by the Rotterdam Criteria (two of three—hyperandrogenism, anovulation, and polycystic appearing ovaries). There were a total of ~8000 cases and ~7500 controls and replication was performed. Interestingly only 3 loci (2p16.3, 2p21, and 9q33.3) were in common in both GWAS studies (Shi et al., 2012). These different findings, along with differing definitions of PCOS must be reconciled for advances to be gained.
3.5. Leiomyomata
Leiomyomata (fibroids) are benign smooth muscle tumors of the uterus in more than one-third of all women that may result in bleeding and hysterectomy. These tumors are clonal and somatic cell in origin, but inheritance patterns are uncertain. When fibroids occur in unusual locations and associated with other anomalies, at least two Mendelian forms should be considered. Hereditary leiomyomatosis and renal cell cancer is an autosomal dominant disorder caused by mutations in the fumarate hydratase (FH gene) (Tomlinson et al., 2002). In this case cutaneous and uterine leiomyomata may be associated with renal tumors. Diffuse leiomyomatosis with Alport syndrome consists of esophageal and vulvar leiomyomata along with renal disease, hearing loss, and ocular anomalies (Alport syndrome). This X-linked dominant contiguous gene deletion syndrome on Xq22 affects two collagen genes (COL4A5 and COL4A6) Zhou et al., 1993.
Somatic cell chromosomal anomalies have been reported in women with fibroids including 7q deletions, trisomy 12, and high mobility group AT-hook 2 (HMGA2) rearrangements of chromosome 12q14. However, since they occur at low frequency, this prompted whole exome sequencing of 18 fibroids from 17 women (Makinen et al., 2011). Mutations in the MED12 gene were initially identified, which led these investigators to study a total of 225 tumors in 80 women. They identified MED12 mutations in 75% patients and in 70% tumors. Interestingly, half of the mutations occurred in exon 2 codon Gly44 (missense, splice site, frameshift); and all were heterozygous, somatic, and clonal (Makinen et al., 2011). The MED12 gene, localized to Xq13.1, encodes for a mediator complex that regulates global and gene-specific transcription. These authors pointed out that two different, allelic Mendelian disorders (Opitz-Kaveggia syndrome and Lujan-Fryns syndrome) are caused by different germline MED12 mutations (Makinen et al., 2011). Both entities include intellectual disability and congenital anomalies, but neither has a predisposition to tumors.
Genome wide association studies have also been performed in women with fibroids. In a study of more than 1600 cases and 1400 controls, 49 SNPs were significantly associated with fibroids (Cha et al., 2011). In the replication phase of more than 3400 cases and 3200 controls, three loci remained significantly associated (10q24.33, 22q13.1, and 11p15.5) (Cha et al., 2011). Recently, two complimentary techniques—genome wide linkage and association—were utilized to identify the molecular basis of fibroids (Eggert et al., 2012). First genome-wide linkage was performed in 261 white affected sister-pair families. Significant linkage was most strongly detected at 10p11 and 3p21 (and less so for 2q37, 5p13, 11p15, 12q14, and 17q25). GWAS was then performed in two independent cohorts of white women, and a meta-analysis was conducted (Eggert et al., 2012). One SNP from the GWAS was found in a block of linkage disequilibrium at 17q25.3, spanning three genes (FASN, CCDC57, and SLC16A3). Using tissue microarray immunohistochemistry, FAS, the protein product of the fatty acid synthase (FASN) gene, was increased threefold in fibroids compared with surrounding myometrial tissue. It is interesting that FASN transcripts and/or FAS protein levels are upregulated in a variety of neoplasms and may play a role in tumor cell survival (Eggert et al., 2012). The combination of genome-wide complimentary techniques provides an unbiased approach that could provide important new clinical information regarding fibroids.
3.6. Steroid hormone resistance syndromes
Complete androgen insensitivity (CAIS) has been long known to occur in 46,XY males with a null mutation in the androgen receptor (AR) gene Brown et al., 1988; Gottlieb et al., 1999. These phenotypic females have normal breast development, but lack a uterus and vagina, and have little or no axillary or pubic hair. Serum testosterone is elevated for a female (and normal for a male). A similar disorder in the estrogen receptor-alpha (ESR1) gene has only been described in one 46,XY male with ESR1 mutation (Smith et al., 1994). These findings suggested that perhaps ESR1 mutations in females would be lethal. The expected phenotype in a female would be absent puberty, high estradiol levels and gonadotropin levels. Recently, such a patient was described; and she had facial acne, absent breast development, markedly elevated estradiol levels (>2000 pg/mL), and mildly elevated/high normal gonadotropins. Interestingly, estrogen induced SHBG, CBG, TBG, prolactin, and triglycerides were all normal. She had a homozygous missense mutation in ESR1 that markedly impaired estrogen signaling (Quaynor SD, Submitted for publication). These findings indicate that complete estrogen insensitivity syndrome (CEIS) does occur in human females and it is not lethal.
3.7. Summary: the genetic basis of female reproductive disorders
The genetic basis of hypogonadism is much better understood than for eugonadal disorders. In patients with hypogonadotropic hypogonadism, at least 35–40% of patients have mutations in 20 genes and digenic disease may affect up to 3–10%. It is important to rule out pituitary insufficiency and realize that additional genes should be considered in these cases. The identification of associated anomalies is also important, as is the possibility of autosomal dominant, autosomal recessive, and X-linked recessive inheritance. For women with hypergonadotropic hypogonadism, it is important to consider Turner syndrome, but in those with 46,XX ovarian failure, about 15% of women may have mutations in any of 14 genes. For most of the genes identified in hypogonadal patients (both hypogonadotropic and hypergonadotropic), known mutations have been supported by functional analyses and many have appropriately segregated within families.
For eugonadal disorders, activating FSHR mutations in sOHSS, inactivating WNT4 mutations in mullerian aplasia, and the one inactivating ESR1 mutation in CEIS similarly have been shown result in impairment in vitro and likely cause their respective phenotypes. For endometriosis, PCOS, and leiomyomata, the genetic basis is not known at present. For endometriosis, CNV analysis of endometriotic lesions has yielded three loci, but for genomic DNA, several regions have been shown to be associated. Interestingly, 1p36 has been shown to be involved in two association studies and in the CNV analysis of endometriosis. For PCOS, there is association only—no specific gene has been identified. For fibroids, MED12 mutations are common somatic cell mutations, found in about three-fourths of all fibroids and women. The FASN locus, identified by the combination of genomic methodologies could play a role in fibroids.
3.8. Practical considerations for the clinician
What testing should be done by the clinician taking care of these patients? For hypogonadotropic hypogonadism, some recommendations can be made. Since FGFR1 (10%) and CHD7 (6%) represent the two most commonly involved genes in either KS or nHH, it is reasonable to test all patients for mutations by DNA sequencing of all protein coding exons and their splice junctions. KAL1 would only be indicated in males as it is X-linked recessive and there is no convincing evidence that heterozygous mutations in females cause disease. For nHH, TACR3 (4–5%) and GNRHR (4–5%) could be added to FGFR1 and CHD7, so that the detection rate approaches 25% of patients. If certain specific anomalies are present, specific gene testing can be considered. The mode of inheritance also needs to be considered since FGFR1 and CHD7 are autosomal dominant, while GNRHR and TACR3 are autosomal recessive. It should be remembered that DNA sequencing can miss intragenic deletions or duplications, so that techniques such as multiplex ligation-dependent probe amplification (MLPA) could be considered. However, this method is not likely to be clinically useful at this time for the autosomal forms of nHH/KS (Pedersen-White et al., 2008).
For hypergonadotropic hypogonadism, Turner syndrome should be excluded with a karyotype counting 30–50 cells to look for a 45,X or 46,XY cell line. For 46,XX females with gonadal failure, FMR1 testing by PCR and Southern blot for the triplet repeat expansion should be considered since 3–4% of isolated and 12–15% of familial POF patients will be premutation carriers. Currently, testing for the other rare single gene defects is not practical. If the prevalence of NR5A1 mutations remains ~8% (2/25) with an increased sample size of POF women, testing for this gene could be clinically useful.
For eugonadal disorders, if sOHSS is present, DNA sequencing for the protein coding exons and splice junctions of the FSHR gene is feasible. WNT4 DNA sequencing could be done for mullerian aplasia patients, but currently it is not practical to do clinical genetic testing on patients with endometriosis, PCOS, or fibroids.
3.9. Future considerations
It is currently feasible to perform targeted deep resequencing of large numbers of genes so that all known genes for either hypogonadotropic hypogonadism or hypergonadotropic hypogonadism could be sequenced. Therefore, all 20 IHH/KS genes (plus 8 more, including those for CPHD and isolated gonadotropin deficiency) could be analyzed simultaneously. It is now also feasible to sequence all exons of all genes (whole exome sequencing) and all nucleotides of the genome (whole genome sequencing). These tests are beginning to make their way into the clinical realm, as currently standards and validation have begun by many clinical molecular laboratories. The cost of the $1000 genome is near, but the interpretation still presents logistic and ethical challenges since several thousand nucleotide changes may be present in each person. Nevertheless, in the near future, we should have the capability to have DNA sequence on every patient.
Footnotes
Disclosure The author has nothing to disclose.
L.C.L. was supported from NIH Grant HD33004.
References
- Abreu AP, Trarbach EB, de Castro M, Frade Costa EM, Versiani B, Matias Baptista MT, et al. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J. Clin. Endocrinol. Metab. 2008;93:4113–4118. doi: 10.1210/jc.2008-0958. [DOI] [PubMed] [Google Scholar]
- Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82:959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, et al. Fragile X premutation is a significant risk factor for premature ovarian failure: the international collaborative POF in fragile X study-preliminary data. Am. J. Med. Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, et al. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J. Clin. Endocrinol. Metab. 2006;91:4237–4245. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Namnoum A, Rosenfield RL, Layman LC. Effects of follicle-stimulating hormone on ovarian androgen production in a woman with isolated follicle-stimulating hormone deficiency. N. Engl. J. Med. 2000;343:1197–1198. doi: 10.1056/NEJM200010193431614. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Namnoum A, Rosenfield RL, Layman LC. The role of LH and FSH in ovarian androgen secretion and ovarian follicular development: clinical studies in a patient with isolated FSH deficiency and multicystic ovaries. Hum. Reprod. 2002;17:88–91. doi: 10.1093/humrep/17.1.88. [DOI] [PubMed] [Google Scholar]
- Bentley GE, Ubuka T, McGuire NL, Calisi R, Perfito N, Kriegsfeld LJ, et al. Gonadotrophin-inhibitory hormone: a multifunctional neuropeptide. J. Neuroendocrinol. 2009;21:276–281. doi: 10.1111/j.1365-2826.2009.01851.x. [DOI] [PubMed] [Google Scholar]
- Bhagavath B, Ozata M, Ozdemir IC, Bolu E, Bick DP, Sherins RJ, et al. The prevalence of gonadotropin-releasing hormone receptor mutations in a large cohort of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2005;84:951–957. doi: 10.1016/j.fertnstert.2005.04.029. [DOI] [PubMed] [Google Scholar]
- Bhagavath B, Podolsky RH, Ozata M, Bolu E, Bick DP, Kulharya A, et al. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2006;85:706–713. doi: 10.1016/j.fertnstert.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46 XX woman. New Engl. J. Med. 2004;351:792–798. doi: 10.1056/NEJMoa040533. [DOI] [PubMed] [Google Scholar]
- Bione S, Sala C, Manzini C, Arrigo G, Zuffardi O, Banfi S, et al. A human homologue of the drosophila melanogaster diaphenous gene is disrupted in a patient with premature ovarian failure: evidence for conserved function in oogenesis and implications for human sterility. Am. J. Hum. Genet. 1998;62:533–541. doi: 10.1086/301761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Engl. J. Med. 2009;360:2742–2748. doi: 10.1056/NEJMoa0900136. [DOI] [PubMed] [Google Scholar]
- Brown TR, Lubahn DB, Wilson EM, Joseph DR, French FS, Migeon CJ. Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: evidence for further genetic heterogeneity in this syndrome. Proc. Nat. Acad. Sci. 1988;85:8151–8155. doi: 10.1073/pnas.85.21.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulun SE. Endometriosis. N. Engl. J. Med. 2009;360:268–279. doi: 10.1056/NEJMra0804690. [DOI] [PubMed] [Google Scholar]
- Cha PC, Takahashi A, Hosono N, Low SK, Kamatani N, Kubo M, et al. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat. Genet. 2011;43:447–450. doi: 10.1038/ng.805. [DOI] [PubMed] [Google Scholar]
- Chan YM, de Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, Tsiaras S, et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA. 2009 doi: 10.1073/pnas.0903449106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology. 2010;151:301–311. doi: 10.1210/en.2009-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- Consortium F-GA. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- Conway GS, Payne NN, Webb J, Murray A, Jacobs PA. Fragile X premutation screening in women with premature ovarian failure. Hum. Reprod. 1998;13:1184–1187. doi: 10.1093/humrep/13.5.1184. [DOI] [PubMed] [Google Scholar]
- Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 2001;27:159–166. doi: 10.1038/84781. [DOI] [PubMed] [Google Scholar]
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 1997;337:1597–1602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. USA. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pasquale E, Beck-Peccoz P, Persani L. Hypergonadotropic ovarian failure associated with an inherited mutation of human bone morphogenetic protein-15 (BMP15) gene. Am. J. Hum. Genet. 2004;75:106–111. doi: 10.1086/422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Dode C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert SL, Huyck KL, Somasundaram P, Kavalla R, Stewart EA, Lu AT, et al. Genome-wide linkage and association analyses implicate FASN in predisposition to uterine leiomyomata. Am. J. Hum. Genet. 2012;91:621–628. doi: 10.1016/j.ajhg.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens KG, Stewart DR, Ankener W, Urbanek M, McAllister JM, Chen C, et al. Family-based analysis of candidate genes for polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2010;95:2306–2315. doi: 10.1210/jc.2009-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 2008;118:2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- Garg A. Clinical review#: lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011;96:3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogusev J, Bouquet de Joliniere J, Telvi L, Doussau M, du Manoir S, Stojkoski A, et al. Detection of DNA copy number changes in human endometriosis by comparative genomic hybridization. Hum. Genet. 1999;105:51–444. doi: 10.1007/s004390051129. [DOI] [PubMed] [Google Scholar]
- Gottlieb B, Pinsky L, Beitel LK, Trifiro M. Androgen insensitivity. Am. J. Med. Genet. 1999;89:210–217. doi: 10.1002/(sici)1096-8628(19991229)89:4<210::aid-ajmg5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Habiby RL, Boepple P, Nachtigall L, Sluss PM, Crowley WF, Jr., Jameson JL. Adrenal hypoplasia congenita with hypogonadotropic hypogonadism: evidence that DAX-1 mutations lead to combined hypothalmic and pituitary defects in gonadotropin production. J. Clin. Invest. 1996;98:1055–1062. doi: 10.1172/JCI118866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchate NK, Giacobini P, Lhuillier P, Parkash J, Espy C, Fouveaut C, et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012;8:e1002896. doi: 10.1371/journal.pgen.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardelin JP, Levilliers J, Blanchard S, Carel JC, Leutenegger M, Pinard-Bertelletto JP, et al. Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum. Mol. Genet. 1993;2:373–377. doi: 10.1093/hmg/2.4.373. [DOI] [PubMed] [Google Scholar]
- Hurley ME, White MJ, Green AJ, Kelleher J. Antley–Bixler syndrome with radioulnar synostosis. Pediatr. Radiol. 2004;34:148–151. doi: 10.1007/s00247-003-1066-7. [DOI] [PubMed] [Google Scholar]
- Ito Y, Fisher CR, Conte FA, Grumbach MM, Simpson ER. Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc. Natl. Acad. Sci. USA. 1993;90:11673–11677. doi: 10.1073/pnas.90.24.11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager RJ, Anvret M, Hall K, Scherer G. A human XY female with a frame shift mutation in the candidate testis-determining gene SRY. Nature. 1990;348:452–454. doi: 10.1038/348452a0. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Bevins CL, Cama A, Ojamaa K, Marcus-Samuels B, Kadowaki H, et al. Two mutant alleles of the insulin receptor gene in a patient with extreme insulin resistance. Science. 1988;240:787–790. doi: 10.1126/science.2834824. [DOI] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr. Rev. 2009;30:790–829. doi: 10.1210/er.2009-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2008;83:511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Bhagavath B, Layman LC. Clinical manifestations of impaired GnRH neuron development and function. Neurosignals. 2008;16:165–182. doi: 10.1159/000111561. [DOI] [PubMed] [Google Scholar]
- Kim HG, Pedersen-White J, Bhagavath B, Layman LC. Genotype and phenotype of patients with gonadotropin-releasing hormone receptor mutations. Front. Horm. Res. 2010;39:94–110. doi: 10.1159/000312696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010;87:465–479. doi: 10.1016/j.ajhg.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress W, Petersen B, Collmann H, Grimm T. An unusual FGFR1 mutation (fibroblast growth factor receptor 1 mutation) in a girl with non-syndromic trigonocephaly. Cytogenet. Cell Genet. 2000;91:138–140. doi: 10.1159/000056834. [DOI] [PubMed] [Google Scholar]
- Laissue P, Lakhal B, Benayoun BA, Dipietromaria A, Braham R, Elghezal H, et al. Functional evidence implicating FOXL2 in non-syndromic premature ovarian failure and in the regulation of the transcription factor OSR2. J. Med. Genet. 2009;46:455–457. doi: 10.1136/jmg.2008.065086. [DOI] [PubMed] [Google Scholar]
- Layman LC. Genetics of human hypogonadotropic hypogonadism. Am. J. Med. Genet. 1999;89:240–248. doi: 10.1002/(sici)1096-8628(19991229)89:4<240::aid-ajmg8>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Layman LC. Human gene mutations causing infertility. J. Med. Genet. 2002;39:153–161. doi: 10.1136/jmg.39.3.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layman LC, Lee EJ, Peak DB, Namnoum AB, Vu KV, van Lingen BL, et al. Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. N. Engl. J. Med. 1997;337:607–611. doi: 10.1056/NEJM199708283370905. [DOI] [PubMed] [Google Scholar]
- Layman LC, Cohen DP, Jin M, Xie J, Li Z, Reindollar RH, et al. Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat. Genet. 1998;18:14–15. doi: 10.1038/ng0198-14. [DOI] [PubMed] [Google Scholar]
- Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V, et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet. 1999;8:2001–2008. doi: 10.1093/hmg/8.11.2001. [DOI] [PubMed] [Google Scholar]
- Lofrano-Porto A, Barra GB, Giacomini LA, Nascimento PP, Latronico AC, Casulari LA, et al. Luteinizing hormone beta mutation and hypogonadism in men and women. N. Engl. J. Med. 2007;357:897–904. doi: 10.1056/NEJMoa071999. [DOI] [PubMed] [Google Scholar]
- Lourenco D, Brauner R, Lin L, De Perdigo A, Weryha G, Muresan M, et al. Mutations in NR5A1 associated with ovarian insufficiency. N. Engl. J. Med. 2009;360:1200–1210. doi: 10.1056/NEJMoa0806228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz B, Karatani S, Rugarli EI, Wawersik S, Wong C, Bieber FR, et al. Expression of the Kallmann syndrome gene in human fetal brain and in the manipulated chick embryo. Hum. Mol. Genet. 1994;3:1717–1723. doi: 10.1093/hmg/3.10.1717. [DOI] [PubMed] [Google Scholar]
- Makinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334:252–255. doi: 10.1126/science.1208930. [DOI] [PubMed] [Google Scholar]
- Mason AJ, Pitts SL, Nikolics K, Szonyi E, Wilcox JN, Seeburg PH, et al. The hypogonadal mouse: reproductive functions restored by gene therapy. Science. 1986;234:1372–1378. doi: 10.1126/science.3097822. [DOI] [PubMed] [Google Scholar]
- Matthews CH, Borgato S, Beck-Peccoz P, Adams M, Tone Y, Gambin G, et al. Primary amenorrhea and infertility due to a mutation in the β-subunit of follicle-stimulating hormone. Nat. Genet. 1993;5:83–86. doi: 10.1038/ng0993-83. [DOI] [PubMed] [Google Scholar]
- Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- Muscatelli F, Strom TM, Walker AP, Zanaria E, Recan D, Meindl A, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat. Genet. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Strick R, Storer M, Huang N, Rad R, Willatt L, et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Mullerian aplasia. J. Med. Genet. 2011;48:197–204. doi: 10.1136/jmg.2010.082412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J, Kamatani Y, Lathrop M. Family-based designs for genome-wide association studies. Nat. Rev. Genet. 2011;12:465–474. doi: 10.1038/nrg2989. [DOI] [PubMed] [Google Scholar]
- Painter JN, Anderson CA, Nyholt DR, Macgregor S, Lin J, Lee SH, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat. Genet. 2011;43:51–54. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen-White JR, Chorich LP, Bick DP, Sherins RJ, Layman LC. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Hum. Reprod. 2008;14:367–370. doi: 10.1093/molehr/gan027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert P, Biason-Lauber A, Rouzier R, Pienkowski C, Paris F, Konrad D, et al. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and mullerian duct abnormalities: a French collaborative study. J. Clin. Endocrinol. Metab. 2008;93:895–900. doi: 10.1210/jc.2007-2023. [DOI] [PubMed] [Google Scholar]
- Phillips JA, 3rd, Poling JS, Phillips CA, Stanton KC, Austin ED, Cogan JD, et al. Synergistic heterozygosity for TGFbeta1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet. Med. 2008;10:359–365. doi: 10.1097/GIM.0b013e318172dcdf. [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr., Meysing AU, Dwyer AA, Hayes FJ, Crowley WF., Jr. Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J. Clin. Endocrinol. Metab. 2005;90:1317–1322. doi: 10.1210/jc.2004-1361. [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 2007;117:457–463. doi: 10.1172/JCI29884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TM. Hypothalamic control of the pituitary-gonadal axis in higher primates: key advances over the last two decades. J. Neuroendocrinol. 2008;20:719–726. doi: 10.1111/j.1365-2826.2008.01708.x. [DOI] [PubMed] [Google Scholar]
- Qin Y, Choi Y, Zhao H, Simpson JL, Chen ZJ, Rajkovic A. NOBOX homeobox mutation causes premature ovarian failure. Am. J. Hum. Genet. 2007;81:576–581. doi: 10.1086/519496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaynor SD, Kim HG, Cappello EM, Williams T, Chorich LP, Bick DP, et al. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 2011;96:1424–30e6. doi: 10.1016/j.fertnstert.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaynor SD. Submitted for publication.
- Reindollar RH, Byrd JR, McDonough PG. Delayed sexual development: study of 252 patients. Am. J. Obstet. Gynecol. 1981;140:371–380. doi: 10.1016/0002-9378(81)90029-6. [DOI] [PubMed] [Google Scholar]
- Reindollar RH, Novak M, Tho SPT, McDonough PG. Adult-onset amenorrhea: a study of 262 patients. Am. J. Obstet. Gynecol. 1986;155:531–543. doi: 10.1016/0002-9378(86)90274-7. [DOI] [PubMed] [Google Scholar]
- Roscioli T, Flanagan S, Kumar P, Masel J, Gattas M, Hyland VJ, et al. Clinical findings in a patient with FGFR1 P252R mutation and comparison with the literature. Am. J. Med. Genet. 2000;93:22–28. doi: 10.1002/1096-8628(20000703)93:1<22::aid-ajmg5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Ballabio A. Kallmann syndrome. From genetics to neurobiology. JAMA. 1993;270:2713–2716. doi: 10.1001/jama.270.22.2713. [DOI] [PubMed] [Google Scholar]
- Schuler AM, Gower BA, Matern D, Rinaldo P, Vockley J, Wood PA. Synergistic heterozygosity in mice with inherited enzyme deficiencies of mitochondrial fatty acid beta-oxidation. Mol. Genet. Metab. 2005;85:7–11. doi: 10.1016/j.ymgme.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr., Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- Shi Y, Zhao H, Cao Y, Yang D, Li Z, Zhang B, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat. Genet. 2012;44:1020–1025. doi: 10.1038/ng.2384. [DOI] [PubMed] [Google Scholar]
- Siegel ET, Kim HG, Nishimoto HK, Layman LC. The molecular basis of impaired follicle-stimulating hormone action: evidence from human mutations and mouse models. Reprod. Sci. 2012 doi: 10.1177/1933719112461184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim H, Argentaro A, Harley VR. Boys, girls and shuttling of SRY and SOX9. Trends Endocrinol. Metab. 2008;19:213–222. doi: 10.1016/j.tem.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. N. Engl. J. Med. 2003;349:760–766. doi: 10.1056/NEJMoa030064. [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am. J. Hum. Genet. 1996;59:983–989. [PMC free article] [PubMed] [Google Scholar]
- Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998;18:213–215. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- Sultan C, Biason-Lauber A, Philibert P. Mayer–Rokitansky–Kuster–Hauser syndrome: recent clinical and genetic findings. Gynecol. Endocrinol. 2009;25:8–11. doi: 10.1080/09513590802288291. [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA. 2010;107:15140–15144. doi: 10.1073/pnas.1009622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobet SA, Schwarting GA. Minireview: recent progress in gonadotropin-releasing hormone neuronal migration. Endocrinology. 2006;147:1159–1165. doi: 10.1210/en.2005-1275. [DOI] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat. Genet. 2009;41:354–358. doi: 10.1038/ng.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu AK, Tello JA, Kotan LD, Ozbek MN, Yilmaz MB, Erdogan S, et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N. Engl. J. Med. 2012;366:629–635. doi: 10.1056/NEJMoa1111184. [DOI] [PubMed] [Google Scholar]
- Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. USA. 2011;108:11524–11529. doi: 10.1073/pnas.1102284108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treloar SA, Wicks J, Nyholt DR, Montgomery GW, Bahlo M, Smith V, et al. Genomewide linkage study in 1176 affected sister pair families identifies a significant susceptibility locus for endometriosis on chromosome 10q26. Am. J. Hum. Genet. 2005;77:365–376. doi: 10.1086/432960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno S, Zembutsu H, Hirasawa A, Takahashi A, Kubo M, Akahane T, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet. 2010;42:707–710. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA, Norman RJ, et al. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc. Natl. Acad. Sci. USA. 1999;96:8573–8578. doi: 10.1073/pnas.96.15.8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur C, Rodien P, Beau I, Desroches A, Gerard C, de Poncheville L, et al. A chorionic gonadotropin–sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. N. Engl. J. Med. 2003;349:753–759. doi: 10.1056/NEJMoa030065. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Vockley J, Rinaldo P, Bennett MJ, Matern D, Vladutiu GD. Synergistic heterozygosity: disease resulting from multiple partial defects in one or more metabolic pathways. Mol. Genet. Metab. 2000;71:10–18. doi: 10.1006/mgme.2000.3066. [DOI] [PubMed] [Google Scholar]
- Weiss J, Axelrod L, Whitcomb RW, Harris PE, Crowley WF, Jr., Jameson JL. Hypogonadism caused by a single amino acid substituion in the β subunit of luteinizing hormone. N. Engl. J. Med. 1992;326:179–183. doi: 10.1056/NEJM199201163260306. [DOI] [PubMed] [Google Scholar]
- Xu N, Kim HG, Bhagavath B, Cho SG, Lee JH, Ha K, et al. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 2011 doi: 10.1016/j.fertnstert.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanase T, Simpson ER, Waterman MR. 17-Alpha-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr. Rev. 1991;12:91–108. doi: 10.1210/edrv-12-1-91. [DOI] [PubMed] [Google Scholar]
- Young J, Metay C, Bouligand J, Tou B, Francou B, Maione L, et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 2012;27:1460–1465. doi: 10.1093/humrep/des022. [DOI] [PubMed] [Google Scholar]
- Zhao H, Chen ZJ, Qin Y, Shi Y, Wang S, Choi Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am. J. Hum. Genet. 2008;82:1342–1348. doi: 10.1016/j.ajhg.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Layman LC. Genetic considerations in the patient with Turner syndrome-45,X with or without mosaicism. Fertil. Steril. 2012;98:775–779. doi: 10.1016/j.fertnstert.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Mochizuki T, Smeets H, Antignac C, Laurila P, de Paepe A, et al. Deletion of the paired alpha 5(IV) and alpha 6(IV) collagen genes in inherited smooth muscle tumors. Science. 1993;261:1167–1169. doi: 10.1126/science.8356449. [DOI] [PubMed] [Google Scholar]