

Cyclotides are fascinating natural plant micro-proteins ranging from 28 to 37 amino acid residues long and exhibit various biological actions including anti-microbial, insecticidal, cytotoxic, antiviral (against HIV), protease inhibitory, and hormone-like activities.[1–4] They share a unique head-to-tail circular knotted topology of three disulfide bridges; one disulfide penetrates through a macrocycle formed by the other two disulfides, thereby inter-connecting the peptide backbone to form what is called a cystine knot topology (Fig. 1). This cyclic cystine knot (CCK) framework gives the cyclotides exceptional rigidity[5], resistance to thermal and chemical denaturation, and enzymatic stability against degradation.[4, 6] In fact, some cyclotides have been shown to be orally bioavailable. For example, the first cyclotide to be discovered, kalata B1, was found to be an orally effective uterotonic,[7] and other cyclotides have been shown to cross the cell membrane through macropinocytosis.[8–10] All of these features make cyclotides ideal tools for drug development.[11–14]
Figure 1.
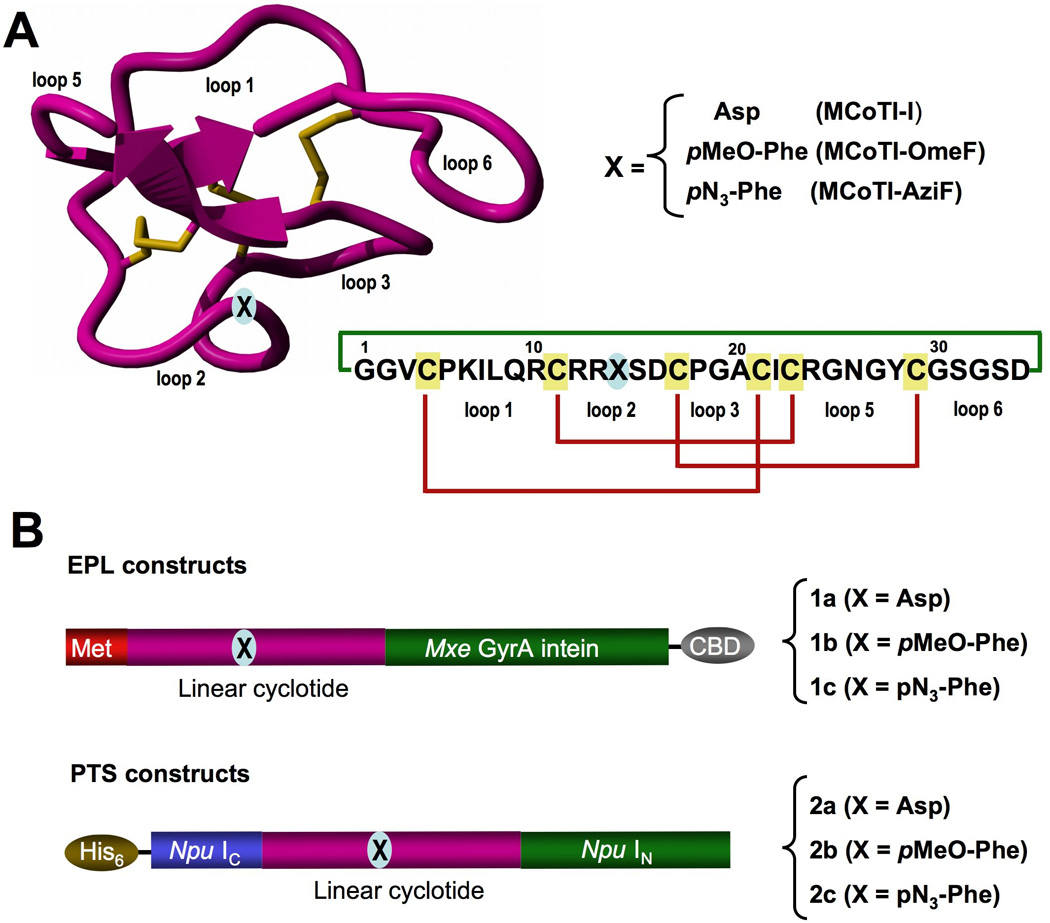
A. Tertiary structure of the cyclotide MCoTI-II (PDB code: 1IB9)[52] and primary structures of cyclotides used in this study. The backbone cyclized peptide (connecting bond shown in green) is stabilized by the three disulfide bonds (shown in red). B. Intein precursors used for the expression of cyclotides produced in this work. The Uaa was introduced at position 14, which is in the middle of loop 2 and is marked with an “X”.
Cyclotides have been isolated from plants in the Rubiaceae, Violaceae, Cucurbitacea,[4, 15] and, most recently, Fabaceae families.[16–18] Around 200 different cyclotide sequences have been reported in the literature,[19, 20] although it has been estimated that ≈ 50,000 cyclotides may exist.[21, 22] Despite sequence diversity, all cyclotides share the same CCK motif (Fig.1A). Hence, these micro-proteins can be considered natural combinatorial peptide libraries that are structurally constrained by the cystine-knot scaffold[2] and head-to-tail cyclization and in which, with the exception of the strictly conserved cysteines comprising the cysteine-knot, hypermutation of essentially all residues is permitted. Cyclotides can be chemically synthesized, thereby permitting the introduction of specific chemical modifications or biophysical probes.[14, 23–25] More importantly, cyclotides can now be biosynthesized in bacterial cells using a biomimetic approach that involves the use of modified protein splicing units.[26–28] These characteristics make them ideal substrates for the production of genetically-encoded libraries based on the cyclotide framework. These cell-based libraries allow in-cell molecular evolution strategies to enable the generation and high throughput selection of compounds with optimal binding and inhibitory characteristics. In contrast to chemically-generated libraries, genetically-encoded libraries enable the facile generation and screening of very large combinatorial libraries of molecules.
The genetic code of most organisms encodes only 20 canonical amino acid building blocks, with the rare exceptions of selenocysteine[29] and pyrrolysine,[30] which limits the chemical complexity of genetically-encoded libraries that can be produced in living cells. The recent development of nonsense suppressing orthogonal tRNA/synthetase technology has allowed the genetic encoding of a large variety of unnatural amino acids (Uaas).[31–34] In this work, we report the in-cell production of natively folded cyclotides containing Uaas by employing in vivo Uaa incorporation in combination with protein splicing to mediate intracellular backbone cyclization. To our knowledge, this is the first time that a natively folded cyclotide containing Uaas has been produced inside living cells. This approach opens the possibility for in-cell generation of cyclotides containing Uaas with new or enhanced biological functions. For example, the introduction of fluorescent amino acids or Uaas able to site-specifically incorporate fluorescent probes should facilitate the in-cell production of fluorescently-labeled cyclotides for screening or probing molecular interactions using optical approaches.
To test the feasibility of introducing Uaas into folded cyclotides in living cells, we used the cyclotide MCoTI-I (Fig. 1A). This cyclotide is a powerful trypsin inhibitor (Ki ≈ 20 pM)[27] that has been recently isolated from dormant seeds of Momordica cochinchinensis, a plant member of the Cucurbitaceae family.[35] Trypsin inhibitor cyclotides are interesting candidates for drug design because their specificity for inhibition can be altered and their structures can be used as natural scaffolds to generate novel binding activities.[36]
Since MCoTI-cyclotides have been expressed inside Escherichia coli cells using an intramolecular version of expressed protein ligation (EPL),[27, 28] we decided to try this approach first for the in-cell generation of MCoTI-based cyclotides containing different Uaas. This method relies on the use of a protein splicing unit in combination with an in-cell intramolecular native chemical ligation reaction to perform the backbone cyclization of the linear cyclotide precursor.[26, 27] The amber stop codon TAG was used to encode the Uaa at the position corresponding to the residue Asp14 in MCoTI-I. This residue is located in the middle of loop 2 (Fig.1A), which has been shown to be tolerant to mutations without affecting the structure and biological activity of the resulting cyclotide.[28] The incorporation of Uaas into the cyclotide framework was tested using the Uaas p-methoxyphenylalanine (OmeF) and p-azidophenylalanine (AziF), which have been successfully incorporated into various recombinant proteins.[33] More importantly, incorporation of AziF into the cyclotide framework should allow the site-specific incorporation of fluorescent probes into this scaffold inside the living cell by using alkyne-containing fluorescent probes and click chemistry.[37]
First, we explored the expression level of the corresponding intein precursors (1b and 1c, Fig. 1B) in BL21(DE3) cells. Expression of the intein precursors of MCoTI-OmeF and MCoTI-AziF was performed in cells co-transformed with a plasmid encoding the corresponding MCoTI-intein precursor for EPL-mediated cyclization and the plasmid encoding the orthogonal amber suppressing tRNACUA/ aminoacyl-tRNA synthetase pair specific for OmeF (pVLOmeRS) or AziF (pERAzi), respectively. In both cases, the expression level of the intein precursors containing Uaas (1b and 1c) was similar (Fig. S1). The suppression efficiency was estimated to be ≈ 10% (MCoTI-OmeF precursor, 1b) and ≈ 20% (MCoTI-AziF precursor, 1c) compared to the expression of the wild-type MCoTI-I intein precursor 1a (≈ 40 mg/L).
Under the expression conditions used in these experiments, all intein precursors showed around 60% in vivo cleavage, indicating that the intein was active and unaffected by the incorporation of the Uaa (Fig. S1). Next, we tested the ability of the different intein-MCoTI precursors to produce the corresponding folded cyclotide by treatment with reduced glutathione (GSH) at pH 7.2 following the conditions optimized for MCoTI-cyclotides.[38] In both cases, the in vitro reaction was clean and efficient in providing the properly folded cyclotides MCoTI-OmeF and MCoTI-AziF (Fig. S1). The final yield after purification was 4 µg/L (MCoTI-OmeF) and 14 µg/L (MCoTI-AziF). The expression yield for the wild-type MCoTI-I using these expression and cyclization conditions was ≈ 48 µg/L after purification. Next, we explored the expression of the MCoTI-OmeF and MCoTI-AziF cyclotides inside bacterial cells using EPL-mediated cyclization.[27, 28, 38] When we tried this approach with the cyclotides MCoTI-OmeF and MCoTI-AziF, however, the amount of folded cyclotides was below the detection limit.
In order to boost the expression of cyclotides in living cells we explored the use of protein trans-splicing (PTS) to facilitate the in-cell cyclization process and to improve the expression yield of Uaa-containing cyclotides (Scheme 1). Protein trans-splicing is a post-translational modification similar to protein splicing with the difference that the intein self-processing domain is split into N- (IN) and C-intein (IC) fragments. The split-intein fragments are not active individually, however, they can bind to each other with high specificity under appropriate conditions to form an active protein splicing or intein domain in trans.[39] PTS-mediated backbone cyclization can be accomplished by rearranging the order of the intein fragments. By fusing the IN and IC fragments to the C- and N-termini of the polypeptide for cyclization, the trans-splicing reaction yields a backbone-cyclized polypeptide (Scheme 1).[40] This approach has been recently used for the biosynthesis of cyclic hexapeptides containinig Uaas.[41] In this work, in-cell cyclization was performed using the naturally occurring Synechocystis sp. (Ssp) PCC6803 DnaE split intein.[42] However, the Ssp DnaE intein requires specific amino acid residues at both intein-extein junctions for efficient protein splicing.[43] To overcome this problem we used the Nostoc puntiforme PCC73102 (Npu) DnaE split-intein. This DnaE intein has the highest reported rate of protein trans-splicing (τ1/2 ≈ 60 s)[44] and has a high splicing yield.[44, 45] First, we explored the ability of the Npu DnaE split-intein to produce folded wild-type MCoTI-I cyclotide inside living E. coli cells. To accomplish this, we designed the split-intein construct 2a (Fig. 1B). In this construct, the MCoTI-I linear precursor was fused in-frame at the C- and N-termini directly to the Npu DnaE IN and IC polypeptides. None of the additional native C- or N-extein residues were added in this construct. We used the native Cys residue located at the beginning of loop 6 of MCoTI-I (Fig. 1) to facilitate backbone cyclization. A His-tag was also added at the N-terminus of the construct to facilitate purification.
Scheme 1.
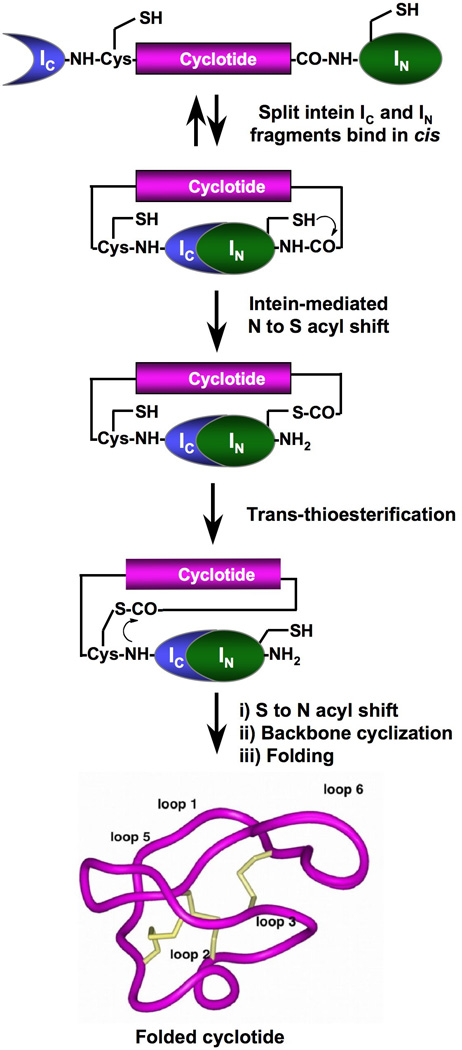
In-cell expression of native folded cyclotides using intein-mediated protein trans-splicing
In-cell expression of wild-type MCoTI-I using PTS-mediated backbone cyclization was achieved by transforming the plasmid encoding the split-precursor 2a into Origami(DE3) cells to facilitate folding.[46] The MCoTI-precursor split-intein was over expressed for 18 h at room temperature. Using these conditions the precursor was expressed at very high levels (≈ 70 mg/L) and was almost completely cleaved (≥ 95% in vivo cleavage, Fig. 2A). The high reactivity of this precursor prevented us from performing a full characterization of the precursor protein including kinetic studies of the trans-splicing induced reaction in vitro. Next, we tried to isolate the natively folded MCoTI-I generated in-cell by incubating the soluble fraction of a fresh cell lysate with trypsin-immobilized sepharose beads. Correctly folded MCoTI-cyclotides are able to bind trypsin with high affinity (Ki ≈ 20–30 pM). Therefore, this step can be used for affinity purification and to test the biological activity of the recombinant cyclotides.[28] After extensive washing, the absorbed products were eluted with a solution containing 8 M guanidinium chloride (GdmCl) and analyzed by HPLC. The HPLC analysis revealed the presence of a major peak that had the expected mass of the native MCoTI-I fold (Figs. 2B and S2). Recombinant MCoTI-I produced by PTS-mediated cyclization was also natively folded as characterized by NMR spectroscopy (Fig. S2 and Table S2).[5] The in-cell expression level of folded MCoTI-I produced by PTS-mediated cyclization was estimated to be ≈ 70 µg/L of bacterial culture, which corresponds to an intracellular concentration of ≈ 7 µM.
Figure 2.
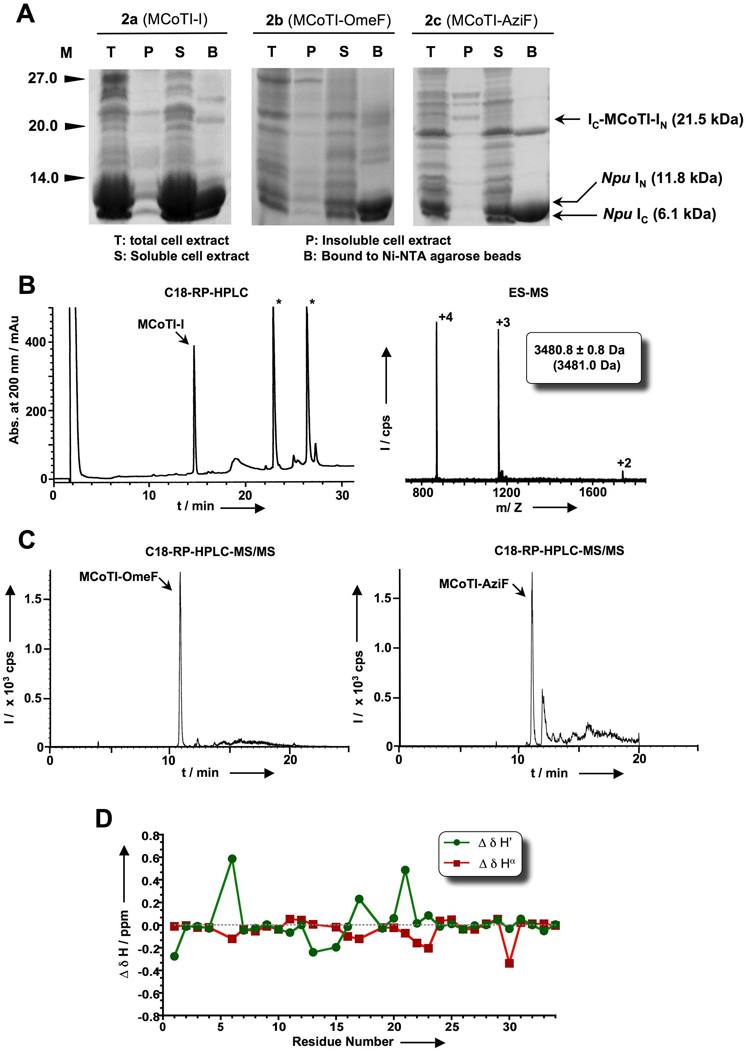
In-cell expression of MCoTI-I based cyclotides in E. coli cells using Npu DnaE intein-mediated PTS. A. SDS-PAGE analysis of the recombinant expression of cyclotide precursors 2a, 2b and 2c in Origami2(DE3) cells for in-cell production of the cyclotides MCoTI-I and MCoTI-OmeF, respectively. B. Analytical HPLC trace (left panel) of the soluble cell extract of bacterial cells expressing precursor 2a (MCoTI-I) after purification by affinity chromatography on a trypsin-sepharose column. Folded MCoTI-I is marked with an arrow. Endogenous bacterial proteins that bind trypsin are marked with an asterisk. Mass spectrum (right panel) of affinity purified MCoTI-I. The expected average molecular weight is shown in parentheses. C. LC-MS/MS analysis of the soluble cell extract of bacterial cells expressing precursors 2b (MCoTI-OmeF) and 2c (MCoTI-AziF). D. Summary of the backbone 1H NMR assignments for the backbone protons of MCoTI-OmeF produced in-cell by PTS: Δδ(1H) are the deviations in the chemical shifts of the main chain protons between the values obtained for MCoTI-OmeF and MCoTI-I (Table S2). Assignments for residue 14 were not included in the graph.
In-cell expression of folded MCoTI-cyclotides by PTS was about 7 times more efficient than intramolecular EPL-mediated backbone cyclization. This improvement may be explained by our choice of the split-intein Npu DnaE. This split-intein is extremely efficient; it exhibits fast kinetics with a good yield of protein trans-splicing. Differences in the cyclization process between the PTS and EPL methods may also contribute to the improvement in the cyclization yield. In PTS, the cyclization is driven by the affinity between the two-intein fragments, IN and IC, which in the case of the Npu DnaE intein is very high (KD ≈ 3 nM).[47] Once the intein complex is formed, the trans-splicing reaction is also extremely fast (τ1/2 ≈ 60 s for the Npu DnaE intein).[48] In contrast, EPL-mediated cyclization follows a slightly more complex mechanism that relies on the formation of the C-terminal thioester at the N-extein-junction and the removal of the N-terminal leading sequence (a Met residue in this case) to provide an N-terminal Cys. These two groups then react to form a peptide bond between the N- and C-termini of the polypeptide. It is also worth noting that in contrast with the Ssp DnaE intein, which requires at least 4 native residues at the N- and C-terminal extein-intein junctions to work efficiently,[49] the Npu ortholog used in this work tolerates different sequences at both junctions as demonstrated by the efficient trans-splicing of precursor 2a (Fig. 2A). The tetrapeptide sequences at both intein-extein junctions in construct 2a have only a 20% sequence homology with the native sequences of both Npu DnaE exteins.
Encouraged by these results, we decided to try in-cell expression of cyclotides MCoTI-OmeF and MCoTI-AziF using PTS. For this purpose precursors 2b and 2c (Fig. 1B) were overexpressed in Origami (DE3) cells by transforming the plasmids pVLOmeRS and pERAzi and growing the bacterial cells in the presence of OmeF or AziF, respectively. Constructs 2b and 2c are similar to 2a but were designed to incorporate Uaas into residue Asp14 in MCoTI-I (Fig. 1B). The expression level of the intein precursors 2b and 2c were ≈ 7 mg/L (10% suppression in comparison to wild-type precursor 2a) and ≈ 20 mg/mL (25% suppression), respectively. In-cell trans-splicing for 2b and 2c was also similar (≥ 95%, Fig. 2A) to that of the wild-type PTS construct 2a. Cyclotides MCoTI-OmeF and MCoTI-AziF were purified by affinity chromatography using trypsin sepharose beads from fresh soluble cell lysates, and the trypsin-bound fractions were analyzed by LC-MS/MS and ES-MS (Figs. 2C and S3). Cyclotide MCoTI-OmeF generated in-cell by PTS was also characterized by NMR, confirming the adoption of a native cyclotide fold (Fig. 2D and S4). The in-cell expression level for cyclotide MCoTI-OmeF and MCoTI-AziF were estimated to be ≈ 1 µg/L and ≈ 2 µg/L corresponding to an intracellular concentration ≈ 0.1 µM and 0.17 µM, respectively.
Next we explored the possibility of using fluorescently-labeled cyclotides to perform screening of protein-cyclotide interactions. To accomplish this we used MCoTI-AziF and trypsin as a model system. Preliminary results showed that MCoTI-AziF can be efficiently (almost quatitatively) labeled in vitro with a dibenzo-cyclooctyne (DBCO)-derivative of the fluorescent dye amino-methyl-coumarin acetate (AMCA) through copper-free click chemistry (Fig. S5). In-cell labeling of MCoTI-AziF with DBCO-AMCA was also very efficient (Figs. 3A and S7). No un-reacted MCoTI-AziF was found after treatment of the cells with DBCO-AMCA as determined by LC-MS/MS (data not shown). As expected, the resulting AMCA-labeled MCoTI-AziF was able to bind commercial porcine pancreatic trypsin efficiently (Fig. S8), indicating that introduction of the fluorophore in the loop 2 of MCoTI-AziF did not have a detrimental effect on its biological activity. To facilitate monitoring of this interaction either in-vitro or in-cell we used rat anionic trypsin, which can be expressed more efficiently in bacterial expression systems.[50] We decided to use fluorescence resonance energy transfer (FRET) to visualize the interaction between trypsin and AMCA-labeled MCoTI-AziF. For this purpose, the protease was fused to the N-terminus of the enhanced green fluorescent protein (EGFP). AMCA and EGFP show a good overlap between the emission band of the donor (AMCA) and the absorption band of the acceptor (EGFP) thus allowing the visualization of the molecular interaction FRET.[51] Moreover, structural analysis of a MCoTI-II-trypsin complex model[28] reveals that the distance between the C-terminus of trypsin and the Cα of residue 15 in MCoTI-I is ≈ 35 Å. This distance is well in range for the visualization of the complex formation by FRET.[51] The catalytic residue Ser195 in trypsin was also mutated to Ala to facilitate the recombinant expression of trypsin-S195A-EGFP by preventing its cellular toxicity. As shown in Figs. 3B and 3C, AMCA-labeled cyclotide MCoTI-AziF was able to efficiently bind trypsin-S195A-EGFP (KD of 1.8 ± 0.7 nM) in vitro, and more importantly the cyclotide-protein interaction could be easily monitored by intermolecular FRET shown by the simultaneous decrease and increase of the fluorescence signal at 445 and 515 nm, respectively.
Figure 3.
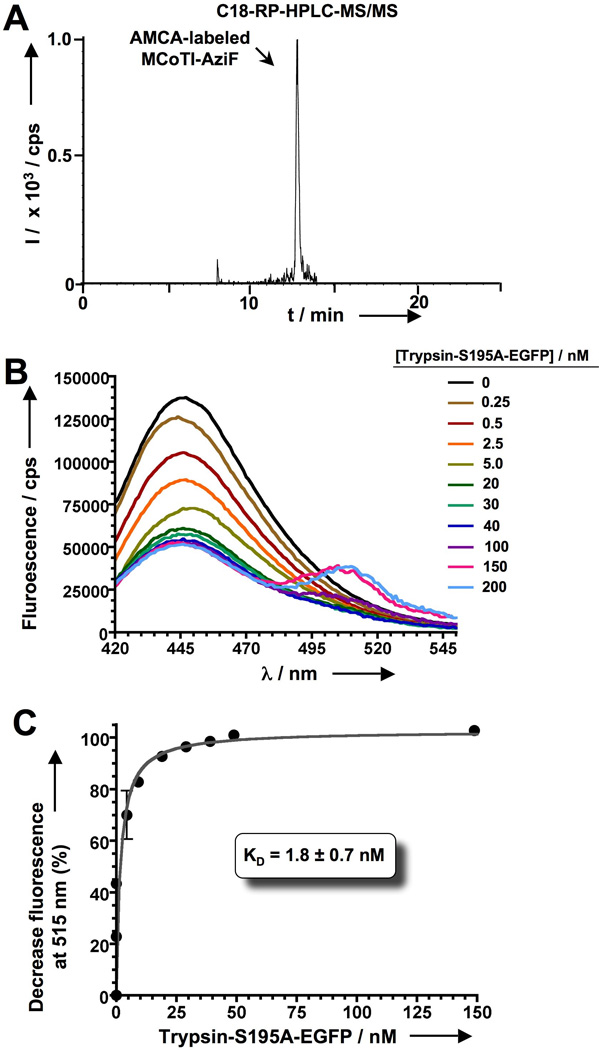
In-cell production and biological activity of AMCA-labeled MCoTI-AziF. A. Analytical HPLC-MS/MS trace of the soluble cell extract of bacterial cells indicating the presence of AMCA-labeled MCoTI-AziF. B. Titration of AMCA-labeled MCoTI-AziF (2 nM) with increasing amounts of trypsin-S195A-EGFP (0 – 200 nM) followed by fluorescence spectroscopy. The formation of the complex can be easily followed by FRET by decrease and concomitant increase of the fluorescence signals at 445 and 515 nm, respectively. Excitation was performed at 360 nm. Background signal from EGFP in the absence of AMCA-labeled MCoTI-AziF was corrected in all spectra. C. Binding isotherm of AMCA-labeled MCoTI-AziF and trypsin-S195A-EGFP by plotting the decrease in fluorescence signal at 445 nm. The dissociation constant was calculated assuming a 1:1 molecular complex.
In summary, we have shown that the biosynthesis of cyclotides containing Uaas can be achieved by using different intein-based methods. EPL-backbone cyclization can provide Uaa-containing cyclotides when the cyclization is carried out in-vitro by GSH-induced cyclization and folding of the corresponding precursor. In-cell production, however, is less efficient using this method. We have shown that PTS-mediated backbone cyclization using the highly efficient Npu DnaE split-intein can be employed for the efficient production of cyclotides inside live E. coli cells. We estimate that the in-cell production of MCoTI-I was around 7 times more efficient using Npu DnaE PTS than EPL, thereby providing an attractive alternative for the production of these types of polypeptides in bacterial cells. The high efficiency of PTS-mediated cyclization combined with nonsense suppressing orthogonal tRNA/synthetase technology made the in-cell production of cyclotides containing Uaas possible. Of particular interest is the introduction azido-containing Uaas, which can react with DBCO-containing fluorescent probes to provide in-cell fluorescently-labeled cyclotides. The classical approach for in-cell production of fluorescent-labeled proteins by fusing a fluorescent protein to the target protein is not applicable to cyclotides due to their small size and restricted backbone-cyclized topology. We have shown that cyclotides containing the Uaa AziF can be expressed in live bacterial cells and easily labeled with DBCO-AMCA to monitor cyclotide-protein interactions. This finding opens the possibility for in-vitro and potentially also in-cell screening of genetically-encoded libraries of cyclotides for the rapid selection of novel cyclotide sequences able to bind a specific bait proteins using high throughput cell-based optical screening approaches.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Research Grants R01-GM090323 (JAC), R01-GM085006 (AS), DP2-OD004744 (LW) and by the Department of Defense Congressionally Directed Medical Research Program Grant PC09305 (JAC). We also like to thank Sam Ulin (Salk Institute) for his contribution in the preparation of plasmid pERAzi.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Craik DJ, Simonsen S, Daly NL. Curr. Opin. Drug Discov. Devel. 2002;5:251. [PubMed] [Google Scholar]
- 2.Craik DJ, Cemazar M, Wang CK, Daly NL. Biopolymers. 2006;84:250. doi: 10.1002/bip.20451. [DOI] [PubMed] [Google Scholar]
- 3.Jagadish K, Camarero JA. Biopolymers. 2010;94:611. doi: 10.1002/bip.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia AE, Camarero JA. Curr Mol Pharmacol. 2010;3:153. doi: 10.2174/1874467211003030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puttamadappa SS, Jagadish K, Shekhtman A, Camarero JA. Angew Chem Int Ed Engl. 2010;49:7030. doi: 10.1002/anie.201002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daly NL, Rosengren KJ, Craik DJ. Adv Drug Deliv Rev. 2009;61:918. doi: 10.1016/j.addr.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Saether O, Craik DJ, Campbell ID, Sletten K, Juul J, Norman DG. Biochemistry. 1995;34:4147. doi: 10.1021/bi00013a002. [DOI] [PubMed] [Google Scholar]
- 8.Greenwood KP, Daly NL, Brown DL, Stow JL, Craik DJ. Int J Biochem Cell Biol. 2007;39:2252. doi: 10.1016/j.biocel.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Contreras J, Elnagar AY, Hamm-Alvarez SF, Camarero JA. J Control Release. 2011;155:134. doi: 10.1016/j.jconrel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cascales L, Henriques ST, Kerr MC, Huang YH, Sweet MJ, Daly NL, Craik DJ. J Biol Chem. 2011;286:36932. doi: 10.1074/jbc.M111.264424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark RJ, Daly NL, Craik DJ. Biochem J. 2006;394:85. doi: 10.1042/BJ20051691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Craik DJ, Cemazar M, Daly NL. Curr Opin Drug Discov Devel. 2006;9:251. [PubMed] [Google Scholar]
- 13.Craik DJ, Daly NL, Mulvenna J, Plan MR, Trabi M. Curr Protein Pept Sci. 2004;5:297. doi: 10.2174/1389203043379512. [DOI] [PubMed] [Google Scholar]
- 14.Thongyoo P, Roque-Rosell N, Leatherbarrow RJ, Tate EW. Org Biomol Chem. 2008;6:1462. doi: 10.1039/b801667d. [DOI] [PubMed] [Google Scholar]
- 15.Cascales L, Craik DJ. Org Biomol Chem. 2010;8:5035. doi: 10.1039/c0ob00139b. [DOI] [PubMed] [Google Scholar]
- 16.Poth AG, Colgrave ML, Philip R, Kerenga B, Daly NL, Anderson MA, Craik DJ. ACS Chem Biol. 2010;6:345. doi: 10.1021/cb100388j. [DOI] [PubMed] [Google Scholar]
- 17.Poth AG, Colgrave ML, Lyons RE, Daly NL, Craik DJ. Proc Natl Acad Sci U S A. 2011;108:1027. doi: 10.1073/pnas.1103660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen GK, Zhang S, Nguyen NT, Nguyen PQ, Chiu MS, Hardjojo A, Tam JP. J Biol Chem. 2011 doi: 10.1074/jbc.M111.229922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulvenna JP, Wang C, Craik DJ. Nucleic Acids Res. 2006;34:D192. doi: 10.1093/nar/gkj005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang CK, Kaas Q, Chiche L, Craik DJ. Nucleic Acids Res. 2008;36:D206. doi: 10.1093/nar/gkm953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulvenna JP, Mylne JS, Bharathi R, Burton RA, Shirley NJ, Fincher GB, Anderson MA, Craik DJ. Plant Cell. 2006;18:2134. doi: 10.1105/tpc.106.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gruber CW, Elliott AG, Ireland DC, Delprete PG, Dessein S, Goransson U, Trabi M, Wang CK, Kinghorn AB, Robbrecht E, Craik DJ. Plant Cell. 2008;20:2471. doi: 10.1105/tpc.108.062331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daly NL, Love S, Alewood PF, Craik DJ. Biochemistry. 1999;38:10606. doi: 10.1021/bi990605b. [DOI] [PubMed] [Google Scholar]
- 24.Avrutina O, Schmoldt HU, Kolmar H, Diederichsen U. Eur. J. Org. Chem. 2004;204:4931. [Google Scholar]
- 25.Thongyoo P, Tate EW, Leatherbarrow RJ. Chem Commun (Camb) 2006:2848. doi: 10.1039/b607324g. [DOI] [PubMed] [Google Scholar]
- 26.Kimura RH, Tran AT, Camarero JA. Angew. Chem. Int. Ed. 2006;45:973. doi: 10.1002/anie.200503882. [DOI] [PubMed] [Google Scholar]
- 27.Camarero JA, Kimura RH, Woo YH, Shekhtman A, Cantor J. Chembiochem. 2007;8:1363. doi: 10.1002/cbic.200700183. [DOI] [PubMed] [Google Scholar]
- 28.Austin J, Wang W, Puttamadappa S, Shekhtman A, Camarero JA. Chembiochem. 2009;10:2663. doi: 10.1002/cbic.200900534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bock A, Forchhammer K, Heider J, Baron C. Trends Biochem Sci. 1991;16:463. doi: 10.1016/0968-0004(91)90180-4. [DOI] [PubMed] [Google Scholar]
- 30.Srinivasan G, James CM, Krzycki JA. Science. 2002;296:1459. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 31.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Parrish AR, Wang L. Chem Biol. 2009;16:323. doi: 10.1016/j.chembiol.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Xie J, Schultz PG. Annu Rev Biophys Biomol Struct. 2006;35:225. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez JF, Gagnon J, Chiche L, Nguyen TM, Andrieu JP, Heitz A, Trinh Hong T, Pham TT, Le Nguyen D. Biochemistry. 2000;39:5722. doi: 10.1021/bi9929756. [DOI] [PubMed] [Google Scholar]
- 36.Gould A, Ji Y, Aboye TL, Camarero JA. Curr Pharm Des. 2011;17:4294. doi: 10.2174/138161211798999438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beatty KE, Fisk JD, Smart BP, Lu YY, Szychowski J, Hangauer MJ, Baskin JM, Bertozzi CR, Tirrell DA. Chembiochem. 2010;11:2092. doi: 10.1002/cbic.201000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin J, Kimura RH, Woo YH, Camarero JA. Amino Acids. 2010;38:1313. doi: 10.1007/s00726-009-0338-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sancheti H, Camarero JA. Adv Drug Deliv Rev. 2009;61:908. doi: 10.1016/j.addr.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aboye TL, Camarero JA. J Biol Chem. 2012;287:27026. doi: 10.1074/jbc.R111.305508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, Schultz PG. Proc Natl Acad Sci U S A. 2011;108:11052. doi: 10.1073/pnas.1108045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott CP, Abel-Santos E, Wall M, Wahnon D, Benkovic SJ. Proc. Natl. Acad. Sci. USA. 1999;96:13638. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tavassoli A, Benkovic SJ. Nat Protoc. 2007;2:1126. doi: 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- 44.Zettler J, Schutz V, Mootz HD. FEBS Lett. 2009;583:909. doi: 10.1016/j.febslet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Iwai H, Zuger S, Jin J, Tam PH. FEBS Lett. 2006;580:1853. doi: 10.1016/j.febslet.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 46.Bessette PH, Aslund F, Beckwith J, Georgiou G. Proc Natl Acad Sci U S A. 1999;96:13703. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah NH, Vila-Perello M, Muir TW. Angew Chem Int Ed Engl. 2011;50:6511. doi: 10.1002/anie.201102909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borra R, Dong D, Elnagar AY, Woldemariam GA, Camarero JA. J Am Chem Soc. 2012;134:6344. doi: 10.1021/ja300209u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwon Y, Coleman MA, Camarero JA. Angew. Chem. Int. Ed. 2006;45:1726. doi: 10.1002/anie.200503475. [DOI] [PubMed] [Google Scholar]
- 50.Vasquez JR, Evnin LB, Higaki JN, Craik CS. J Cell Biochem. 1989;39:265. doi: 10.1002/jcb.240390306. [DOI] [PubMed] [Google Scholar]
- 51.Kuhn SM, Rubini M, Muller MA, Skerra A. J Am Chem Soc. 2011;133:3708. doi: 10.1021/ja1099787. [DOI] [PubMed] [Google Scholar]
- 52.Felizmenio-Quimio ME, Daly NL, Craik DJ. J Biol Chem. 2001;276:22875. doi: 10.1074/jbc.M101666200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.