

Abstract
Platinum-based chemotherapy, such as cisplatin, oxaliplatin, and carboplatin, is one of the most widely utilized classes of cancer therapeutics. While highly effective, the clinical applications of platinum-based drugs are limited by their toxicity profiles as well as suboptimal pharmacokinetic properties. Therefore, one of the key research areas in oncology has been to develop novel platinum analog drugs and engineer new platinum drug formulations to improve the therapeutic ratio further. Such efforts have led to the development of platinum analogs including nedaplatin, heptaplatin, and lobaplatin. Moreover, reformulating platinum drugs using liposomes has resulted in the development of L-NDPP (Aroplatin™), SPI-77, Lipoplatin™, Lipoxal™, and LiPlaCis®. Liposomes possess several attractive biological activities, including biocompatibility, high drug loading, and improved pharmacokinetics, that are well suited for platinum drug delivery. In this review, we discuss the various platinum drugs and their delivery using liposome-based drug delivery vehicles. We compare and contrast the different liposome platforms as well as speculate on the future of platinum drug delivery research.
Keywords: liposome, platinum analog, drug delivery, cancer
Introduction
Platinum-based chemotherapy is one of the most widely used classes of cancer therapeutics. Today, there are three platinum chemotherapeutics approved by the US Food and Drug Administration, cisplatin, carboplatin, and oxaliplatin. Together, these drugs are used to treat a wide variety of cancers, including non-small and small cell lung, breast, colorectal, gastric, esophageal, testicular, cervical, and ovarian cancers, and non-Hodgkin’s lymphoma.1 Although the compound cis-[Pt(NH3)2(Cl)2] was described in the 1840s, its ability to inhibit cell division (in Escherichia coli) was not discovered until 1965.2 Subsequent clinical development of cis-dichloro-diammine-platinum (II), or cisplatin, eventually led to its approval for the treatment of testicular and ovarian cancers in 1978.1 The efficacy of cisplatin in testicular cancer was dramatic, with improvement in the cure rate from 5%–10% to 75%–80%.3 Following the clinical development of cisplatin, carboplatin was developed in the 1980s and oxaliplatin was developed in the 1990s. Carboplatin is used to treat similar types of cancers as cisplatin, although its toxicity, especially nephrotoxicity, is much lower than that of cisplatin. Oxaliplatin, on the other hand, has been shown to be effective against most gastrointestinal cancers, including colorectal, pancreatic, and gastric cancers.4
The mechanism of action of platinum chemotherapeutics is through DNA damage.5 For example, cisplatin undergoes aquation to form more reactive [Pt(NH3)2Cl(OH2)]+ and [Pt(NH3)2(OH2)2]2+ species after being internalized into cells. The more reactive platinum species then bind to their primary biological target, DNA, by forming coordination bonds with purine bases at the N7 positions. Such a reaction results in primarily 1,2-intrastrand or 1,3-intrastrand crosslinks and few interstrand crosslinks or adducts.6 These adducts can cause bending of the DNA duplex and facilitate binding of various proteins, such as high-mobility group box proteins. Protein-bound DNA adducts induce a number of cellular responses, including cell cycle arrest, inhibition of DNA replication and the transcription process, and cell apoptosis and necrosis. Cisplatin-bound DNA can also be recognized by repair proteins, such as xeroderma pigmentosum group A, xeroderma pigmentosum group F, and DNA excision repair protein ERCC1, leading to lesion removal and DNA recovery.7,8 Although the exact mechanisms and pathways that lead to cell death still require further investigation, the nucleotide excision repair pathway and several signal transduction pathways which control the ultimate fate of tumor cells, including those of the AKT, c-ABL, p53, and mitogen-activated protein/Jun N-terminal kinase/extracellular signal-regulated kinase pathways, are well documented and summarized in the literature.9
Despite being one of the most effective classes of chemotherapeutics, platinum drugs do have several significant shortcomings. First, all of the platinum chemotherapeutics are neurotoxic. The toxicity to the peripheral nervous system is one of the key dose-limiting toxicities.10 All three drugs also have relatively short blood circulation times, resulting in suboptimal pharmacokinetics. For cisplatin, nephrotoxicity as well as nausea and vomiting have significantly limited its clinical use.11 Although carboplatin has less toxicity than cisplatin, it is also much less potent.4,12 Myelotoxicity is also more profound with carboplatin, which is a dose-limiting toxicity.13 Because of these limitations, there has been strong interest in the development of novel platinum-based therapeutics to not only lower toxicity but also improve therapeutic efficacy. Two main strategies are employed. One is to develop new platinum analog drugs and the other is to utilize drug delivery technologies to engineer novel platinum drug formulations.14
Over the past several decades, researchers have developed over 3,000 platinum analogs or formulations. Unfortunately, only about 35 compounds exhibit adequate biological and pharmacologic activity to justify further preclinical and clinical investigations.12 Besides carboplatin and oxaliplatin, several other platinum analogs have been approved or entered clinical trials in some countries (structures are shown in Table 1).15 Nedaplatin has been registered in Japan for the treatment of head and neck, testicular, lung, ovarian, cervical, and non-small cell lung cancers. Heptaplatin (SKI2053R), which shows less anticancer activity than cisplatin in gastric cancer, has also been approved in South Korea because of its decreased toxicity profile. Lobaplatin has been used to treat chronic myelogenous leukemia and inoperable metastatic breast and small cell lung cancer in the People’s Republic of China.
Table 1.
Platinum-based anticancer drugs used in the clinic
| Molecular formula and structure | Manufacturer/distributor | Dose-limiting toxicity | Clinical status and indications | |
|---|---|---|---|---|
| Cisplatin |

|
Generic | Nephrotoxicity | Approved worldwide (sarcomas, small cell lung cancer, ovarian cancer, lymphomas, and germ cell tumors) |
| Carboplatin |
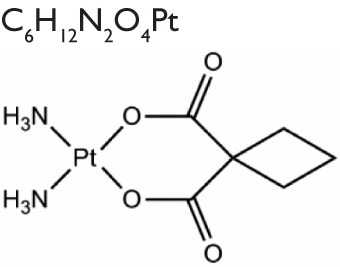
|
Generic | Myelosuppression | Approved worldwide (ovarian carcinoma, lung, head and neck cancers) |
| Oxaliplatin |

|
Sanofi S.A., Paris, France | Neurotoxicity | Approved worldwide (colorectal cancer, advanced gastric and ovarian cancers) |
| Nedaplatin |

|
Shionogi Pharmaceuticals, Osaka, Japan | Myelosuppression | Approved in Japan (head and neck, lung small cell, bladder, ovary, esophagus and cervix cancer) |
| Heptaplatin |

|
SK Chemicals Life Sciences, Seongnam, South Korea | Nephrotoxicity, intra-abdominal bleeding | Approved in Korea (gastric cancer) |
| Lobaplatin |

|
Asta-Medica GmbH, Dresden, Germany | Thrombocytopenia | Approved in the People’s Republic of China (chronic myelogenous leukemia, inoperable, metastatic breast, small cell lung cancer) |
Liposomal nanocarriers in drug delivery
Another strategy to improve the platinum drugs has been to improve the delivery of platinum therapeutics to tumors by use of nanoparticle drug delivery technology. A key challenge in cancer therapy is to deliver anticancer drugs and other chemotherapeutics selectively to tumors while minimizing accumulation in normal tissues. Such targeted delivery can improve therapeutic efficacy while reducing toxicity. Although such differential drug delivery is generally not possible with small molecular drugs, nanocarrier-based delivery can overcome this challenge via the enhanced permeability and retention effect.16,17 A distinct feature of tumor tissue compared with normal tissue is its rapid formation of vasculature triggered by vascular endothelial growth factor and other growth factors overexpressed in various cancerous cells. These newly formed vessels do not have a smooth muscle layer so are defective and have a wider lumen, leading to irregular and leaky boundaries. The other key feature of tumor tissue is dysfunctional lymphatic drainage, resulting in ineffective clearance of extravascular proteins, particles, and white blood cells.18 Due to their large size, nanocarriers are not able to penetrate through the normal vasculature, but can penetrate through the leaky vasculature around tumor regions. Together with ineffective lymphatic drainage of tumor tissues, differential delivery/accumulation could be realized.19 Thus, incorporation of small molecular drugs into a nanoplatform could lead to improved efficacy due to their favorable pharmacokinetic profiles.
Liposomes were engineered in 1965,20 and were soon appreciated by pioneers such as Gregory Gregoriadis as promising drug delivery vehicles.21 Liposomes are spherical vesicles with an aqueous interior surrounded by one or more concentric bilayers (called lamellae) of phospholipids with a diameter ranging from 30 nm to several microns. Liposomes can be divided into several subtypes according to their size and layered structures. Multilamellar liposomes usually have a diameter ranging from 500 to 10,000 nm, while small unilamellar liposomes normally have a diameter smaller than 50 nm and large unilamellar liposomes have a diameter greater than 50 nm. Liposomes are formed when a thin lipid film is hydrated with aqueous buffer solution, and are typically sonicated or repeatedly extruded through a 100 nm polycarbonate membrane to reduce their size and narrow their size distribution to afford small or large unilamellar liposomes, respectively.22 The physical and chemical properties of liposomes, such as surface charge, size, and stability, can be tuned using different lipid compositions. For instance, cationic, neutral, or anionic lipids can be used to control the surface charge of liposomes.23 Unsaturated phosphatidylcholine from natural sources (egg or soybean) generally produces less stable liposomes when compared with liposomes constructed using saturated phospholipids with long acyl chains, such as dipalmitoylphosphatidylcholine. Liposomes are biocompatible and can encapsulate hydrophilic and hydrophobic pharmaceutical agents in their internal water compartment and membrane, respectively. Encapsulation of hydrophobic drugs inside liposomes significantly increases their solubility in aqueous solution. The size, charge, and surface properties of liposomes that influence the pharmacokinetic profile of the encapsulated drug can be easily manipulated by adding other ingredients to the lipid mixture before liposome preparation and/or by altering preparation parameters.24 It is thus possible to prolong the half-life of a cytotoxic drug in the systemic circulation and alter its biodistribution pattern, leading to elevated accumulation in tumor tissue and a decreased dose to normal tissues. Formulation of therapeutics with liposomes can significantly reduce their side effect profile by avoiding non-targeted systemic drug exposure in the body. Upon accumulation at tumor sites, liposomes can also provide a unique opportunity to facilitate drug uptake into targeted cells or even localize the drugs to specific cellular compartments.
The efficacy of liposomal drugs has been further enhanced using a number of innovative strategies, such as remote drug loading,25–27 extrusion for homogeneous size,28 long-circulating (PEGylated) liposomes,29,30 triggered-release liposomes,31–34 and ligand-targeted liposomes.35–37 These advanced techniques have indeed led to several liposomal formulations in clinical use, with AmBisome® (Astellas Pharma US Inc, Northbrook, IL, USA) and Doxil® (Johnson and Johnson, New Brunswick, NJ, USA) being the most successful examples. Doxil, a pegylated liposomal formulation of doxorubicin, is the first liposomal anticancer formulation approved by the US Food and Drug Administration. In human studies, Doxil was found to have pharmacokinetics dramatically different to those of doxorubicin. Doxil has a half-life of approximately 90 hours, whereas doxorubicin has an initial distribution half-life of approximately 5 minutes followed by a terminal half-life of 20–48 hours. The area under the concentration-time curve (AUC) after a dose of 50 mg/m2 is about 300-fold greater with Doxil than with doxorubicin. More importantly, as the very first proof of the enhanced permeability and retention effect observed in humans, Doxil was found to accumulate preferentially in tumor tissue through passive targeting.38 The differential pharmacokinetic profiles between Doxil and doxorubicin also led to differing toxicity profiles. Doxil has significantly reduced cardiotoxicity, which is a dose-limiting toxicity using doxorubicin.39 On the other hand, Doxil causes more pronounced palmar-plantar erythrodysesthesia (hand-foot syndrome) than doxorubicin.40 The lower cardiotoxicity of Doxil is significant, because it allows prolonged and repeated treatments with Doxil that were previously not possible with doxorubicin. In addition to optimized biodistribution, tumor accumulation, and reduced cardiac toxicity, superior efficacy was observed in Kaposi’s sarcoma associated with acquired immune deficiency syndrome and recurrent ovarian cancer, and equivalent efficacy was observed in metastatic breast cancer and multiple myeloma.41 The most recent liposomal drug to be approved (in August 2012) by the US Food and Drug Administration is Marqibo® (Talon Therapeutics Inc, South San Francisco, CA, USA), a liposomal formulation of vincristine for treatment of relapsed Philadelphia chromosome-negative acute lymphoblastic leukemia.42
Clinically evaluated liposomal formulations for platinum drug delivery
In this section, we review several liposome particles that have been evaluated clinically (see Table 2). We compare their lipid compositions, physical properties, loading methods, and drug-to-lipid ratios. We also discuss their pharmacokinetics, biodistribution, toxicity profiles, and therapeutic efficacy, both in preclinical animal models and in patients.
Table 2.
Clinically evaluated liposomal formulations of platinum drugs
| Formulation | L-NDDP | SPI-77 | Lipoplatin | Lipoxal | LiPlaCis |
|---|---|---|---|---|---|
| Encapsulated drug | NDDP | Cisplatin | Cisplatin | Oxaliplatin | Cisplatin |
| Lipid composition | DMPC/DMPG | HSPC/cholesterol/DSPE-PEG2000 | HSPC/DPPG/DSPE-PEG2000 | NA | DSPC/DSPG/DSPE-PEG2000 |
| Particle size | 1–5 μm | 110 nm | 110 nm | NA | NA |
| Drug-to-lipid weight ratio | 1:15 | 1:70 | 1:10 | NA | NA |
| Half-life in animals (hours) | NA | 16 | 7 | NA | NA |
| Half-life in humans (hours) | t1/2α, 0.8–21 min t1/2β, 14–36 |
80–145 | 60–117 | 24–35 | t1/2α, 3–5.5 t1/2β, 80–141 |
| MTD (mg/m2) | 312.5 | 420 | 300 | 300 | 120 |
| Clinical status | Phase II | Phase II | Phase II, III | Phase I | Phase I |
| Indications | Colorectal cancer, malignant pleural mesothelioma | Ovarian, non-small cell lung, and head and neck cancer | Pancreatic cancer, head and neck cancer, mesothelioma, breast, gastric and non-small cell lung | Advanced gastrointestinal tract cancer | NA |
| References | 43–50 | 52–58 | 59–66 | 67 | 68 |
Abbreviations: MTD, maximum tolerated dose; NDDP, cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum (II); DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DMPG, 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt); HSPC, hydrogenated soy phosphatidylcholine; DSPE-PEG2000, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt); DPPG, 1,2-dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt); NA, not available; DSPC, 1,2-distearoyl-sn-glycero-3-phosphocholine; DSPG, 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt); min, minutes.
L-NDDP (Aroplatin™, Antigenics Inc, Lexington, MA, USA) was the first liposomal formulation studied in the clinic for the delivery of a cisplatin analog (cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum II, NDDP). Multilamellar liposomes encapsulating NDDP are formed after reconstitution using a mixture of 1,2-dimyristoylphosphatidylcholine (DMPC) and 1,2-dimyristoylphosphatidylglycerol (DMPG) lipids with acidified saline solution.4 Preclinical data showed that L-NDDP had a dramatically different biodistribution from that of NDDP, with accumulation of platinum in major organs, such as the liver, spleen, and lymph nodes.43,44 In a preclinical toxicology and antitumor activity study, it was found that L-NDDP did not induce nephrotoxicity, but myelosuppression was the major toxicity in mice. However, in canine models, L-NDDP also caused diffuse hemorrhagic syndrome.45–47 L-NDDP was found to be more active against liver and spleen metastases of M5076 reticulosarcoma and RAW 117 H-10 lymphoma in mice.48 A Phase I study of L-NDDP was performed using a single intravenous injection every 4 weeks.49 The maximum tolerated dose of L-NDDP was found to be 312.5 mg/m2 and the dose-limiting toxicity to be myelosuppression. A two-compartment pharmacokinetic model was found at lower doses but a single-compartment model at the maximum tolerated dose, suggesting that saturation occurs in the organs of the reticuloendothelial system. A Phase II study explored the antitumor activity and tolerability of L-NDDP in patients with refractory advanced colorectal carcinoma.50 The response was modest, with 5.6% having a partial response, 16.7% achieving stable disease, and 77.8% developing disease progression. L-NDDP was found to be well tolerated, and 9/20 patients (45%) were able to receive an escalated dose of 375 mg/m2 during the course of their treatment.
Studies using non-PEGylated multilamellar DMPC/DMPG liposomes showed accumulation of these particles in the liver, which limits their clinical utility. In order to avoid particle uptake by organs of the reticuloendothelial system, Mori et al formulated NDDP-containing unilamellar phosphatidylcholine/cholesterol-based liposomes with either monosialoganglioside (GM1) or polyethylene glycol (PEG)-phosphatidylethanolamine as the surface coating.51 Indeed, these long-circulating NDDP-containing liposomes showed an approximately three-fold increase in tumor accumulation as compared with conventional phosphatidylcholine/cholesterol-based liposomes. In vitro cytotoxicity studies using RIF-1 fibrosarcoma tumor cells showed that the presence of PEG-phosphatidylethanolamine, but not GM1, significantly enhanced the cytotoxicity of liposomal NDDP. In an in vivo RIF-1 tumor model in mice, a significant reduction in tumor growth rate was observed when NDDP was formulated in phosphatidylcholine/cholesterol/PEG3000/phosphatidylethanolamine liposomes. These results indicate the potential utility of long-circulating NDDP-containing liposomes for cancer treatment.
SPI-77 is a formulation of sterically stabilized, long-circulating liposomes encapsulating cisplatin. Unlike cisplatin, which has two-compartment pharmacokinetics with linear elimination, the pharmacokinetics of SPI-77 are best characterized according to a one-compartment model with nonlinear elimination. The half-life was estimated to be 16 hours in mice, compared with cisplatin which has a halflife of 0.24 hours. In addition to a longer blood circulation time, SPI-77 exhibits a 60-fold larger plasma AUC, three-fold higher peak plasma levels, a four-fold reduction in the amount of platinum delivered to the kidneys, and a 28-fold higher tumor AUC compared with cisplatin.52 SPI-77 was also shown to be more effective and better tolerated than free cisplatin in a variety of treatment schedules and cumulative doses in C26 and Lewis lung tumor xenograft models.52
Despite its superior pharmacokinetic properties, SPI-77 did not demonstrate enhanced therapeutic efficacy over cisplatin in preclinical experiments in a separate study of M-109 lung carcinoma, J-6456 lymphoma, and A-375 melanoma.53 In vitro release experiments showed that less than 10% of cisplatin was released from the liposomes, and a cytotoxicity assay also indicated reduced cytotoxic activity of SPI-77 in vitro when compared with cisplatin. It is believed that SPI-77 is delivered to tumor sites, but with extremely slow release kinetics. Similar results were obtained by Zamboni et al using microdialysis technology, showing that more SPI-77 distributes into tumors but releases less platinum into the extracellular tumoral fluid and forms fewer platinum-DNA adducts than cisplatin.54
Not surprisingly, SPI-77 was shown to have a long circulation time, with a plasma circulation half-life as long as 145 ± 107 hours in patients given a 420 mg/m2 dose in a Phase I study.55 More importantly, SPI-77 did not induce any of the toxicities commonly associated with platinum-based chemotherapy, such as nephrotoxicity and neutropenia. The formulation was shown to be well tolerated in patients at a dose range of 40–420 mg/m2. Side effects include mild gastrointestinal toxicity and mild anemia and muscle weakness. However, SPI-77 did not produce significant clinical response rates in several Phase II studies of patients with inoperable head and neck cancer, advanced non-small-cell lung cancer, or platinum-sensitive recurrence of ovarian cancer.56–58 The lack of therapeutic efficacy is likely due to slow and inefficient release of platinum from SPI-77, as shown in preclinical studies.
Lipoplatin™ (Regulon Inc, Mountain View, CA, USA) is another cisplatin-containing long-circulating liposomal formulation. It is composed of soy phosphatidylcholine, cholesterol, dipalmitoyl phosphatidyl glycerol, and methoxy-PEG-distearoyl phosphatidylethanolamine.59 The Lipoplatin formulation differs from SPI-77 in several ways. First, the loading method used in Lipoplatin is based on formation of reverse micelles between cisplatin and dipalmitoyl phosphatidyl glycerol, while the mechanism of cisplatin encapsulation in SPI-77 is totally passive. Second, the Lipoplatin formulation uses anionic dipalmitoyl phosphatidyl glycerol lipid and neutral soy phosphatidylcholine lipid, whereas SPI-77 uses only neutral lipids. Third, the cisplatin to total lipid ratio is around 1:10 in the case of Lipoplatin, but SPI-77 has a much lower drug-to-lipid ratio of 1:70.60
Preclinical studies of Lipoplatin have shown lower nephrotoxicity and other side effects in mice and rats when compared with free cisplatin but with higher antitumor activity in breast MCF-7 and prostate LNCaP human tumor xenograft models.61,62 Treatment of dogs with Lipoplatin led to the conclusion that the drug can be safely administered to healthy dogs at dosages of up to 150 mg/m2 without the need for concurrent hydration protocols.63 In several Phase I, II, and III studies, Lipoplatin was shown to reduce renal toxicity, peripheral neuropathy, ototoxicity, and myelotoxicity substantially but with enhanced or comparable efficacy to cisplatin.64 A 10–200-fold higher accumulation of Lipoplatin in solid tumors compared with adjacent normal tissues was found in patients.65 A Phase II study showed that Lipoplatin has lower renal toxicity as well as higher efficacy than cisplatin when combined with gemcitabine in advanced non-small cell lung cancer. A statistically significant higher response rate was also observed for a combination of Lipoplatin and paclitaxel when compared with the combination of cisplatin and paclitaxel in advanced non-small cell lung cancer in one randomized trial.66 However, there was no significant increase in survival. Lipoplatin has also been tested in a number of malignancies in several Phase II and III trials, including pancreatic cancer, head and neck cancer, and breast and gastric cancer, and preliminary results collected from these studies seem encouraging.59
Like Lipoplatin, Lipoxal™ is a liposomal formulation that carries oxaliplatin. In a Phase I study using six Lipoxal dose levels (100, 150, 200, 250, 300, and 350 mg/m2), no serious side effects were observed at doses of 100–250 mg/m2. Mild myelotoxicity, nausea, and grade 2–3 peripheral neuropathy were observed at doses of 300–350 mg/m2.67 With the reduction of many of the side effects of oxaliplatin, including myelotoxicity and gastrointestinal tract toxicity, and adequate antitumor activity, further clinical tests are warranted to demonstrate the superiority of Lipoxal over free oxaliplatin.
The lack of antitumor activity of SPI-77 in clinical trials suggests that hydrophilic chemotherapeutic agents like cisplatin cannot pass readily through the lipid membrane. The release of encapsulated drugs from liposome carriers upon their deposition in tumor tissues is critical to confer antitumor activity. LiPlaCis®, a novel liposomal formulation of cisplatin, is designed to be degraded by secretory phospholipase A2. Given that secretory phospholipase A2 is relatively abundant in tumor sites, triggered drug release in tumor tissue is expected. As shown in a Phase I study, the pharmacokinetic profile of LiPlaCis could be best fitted into a two-compartment model with the initial half-life (t1/2a) reflecting the half-life of the intact liposome, and the secondary half-life (t1/2B) reflecting the half-life of plasma protein-bound platinum.68 Although LiPlaCis was designed to be decomposed by secretory phospholipase A2 specifically, other factors also contributed to the degradation of the particles because no correlation between the baseline levels of secretory phospholipase A2 and the initial half-life of LiPlaCis was observed in patients. In addition, renal toxicity was not prevented by treatment by LiPlaCis and acute infusion reactions were observed in many patients even with premedication. The poor safety profile of LiPlaCis led to early cessation of this particular formulation in the Phase I stage and LiPlaCis requires reformulation to enable further development.
Preclinical liposomal formulations of platinum drugs with enhanced loading efficiency and active targeting capability
The clinical promise of liposomes in platinum drug delivery has encouraged many researchers to explore other possibilities to enhance delivery efficiency further. New technologies and novel systems with improved encapsulation efficiency, drug loading capacity, and active targeting capability have been reported. Although many of these platforms are still in the early development stage, their in vitro and in vivo data show great promise for further clinical evaluations. Here we review several of these platforms.
By repeated freezing and thawing of a concentrated solution of cisplatin, Burger et al achieved significantly more efficient cisplatin encapsulation in liposomes.69 Their method involves hydration of a dry lipid film composed of equimolar amounts of dioleoyl-phosphatidylserine and dioleoyl-phosphatidylcholine with a buffered solution of 5 mM cisplatin, followed by ten freeze-thaw cycles. This method generated nanocapsules with an unprecedented drug-to-lipid molar ratio of 0.5 ± 0.1, corresponding to about 30 mM cisplatin, which far exceeded the solubility limit of cisplatin (8 mM). More impressively, in vitro cytotoxicity up to 1000-fold higher than that of the free drug was observed in human-derived ovarian (IGROV-1) tumor cells (Figure 1A and B). The authors attributed the formation of nanocapsules mainly to the solubility differences among neutral and charged aquo species of cisplatin, formation of ice phase during the freeze-thaw cycles, and electrostatic interactions between cisplatin aggregates and negatively charged lipids (Figure 1C).
Figure 1.

Cytotoxicity towards human ovarian carcinoma cells. (A) Lipid suspension of cisplatin (cisPt-phosphatidylserine/phosphatidylcholine) (▴), conventional cisplatin (∎), conventional cisplatin mixed with a blank lipid suspension (◻), and blank lipid suspension (dashed line). (B) Variations on the standard protocol, omitting freeze-thaw (▵), omitting phosphatidylserine (엯). (C) Proposed mechanism of nanocapsule formation and cell interaction.
Note: Adapted by permission from Macmillan Publishers Ltd: Nature Medicine,69 copyright 2002.
Khiati et al reported another approach to encapsulating cisplatin efficiently into nucleoside lipids.70 Nucleoside lipids were used in this case to enhance the electrostatic interactions between negatively charged phospholipids and positively charged aquated platinum species in order to control and guide the precipitation and self-assembly process to form highly loaded and stable nanoparticles. This method involved a two-step layer-by-layer strategy, as shown in Figure 2A, where encapsulation of cisplatin was achieved via an anionic nucleotide lipid, diC16-3′-dT (thymidine 3′-[1,2-dipalmitoyl-sn-glycero-3-phosphate]) and further stabilization of the resulting anionic nanoparticles was realized by another bilayer of a cationic nucleoside lipid, DOTAU (2′,3′-dioleyl-5′-deoxy-5′-trimethylammoniumuridine). It was shown that these particles were more efficiently internalized and more potent against a variety of cancer cell lines in vitro (Figure 2D and E). The present nucleolipid-based multilayer nanoparticles can thus overcome some of the disadvantages or limitations associated with other loading techniques, such as low drug-to-lipid ratio and instability of the assembly.
Figure 2.

(A) Layer-by-layer assembly of nanoparticles and chemical structures of the anionic and cationic lipids. (B) Transmission electron microscopic image of anionic nanoparticles (NP−) after uranyl acetate negative staining. (C) Transmission electron microscopic images of cationic nanoparticles (NP+) after uranyl acetate negative staining. Arrows indicate the multilayer. Scale bar, 50 nm. (D) Cytotoxic effect on IGROV-1 cancer cell line. (E) Comparison of cytotoxicity of cisplatin-loaded NP+ nanoparticles compared with free cisplatin in various human carcinoma cell lines.
Note: Adapted with permission from Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside-lipid-based nanoparticles for cisplatin delivery. ACS Nano. 2011;5(11):8649–8655. Copyright (2011) American Chemical Society.70
Aryal et al reported a novel platform for delivery of platinum-based drugs by using a synthetic phospholipid-like platinum compound to allow its self-assembly into a liposome-like nanostructure (the Ptsome).71 Two hydrophobic acyl chains were attached to the Pt(II) center to endow amphiphilic properties. The authors hypothesized that the chloride and hydrazide moieties next to the platinum atom can readily form hydrogen bonds in aqueous solution to increase hydrophilicity. With the hydrophobic acyl chains, a liposomal structure should form with these compounds in a manner similar to the self-assembly of phospholipids into liposomes. After extruding the particle solution through a 100 nm pore size membrane, particles with a size of 100 nm in diameter was obtained. Scanning electron microscopic imaging confirmed formation of spherical Ptsomes with nearly uniform size. This delivery vehicle is unique in a sense, in that unlike other liposomal delivery systems that utilize the aqueous space inside the liposome to store platinum drugs, the current study integrates drugs into the lipid composition, leading to an extremely high drug-to-lipid ratio. The Ptsome should also allow integration of other chemotherapeutics for combination therapy.
A novel chondroitin sulfate-binding cationic liposome loaded with cisplatin was reported by Lee et al.72 In their study, a new formulation of long-circulating PEG-coated liposomes comprising a new cationic lipid (3, 5-dipentadecycloxybenzamidine hydrochloride, TRX-20) was evaluated in vitro and in vivo against highly metastatic tumor cells expressing an increased level of chondroitin sulfate. It was shown that PEG-coated TRX-20 liposomes bound preferentially to certain chondroitin sulfates, such as B, D, and E. Confocal microscopy revealed efficient internalization of TRX-20 liposomes but not plain PEG liposomes by human ACHN renal adenocarcinoma and murine LM8G5 osteosarcoma cells. The cisplatin-loaded TRX-20 liposomes had higher cellular toxicity in vitro compared with cisplatin-PEG liposomes without TRX-20. Cisplatin-loaded TRX-20 liposomes, after intravenous injection, preferentially accumulated in the liver and tumor region, inhibited tumor growth, and suppressed metastasis to the liver more effectively than plain cisplatin-loaded PEG liposomes or free cisplatin in an LM8G5 liver metastasis model. These novel cationic liposomes thus provide a promising vehicle to deliver many other anticancer drugs to solid tumors and metastases with enhanced expression of chondroitin sulfate.
Suzuki et al developed a transferrin-conjugated PEG liposome formulation for tumor-selective delivery of oxaliplatin (L-OHP).73 This delivery system achieved a significantly longer blood circulation time compared with free oxaliplatin and higher L-OHP concentration in tumors compared with liposomes modified by PEG (Figure 3A and B). In a murine colon-26 tumor model, intravenous injection of L-OHP encapsulated within transferrin-conjugated PEG liposomes (L-OHP 5 mg/kg) suppressed tumor growth more effectively than PEG liposomes, bare liposomes, and free L-OHP (Figure 3C). Given that transferrin receptors are overexpressed in various types of tumors, this targeting strategy should allow more efficient delivery of active agents to tumor sites through both passive targeting and active targeting pathways.
Figure 3.

Plasma clearance (A) and biodistribution (B) of L-OHP, bare liposomes, PEG liposomes, and TF-PEG liposomes after intravenous injection. (C) Comparison of tumor growth suppression with free L-OHP and liposomes encapsulating L-OHP in a Colon-26 mouse model.
Note: Reprinted from International Journal of Pharmaceutics. Vol 346(1–2). Suzuki R, Takizawa T, Kuwata Y, et al. Effective anti-tumor activity of oxaliplatin encapsulated in transferrin PEG-liposome, pages 143–150. Copyright (2008), with permission from Elsevier.73
Abbreviations: PEG, polyethylene glycol; TF, transferrin; L-OHP, oxaliplatin; h, hours.
Lin and coworkers have developed a novel platform based on a nanoscale metal-organic framework or nanoscale coordination polymer for cancer-specific imaging and drug delivery.74–80 A nanoscale coordination polymer formulation based on a cisplatin prodrug, disuccinatocisplatin, and a La3+ metal ion was recently designed for targeted delivery to non-small cell lung cancer cell lines.81 The nanoscale coordination polymer particles were stabilized with a cholesterol/dioleoyl-phosphatidylcholine/1,2-distearoyl-sn-glycero-3-phosphoethanolamine-PEG lipid coating and further doped with a DSPE-PEG-anisamide conjugate to render them cancer-specific. This formulation showed higher potency than free cisplatin against non-small cell lung cancer cell lines, and enhanced uptake was confirmed by confocal microscopy and a competitive binding assay. The nanoscale coordination polymer delivery strategy is general and should allow incorporation of many other chemotherapeutics and imaging agents for cancer diagnosis and therapy.
Concluding remarks and perspectives
Nearly half a century of clinical research on platinum analogs has yielded remarkable anticancer agents. However, of the thousands of platinum compounds, only a very small fraction has shown sufficient promise during preclinical evaluation to enter human clinical trials. Harnessing their potency while reducing unwanted side effects and expanding the activity spectrum of platinum drugs while avoiding cross-resistance are important goals for the near future. A detailed understanding of how platinum drugs induce DNA damage, how the signals of DNA damage are transduced, how cell cycle arrest occurs, and how DNA repair and apoptosis are activated provide us with invaluable knowledge. With a better understanding of the mechanism of action, improved designs of new platinum-based compounds are expected. Among several emerging chemogenotherapeutic strategies, disruptions of certain pathways by RNA interference that modulate cellular sensitivity to platinum drugs are likely to lead to clinical benefits. The combination of platinum analogs and other chemotherapeutics, such as gemcitabine, paclitaxel, doxorubicin, and 5-fluorouracil, will also contribute to the increased spectrum of activity and anticancer efficacy.
At the same time, targeted delivery of platinum drugs with long-circulating liposomes also provides another efficient strategy to improve platinum drug efficacy with reduced toxicity. Liposomal drugs are highlighted for their abilities to “passively” accumulate at tumor sites via the enhanced permeability and retention effect and to reduce the side effects of encapsulated drugs by lowering unspecific cytotoxic drug distribution in normal tissues. These two features endow liposomal drugs with an increased therapeutic ratio. The current liposomal formulations have primarily taken advantage of reduced systemic toxicity rather than increased efficacy. For example, the irreversible cardiotoxicity induced by free doxorubicin could be significantly alleviated by entrapping the drug in liposomes.82,83 In order to increase drug bioavailability further and improve drug biodistribution at tumor sites, drug-loaded liposomes have been further modified with internalizing receptors, such as small molecules,84 sugar molecules,85 and antibodies86 or antibody fragments.87 However, liposomal formulations with active-targeting groups have yet to enter the clinic, owing to the high development costs (manufacturing, source of good, intellectual property) and batch-to-batch variation.
Despite the numerous successes in overcoming barriers to liposomal drug delivery, it is still difficult to achieve an optimized balance between high and specific drug bioavailability in tumor tissue and prolonged liposome stability in systemic circulation. Drug release from stable formulations such as the Stealth® formulation is at best slow.88 The release of hydrophilic drugs such as cisplatin is dependent on degradation of the liposome vehicle. Therefore, a long systemic circulation and minimal side effects of chemotherapy could be achieved at the expense of lowered efficacy in vivo. More detailed knowledge is needed in order to reach a balance between obtaining stable formulations with long circulation times to minimize undesired systemic exposure and adequate drug release kinetics in order to achieve enhanced chemotherapeutic efficacy.89 The development of liposomes from which drug release can be actively triggered is therefore of crucial importance for guiding liposomal drug delivery technologies to wide clinical applications in the treatment of cancer.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg B, Van Camp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205(4972):698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 3.Einhorn LH. Treatment of testicular cancer: a new and improved model. J Clin Oncol. 1990;8(11):1777–1781. doi: 10.1200/JCO.1990.8.11.1777. [DOI] [PubMed] [Google Scholar]
- 4.Lebwohl D, Canetta R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer. 1998;34(10):1522–1534. doi: 10.1016/s0959-8049(98)00224-x. [DOI] [PubMed] [Google Scholar]
- 5.Wong E, Giandomenico CM. Current status of platinum-based antitumor drugs. Chem Rev. 1999;99(9):2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- 6.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107(5):1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 7.Welsh C, Day R, McGurk C, Masters JRW, Wood RD, Köberle B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004;110(3):352–361. doi: 10.1002/ijc.20134. [DOI] [PubMed] [Google Scholar]
- 8.Köberle B, Masters JRW, Hartley JA, Wood RD. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr Biol. 1999;9(5):273–278. doi: 10.1016/s0960-9822(99)80118-3. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4(4):307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 10.Amptoulach S, Tsavaris N. Neurotoxicity caused by the treatment with platinum analogues. Chemother Res Pract. 2011;2011:5. doi: 10.1155/2011/843019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol. 2008;6(1):1–18. doi: 10.1111/j.1476-5829.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 12.Boulikas T, Pantos A, Bellis E, Christofis P. Designing platinum compounds in cancer: structures and mechanisms. Cancer Ther. 2007;5:537–583. [Google Scholar]
- 13.Calvert A, Harland S, Newell D, et al. Early clinical studies with cis-diammine-1,1-cyclobutane dicarboxylate platinum II. Cancer Chemother Pharmacol. 1982;9(3):140–147. doi: 10.1007/BF00257742. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Guo Z. Targeting and delivery of platinum-based anticancer drugs. Chem Soc Rev. 2013;42(1):202–224. doi: 10.1039/c2cs35259a. [DOI] [PubMed] [Google Scholar]
- 15.Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39(35):8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 16.Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 17.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond) 2012;7(10):1577–1590. doi: 10.2217/nnm.12.128. [DOI] [PubMed] [Google Scholar]
- 19.Maeda H, Matsumura Y. EPR effect based drug design and clinical outlook for enhanced cancer chemotherapy. Adv Drug Deliv Rev. 2011;63(3):129–130. doi: 10.1016/j.addr.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1):238–252. doi: 10.1016/s0022-2836(65)80093-6. [DOI] [PubMed] [Google Scholar]
- 21.Gregoriadis G, Swain CP, Wills EJ, Tavill AS. Drug-carrier potentialof lipsomes in cancer chemotherapy. Lancet. 1974;303(7870):1313–1316. doi: 10.1016/s0140-6736(74)90682-5. [DOI] [PubMed] [Google Scholar]
- 22.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. doi: 10.1186/1556-276X-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105(4):561–567. doi: 10.1002/ijc.11108. [DOI] [PubMed] [Google Scholar]
- 24.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 25.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 26.Bolotin EM, Cohen R, Bar LK, et al. Ammonium sulfate gradients for efficient and stable remote loading of amphipathic weak bases into liposomes and ligandoliposomes. J Liposome Res. 1994;4(1):455–479. [Google Scholar]
- 27.Mayer LD, Tai LCL, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta. 1990;1025(2):143–151. doi: 10.1016/0005-2736(90)90091-2. [DOI] [PubMed] [Google Scholar]
- 28.Olson F, Hunt CA, Szoka FC, Vail WJ, Papahadjopoulos D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim Biophys Acta. 1979;557(1):9–23. doi: 10.1016/0005-2736(79)90085-3. [DOI] [PubMed] [Google Scholar]
- 29.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 30.Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88(24):11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bibi S, Lattmann E, Mohammed AR, Perrie Y. Trigger release liposome systems: local and remote controlled delivery? J Microencapsul. 2011;29(3):262–276. doi: 10.3109/02652048.2011.646330. [DOI] [PubMed] [Google Scholar]
- 32.Davidsen J, Jørgensen K, Andresen TL, Mouritsen OG. Secreted phospholipase A2 as a new enzymatic trigger mechanism for localised liposomal drug release and absorption in diseased tissue. Biochim Biophys Acta. 2003;1609(1):95–101. doi: 10.1016/s0005-2736(02)00659-4. [DOI] [PubMed] [Google Scholar]
- 33.Huang S-L, MacDonald RC. Acoustically active liposomes for drug encapsulation and ultrasound-triggered release. Biochim Biophys Acta. 2004;1665(1–2):134–141. doi: 10.1016/j.bbamem.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Smith B, Lyakhov I, Loomis K, et al. Hyperthermia-triggered intracellular delivery of anticancer agent to HER2+ cells by HER2-specific affibody (ZHER2-GS-Cys)-conjugated thermosensitive liposomes (HER2+ affisomes) J Control Release. 2011;153(2):187–194. doi: 10.1016/j.jconrel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad I, Longenecker M, Samuel J, Allen TM. Antibody-targeted delivery of doxorubicin entrapped in sterically stabilized liposomes can eradicate lung cancer in mice. Cancer Res. 1993;53(7):1484–1488. [PubMed] [Google Scholar]
- 36.Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62(24):7190–7194. [PubMed] [Google Scholar]
- 37.Allen TM, Moase EH. Therapeutic opportunities for targeted liposomal drug delivery. Adv Drug Deliv Rev. 1996;21(2):117–133. [Google Scholar]
- 38.Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–992. [PubMed] [Google Scholar]
- 39.Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal doxorubicin (Doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 40.Lorusso D, Di Stefano A, Carone V, Fagotti A, Pisconti S, Scambia G. PEGylated liposomal doxorubicin-related palmar-plantar erythrodysesthesia (‘hand-foot’ syndrome) Ann Oncol. 2007;18(7):1159–1164. doi: 10.1093/annonc/mdl477. [DOI] [PubMed] [Google Scholar]
- 41.Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 42.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 43.Vadiei K, Siddik Z, Khokhar A, Al-Baker S, Sampedro F, Perez-Soler R. Pharmacokinetics of liposome-entrapped cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum(II) and cisplatin given i.v. and i.p. in the rat. Cancer Chemother Pharmacol. 1992;30(5):365–369. doi: 10.1007/BF00689964. [DOI] [PubMed] [Google Scholar]
- 44.Khokhar AR, Wright K, Siddik ZH, Perez-Soler R. Organ distribution and tumor uptake of liposome entrapped cis-bis-neodecanoato trans-R, R-1,2 diaminocyclohexane platinum (II) administered intravenously and into the proper hepatic artery. Cancer Chemother Pharmacol. 1988;22(3):223–227. doi: 10.1007/BF00273415. [DOI] [PubMed] [Google Scholar]
- 45.Han I, Ling Y-H, Al-Baker S, Khokhar AR, Perez-Soler R. Cellular pharmacology of liposomal cis-bis-neodecanoato-trans-r,r-1,2-diaminocyclohexaneplatinum(II) in A2780/S and A2780/PDD cells. Cancer Res. 1993;53(20):4913–4919. [PubMed] [Google Scholar]
- 46.Perez-Soler R, Lautersztain J, Stephens LC, Wright K, Khokhar AR. Preclinical toxicity and pharmacology of liposome-entrapped cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexane platinum(II) Cancer Chemother Pharmacol. 1989;24(1):1–8. doi: 10.1007/BF00254097. [DOI] [PubMed] [Google Scholar]
- 47.Perez-Soler R, Yang LY, Drewinko B, Lauterzstain J, Khokhar AR. Increased cytotoxicity and reversal of resistance to cis-diamminedichloroplatinum(II) with entrapment of cis-bis-neodecanoato-trans-r,r-1,2-diaminocyclohexaneplatinum(II) in multilamellar lipid vesicles. Cancer Res. 1988;48(16):4509–4512. [PubMed] [Google Scholar]
- 48.Perez-Soler R, Khokhar AR, Lopez-Berestein G. Treatment and prophylaxis of experimental liver metastases of M5076 reticulosarcoma with cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexaneplatinum(II) encapsulated in multilamellar vesicles. Cancer Res. 1987;47(24 Part 1):6462–6466. [PubMed] [Google Scholar]
- 49.Perez-Soler R, Lopez-Berestein G, Lautersztain J, et al. Phase I clinical and pharmacological study of liposome-entrapped cis-bis-neodecanoato-trans-R,R,-1,2-diaminocyclohexane platinum(II) Cancer Res. 1990;50(14):4254–4259. [PubMed] [Google Scholar]
- 50.Dragovich T, Mendelson D, Kurtin S, Richardson K, Hoff D, Hoos A. A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother Pharmacol. 2006;58(6):759–764. doi: 10.1007/s00280-006-0235-4. [DOI] [PubMed] [Google Scholar]
- 51.Mori A, Wu S-P, Han I, Khokhar A, Perez-Soler R, Huang L. In vivo antitumor activity of cis-bis-neodecanoato-trans-R,R-l,2-diaminocyclohexane platinum(II) formulated in long-circulating liposomes. Cancer Chemother Pharmacol. 1996;37(5):435–444. doi: 10.1007/s002800050409. [DOI] [PubMed] [Google Scholar]
- 52.Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, PEGylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother Pharmacol. 1999;43(1):1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- 53.Bandak S, Goren D, Horowitz A, Tzemach D, Gabizon A. Pharmacological studies of cisplatin encapsulated in long-circulating liposomes in mouse tumor models. Anticancer Drugs. 1999;10(10):911–920. doi: 10.1097/00001813-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 54.Zamboni W, Gervais A, Egorin M, et al. Systemic and tumor disposition of platinum after administration of cisplatin or STEALTH liposomal-cisplatin formulations (SPI-077 and SPI-077 B103) in a preclinical tumor model of melanoma. Cancer Chemother Pharmacol. 2004;53(4):329–336. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- 55.Meerum Terwogt J, Groenewegen G, Pluim D, et al. Phase I and pharmacokinetic study of SPI-77, a liposomal encapsulated dosage form of cisplatin. Cancer Chemother Pharmacol. 2002;49(3):201–210. doi: 10.1007/s002800100371. [DOI] [PubMed] [Google Scholar]
- 56.Harrington KJ, Lewanski CR, Northcote AD, et al. Phase I-II study of PEGylated liposomal cisplatin (SPI-077 TM) in patients with inoperable head and neck cancer. Ann Oncol. 2001;12(4):493–496. doi: 10.1023/a:1011199028318. [DOI] [PubMed] [Google Scholar]
- 57.White SC, Lorigan P, Margison GP, et al. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br J Cancer. 2006;95(7):822–828. doi: 10.1038/sj.bjc.6603345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seetharamu N, Kim E, Hochster H, Martin F, Muggia F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. Anticancer Res. 2010;30(2):541–545. [PubMed] [Google Scholar]
- 59.Stathopoulos GP, Boulikas T. Lipoplatin formulation review article. J Drug Deliv. 2012;2012:581363. doi: 10.1155/2012/581363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Veal GJ, Griffin MJ, Price E, et al. A phase I study in paediatric patients to evaluate the safety and pharmacokinetics of SPI-77, a liposome encapsulated formulation of cisplatin. Br J Cancer. 2001;84(8):1029–1035. doi: 10.1054/bjoc.2001.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boulikas T. Low toxicity and anticancer activity of a novel liposomal cisplatin (Lipoplatin) in mouse xenografts. Oncol Rep. 2004;12(1):3–12. [PubMed] [Google Scholar]
- 62.Devarajan P, Tarabishi R, Mishra J, et al. Low renal toxicity of lipoplatin compared to cisplatin in animals. Anticancer Res. 2004;24(4):2193–2200. [PubMed] [Google Scholar]
- 63.Marr AK, Kurzman ID, Vail DM. Preclinical evaluation of a liposome-encapsulated formulation of cisplatin in clinically normal dogs. Am J Vet Res. 2004;65(11):1474–1478. doi: 10.2460/ajvr.2004.65.1474. [DOI] [PubMed] [Google Scholar]
- 64.Boulikas T. Clinical overview on Lipoplatin™: a successful liposomal formulation of cisplatin. Expert Opin Investig Drugs. 2009;18(8):1197–1218. doi: 10.1517/13543780903114168. [DOI] [PubMed] [Google Scholar]
- 65.Boulikas T, Stathopoulos GP, Volakakis N, Vougiouka M. Systemic Lipoplatin infusion results in preferential tumor uptake in human studies. Anticancer Res. 2005;25(4):3031–3039. [PubMed] [Google Scholar]
- 66.Stathopoulos GP, Antoniou D, Dimitroulis J, Stathopoulos J, Marosis K, Michalopoulou P. Comparison of liposomal cisplatin versus cisplatin in non-squamous cell non-small-cell lung cancer. Cancer Chemother Pharmacol. 2011;68(4):945–950. doi: 10.1007/s00280-011-1572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stathopoulos GP, Boulikas T, Kourvetaris A, Stathopoulos J. Liposomal oxaliplatin in the treatment of advanced cancer: a Phase I study. Anticancer Res. 2006;26(2B):1489–1493. [PubMed] [Google Scholar]
- 68.de Jonge MJ, Slingerland M, Loos WJ, et al. Early cessation of the clinical development of LiPlaCis, a liposomal cisplatin formulation. Eur J Cancer. 2010;46(16):3016–3021. doi: 10.1016/j.ejca.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 69.Burger KN, Staffhorst RW, de Vijlder HC, et al. Nanocapsules: lipid-coated aggregates of cisplatin with high cytotoxicity. Nat Med. 2002;8(1):81–84. doi: 10.1038/nm0102-81. [DOI] [PubMed] [Google Scholar]
- 70.Khiati S, Luvino D, Oumzil K, Chauffert B, Camplo M, Barthélémy P. Nucleoside-lipid-based nanoparticles for cisplatin delivery. ACS Nano. 2011;5(11):8649–8655. doi: 10.1021/nn202291k. [DOI] [PubMed] [Google Scholar]
- 71.Aryal S, Hu C-MJ, Zhang L. Synthesis of Ptsome: a platinum-based liposome-like nanostructure. Chem Commun (Camb) 2012;48(20):2630–2632. doi: 10.1039/c2cc18176b. [DOI] [PubMed] [Google Scholar]
- 72.Lee CM, Tanaka T, Murai T, et al. Novel chondroitin sulfate-binding cationic liposomes loaded with cisplatin efficiently suppress the local growth and liver metastasis of tumor cells in vivo. Cancer Res. 2002;62(15):4282–4288. [PubMed] [Google Scholar]
- 73.Suzuki R, Takizawa T, Kuwata Y, et al. Effective anti-tumor activity of oxaliplatin encapsulated in transferrin PEG-liposome. Int J Pharm. 2008;346(1–2):143–150. doi: 10.1016/j.ijpharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 74.Rieter WJ, Taylor KM, An H, Lin W, Lin W. Nanoscale metal-organic frameworks as potential multimodal contrast enhancing agents. J Am Chem Soc. 2006;128(28):9024–9025. doi: 10.1021/ja0627444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rieter WJ, Pott KM, Taylor KM, Lin W. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J Am Chem Soc. 2008;130(35):11584–11585. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]
- 76.Taylor KM, Jin A, Lin W. Surfactant-assisted synthesis of nanoscale gadolinium metal-organic frameworks for potential multimodal imaging. Angew Chem Int Ed. 2008;47(40):7722–7725. doi: 10.1002/anie.200802911. [DOI] [PubMed] [Google Scholar]
- 77.Liu D, Huxford RC, Lin W. Phosphorescent nanoscale coordination polymers as contrast agents for optical imaging. Angew Chem Int Ed. 2011;50(16):3696–3700. doi: 10.1002/anie.201008277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huxford RC, deKrafft KE, Boyle WS, Liu D, Lin W. Lipid-coated nanoscale coordination polymers for targeted delivery of antifolates to cancer cells. Chem Sci. 2012;3(1):198–204. doi: 10.1039/C1SC00499A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu D, Kramer SA, Huxford-Phillips RC, Wang S, Della Rocca J, Lin W. Coercing bisphosphonates to kill cancer cells with nanoscale coordination polymers. Chem Commun (Camb) 2012;48(21):2668–2670. doi: 10.1039/c2cc17635a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.deKrafft KE, Boyle WS, Burk LM, Zhou OZ, Lin W. Zr- and Hf-based nanoscale metal-organic frameworks as contrast agents for computed tomography. J Mater Chem. 2012;22(35):18139–18144. doi: 10.1039/C2JM32299D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huxford-Phillips RC, Russell SR, Liu D. Lipid-coated nanoscale coordination polymers for targeted cisplatin delivery. RSC Adv. 2013;3:14438–14443. doi: 10.1039/C3RA42033G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Batist G, Ramakrishnan G, Rao CS, et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J Clin Oncol. 2001;19(5):1444–1454. doi: 10.1200/JCO.2001.19.5.1444. [DOI] [PubMed] [Google Scholar]
- 83.Berry G, Billingham M, Alderman E, et al. The use of cardiac biopsy to demonstrate reduced cardiotoxicity in AIDS Kaposi’s sarcoma patients treated with pegylated liposomal doxorubicin. Ann Oncol. 1998;9(7):711–716. doi: 10.1023/a:1008216430806. [DOI] [PubMed] [Google Scholar]
- 84.Lee RJ, Low PS. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J Biol Chem. 1994;269(5):3198–3204. [PubMed] [Google Scholar]
- 85.Banerjee G, Nandi G, Mahato SB, Pakrashi A, Basu MK. Drug delivery system: targeting of pentamidines to specific sites using sugar grafted liposomes. J Antimicrob Chemother. 1996;38(1):145–150. doi: 10.1093/jac/38.1.145. [DOI] [PubMed] [Google Scholar]
- 86.Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res. 1998;58(15):3320–3330. [PubMed] [Google Scholar]
- 87.Kirpotin D, Park JW, Hong K, et al. Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry. 1997;36(1):66–75. doi: 10.1021/bi962148u. [DOI] [PubMed] [Google Scholar]
- 88.Allen TM, Mehra T, Hansen C, Chin YC. Stealth liposomes: an improved sustained release system for 1-β-d-arabinofuranosylcytosine. Cancer Res. 1992;52(9):2431–2439. [PubMed] [Google Scholar]
- 89.Bally MB, Lim H, Cullis PR, Mayer LD. Controlling the drug delivery attributes of lipid-based drug formulations. J Liposome Res. 1998;8(3):299–335. [Google Scholar]