

Abstract
The major facilitator superfamily (MFS) is the largest family of secondary active transporters and is present in all life kingdoms. Detailed structural basis of the substrate transport and energy-coupling mechanisms of these proteins remain to be elucidated. YajR is a putative proton-driven MFS transporter found in many Gram-negative bacteria. Here we report the crystal structure of Escherichia coli YajR at 3.15 Å resolution in an outward-facing conformation. In addition to having the 12 canonical transmembrane helices, the YajR structure includes a unique 65-residue C-terminal domain which is independently stable. The structure is unique in illustrating the functional role of “sequence motif A.” This highly conserved element is seen to stabilize the outward conformation of YajR and suggests a general mechanism for the conformational change between the inward and outward states of the MFS transporters.
Keywords: membrane potential, protonation, charge-dipole interaction, charge relay
Transporters are a type of membrane protein essential for all living cells that actively up-take nutrition and export metabolic substances and toxic materials across cellular membranes. Transporters are divided into two major types based on their energy sources. Although primary active transporters directly consume energy from ATP hydrolysis to drive substrate transport, secondary active transporters use energy derived from the electrochemical potential across the cell membrane. The major facilitator superfamily (MFS) is the largest class of secondary transporters and is present in all life kingdoms (1). For example, 25% of prokaryotic transporters belong to the MFS family (2), and the human genome contains over 110 MFS proteins (3). Currently, 3D crystal structures of nine bacterial MFS transporters (4–13) and one from fungi (9) have been reported at medium-high resolution. These studies have shown that MFS proteins contain a 12-transmembrane (TM) helix core composed of two six-helix rigid domains forming a central TM channel, which transports substrates using a rocker-switch mechanism (5). In such a mechanism, MFS proteins are believed to switch between two major conformations, inward and outward, which differ by an ∼40° rotation of one domain relative to the other. Both conformations have been captured in MFS crystal structures. However, many questions remain to be addressed, particularly those related to energy coupling and functional roles of conserved motifs.
YajR, a 49-kDa transporter of the MFS family, has putatively been classified as a drug efflux protein solely on the basis of amino acid sequence analysis (14). In Escherichia coli, YajR consists of 454 amino acid residues. Besides containing 12 TM helices, YajR is predicted to possess an extra domain of about 65 residues at the C-terminal. Of the MFS proteins with reported 3D structures, the TM core of YajR shares highest sequence homology (21% identity) with EmrD (SI Appendix, Fig. S1A), which belongs to the 12-TM drug-resistance H+-driven antiporter (DHA12) subfamily (15). The YajR gene is found in a number of Gram-negative bacteria, and it shows high sequence homology (identity ranging from 47–85%) with a group of arabinose efflux permeases, AraEP, also from Gram-negative bacteria (SI Appendix, Fig. S1B). In addition, YajR homolog genes (TM-core coding region only) are found in genomes of archaea as well as Gram-positive bacteria.
To address questions about the general mechanisms of MFS proteins and specific questions related to YajR, such as properties of potential substrates and functions of the small domain at the C terminal, we determined the crystal structure of full-length E. coli YajR. The structure showed an outward conformation, and the small C-terminal domain forms a compact structure outside of the TM core. More importantly, the crystal structure of YajR enabled us to observe the most conserved sequence motif in the MFS family (i.e., motif A, stabilizing the outward conformation through an interaction network centered at a charge-helix dipole interaction). This observation prompts us to propose a general mechanism of conformational changes of MFS proteins, which is supported by many existing data of structure and function studies on MFS proteins.
Results
Crystal Structure of YajR.
Recombinant full-length E. coli YajR protein was expressed in E. coli, and the melting temperature (Tm) of the native protein was determined to be approximately 63.9 (±0.5) °C using thermofluor assays (16) (Fig. 1A). The crystal structure of full-length YajR protein was solved using a mercury-based multiwavelength anomalous dispersion (MAD) method (17). The final refined model had an Rwork of 27% (Rfree 29%) and contained native residues 3−454. Statistics from data collection and refinement are summarized in SI Appendix, Table S1.
Fig. 1.

Thermal stability and overall structure of YajR. (A) Thermal denaturation curves of YajR variants. Thermal denaturation of full-length WT YajR, the G51W G69W, and D73R mutants, and truncation YajR-ΔYAM (i.e., residues 1−394 of WT) were measured using the blue fluorescence of the cpm dye (the left vertical axis), and that of the YAM-alone sample, which does not contain any Cys residue, was measured using the green fluorescence of Sypro orange (the right vertical axis). Raw data were analyzed with Prism (GraphPad). Multiple experiments were performed with good reproducibility and representative results are shown. (B) Cartoon representation of the YajR crystal structure. In the TM core, central helices (i.e., TMs 1, 4, 7, and 10), rocker helices (TMs 2, 5, 8, and 11), and supporting helices (TMs 3, 6, 9, and 12) are shown in green, yellow, and red, respectively. The short helix α6–7 is colored in blue and the YAM domain in orange.
Similar to previously reported MFS transporter structures, the crystal structure of YajR contains 12 TM helices (TMs 1−12), which form the N (residues 3−184) and C domains (residues 215−386) of the TM core (Fig. 1B). The two domains are connected by a 30-residue linker containing a three-turn amphipathic α-helix (i.e., residues 201−209 and termed as α6–7) (SI Appendix, Fig. S2) as well as an extended loop. The overall charge distribution of the TM core is consistent with the “positive inside rule” (18) (SI Appendix, Fig. S3). Unlike MFS structures reported earlier, however, YajR contains a 65-residue, highly negatively charged domain (residues 389−454 and pI 4.75) located C-terminally to the TM core and possessing a ferredoxin-like fold of a metal-binding protein (termed the YajR/AraEP/MBD domain or YAM) (Fig. 1B and SI Appendix,Fig. S4). As predicted by amino acid sequence analysis, both N and C termini of YajR are located on the cytoplasmic side of the plasma membrane. There is a central cavity in the TM core between the N and C domains, with the cytoplasmic side closed and the periplasmic side accessible to solvent. The current crystal structure of YajR therefore assumes an outward conformation with the novel, small, soluble domain at the C terminal located in cytoplasm. More detailed structural descriptions of YajR can be found in SI Appendix.
The Conserved Motif A.
Most MFS transporters contain the signature motif A, “G(+1)xlaD(+5) rxGR(+9)kp” (19). In YajR, motif A is “G69LLSD73RIGR77KP” and is located in the loop connecting TMs 2 and 3 (L2-3) (Fig. 2A). In particular, Gly69(+1) of TM2 forms a close helix-helix contact with conserved Gly337 and Gly341 of TM11. The formation of this interdomain helical bundle between TMs 2 and 11 appears to be essential for the outward conformation of YajR. Asp73(+5) was observed to be completely buried in the domain interface in the outward conformation (with a zero side-chain solvent-accessible surface). In comparison, in the absence of the C domain (thus roughly equivalent to an inward conformation), the side chain of Asp73 has a solvent-accessible surface of 61 Å2. Asp73(+5) plays the role of an N-cap for TM11, presumably stabilizing the latter helix and the domain interface through a charge-helix dipole interaction. An Asp73-to-Arg (D73R) point mutation decreased the melting temperature of YajR by nearly 20 °C [Tm 45.3 (±0.2) °C] (Fig. 1A). This interdomain interaction can only be clearly illustrated in the outward conformation, and were not identified in previously reported MFS structures of inward conformations. In addition, Arg74(+6) may interact with the surrounding lipid molecules. Regarding intradomain interactions, Gly76(+8) is favored by the S-shaped backbone conformation of the short loop L2-3; Arg77(+9) interacts with side chains of both Asp73(+5) and conserved Asp126 at the C-terminal end of TM4, forming a charge-relay system; and Lys78(+10) interacts with the C-terminal end of TM6.
Fig. 2.
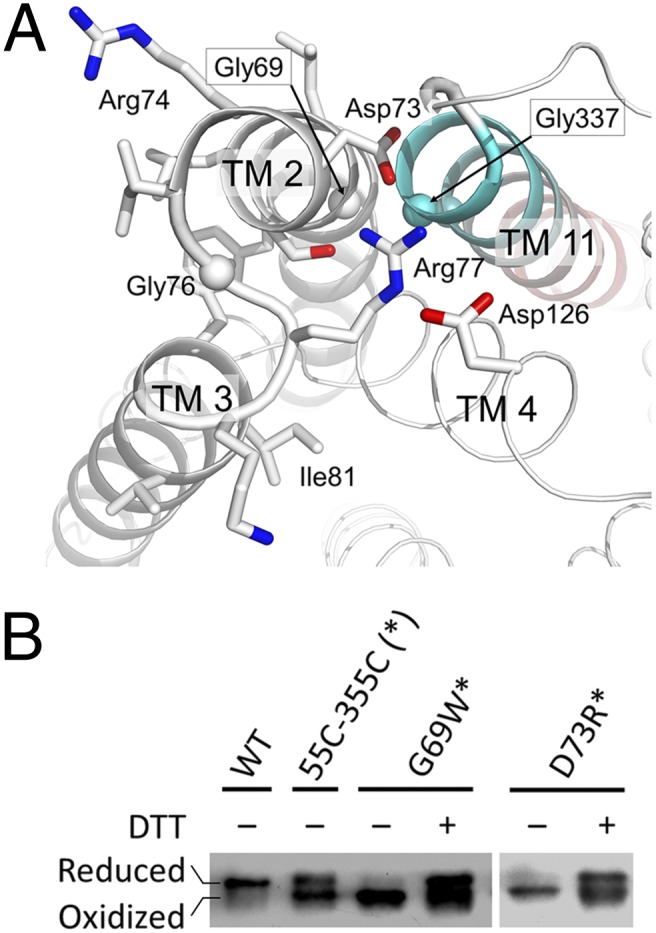
Functional roles of motif A. (A) Active conformation of motif A from the MFS family. Side chains in motif A and Asp126 are shown as stick models. Cα atoms of conserved Gly residues are shown as spheres. TM11 is colored from cyan at the N-terminal end to red at the C-terminal end. The figure was generated using PyMol. (B) Disulfide-bond formation assay. Protein samples were subjected to SDS/PAGE in the absence and presence of DTT, followed by immunoblot against the His-tag at the C termini of recombinant proteins. A double-point mutation G55C-G355C in the WT background, which presumably forms a disulfide bond in the inward conformation but not in the outward conformation, showed both reduced (Upper band) and oxidized (Lower band) forms. Additional G69W and D73R point mutations in the double Cys-mutation background (marked with an asterisk) showed predominantly oxidized forms. The WT YajR was included as a negative control. The results demonstrated that mutations in the motif A increase occupancy of the inward conformation.
To further confirm the importance of motif A in YajR stability, we constructed a point mutation of Gly69(+1)-to-Trp (i.e., G69W), which introduced a bulky side chain to the domain interface of the outward conformation. This G69W mutation dramatically destabilized YajR in solution (Fig. 1A). As an experimental control, a similar Gly-to-Trp point mutation, G51W, was constructed at the opposite end of TM2 from Gly69. Positions equivalent to YajR Gly51 are found to be located in the domain interfaces of reported MFS structures of the inward conformation (e.g., Gly46 in LacY/1PV7) and are most populated by Gly residues (19). This G51W variant of YajR had a Tm similar to that of WT [60.1 (±1.8)°C] (Fig. 1A). Taken together, these data indicate that, under the experimental condition, the dominant, low-energy conformation of YajR is an outward one. In addition, point mutations R77E [Tm 57.6 (±0.3) °C] and D126R [Tm 51.3 (±0.5) °C] in the charge relay system also resulted in stability decrease (with ΔTm of −6 °C and −13 °C, respectively), consistent with their roles in regulating the charge-dipole interaction.
In addition, we performed a disulfide-bond formation experiment to confirm the role of motif A in stabilizing the outward conformation of YajR. First, a double-point mutation G55C-G355C was introduced into the WT YajR protein. Based on available structural information, these two cysteine residues are located in the N-terminal half of TM2 and C-terminal half of TM11, respectively, and were predicted to form an interdomain disulfide bond in the inward conformation but not in the outward conformation. SDS/PAGE showed that this mutant existed in both the reduced and oxidized forms (Fig. 2B). Furthermore, both G69W and D73R point mutations, which presumably disturbs the motif A, were introduced individually into the G55C-G355C variant. In both cases, SDS/PAGE showed that the triple-point mutation existed predominantly in the oxidized form (Fig. 2B). These results were consistent with a prediction that disrupting motif A at the +1 and +5 positions would allow the inward conformation to occur at a higher frequency.
Discussion
Two States of MFS Transporters.
Transporters of the MFS family share a similar folding topology as well as a conserved motif A. These structural features are likely to be the basis for the functions common to most members of the MFS family. Such common functions include protonation (or binding to other charged ions) and conformational alternation between the outward and inward states (i.e., the rocker switch process) (5). The outward conformation of a H+-driven, electrogenic, MFS protein can be considered as a ground state, and the inward conformation can be considered as an excited state (Fig. 3A). From an energetic point of view, the transition from the ground state to the excited state is initiated by protonation inside the central cavity, whereas the transition from the excited state back to the ground state is more or less automatic. The conformational changes can be further regulated by substrate binding inside the cavity or at allosteric sites on either side of the TM core, depending on the direction of the substrate transport. In terms of the energy source, it is shown that in the presence of ΔμH+, LacY—the most thoroughly studied MFS protein—can accumulate lactose against a 100-fold concentration gradient (20). Because of the limited [H+] gradient across the membrane (i.e., ΔpH of ∼0.6, corresponding to a fourfold difference), the associated free energy alone would not be sufficient to build up a 100-fold concentration gradient of the substrate. Therefore, other energy sources must function in the transporting process.
Fig. 3.
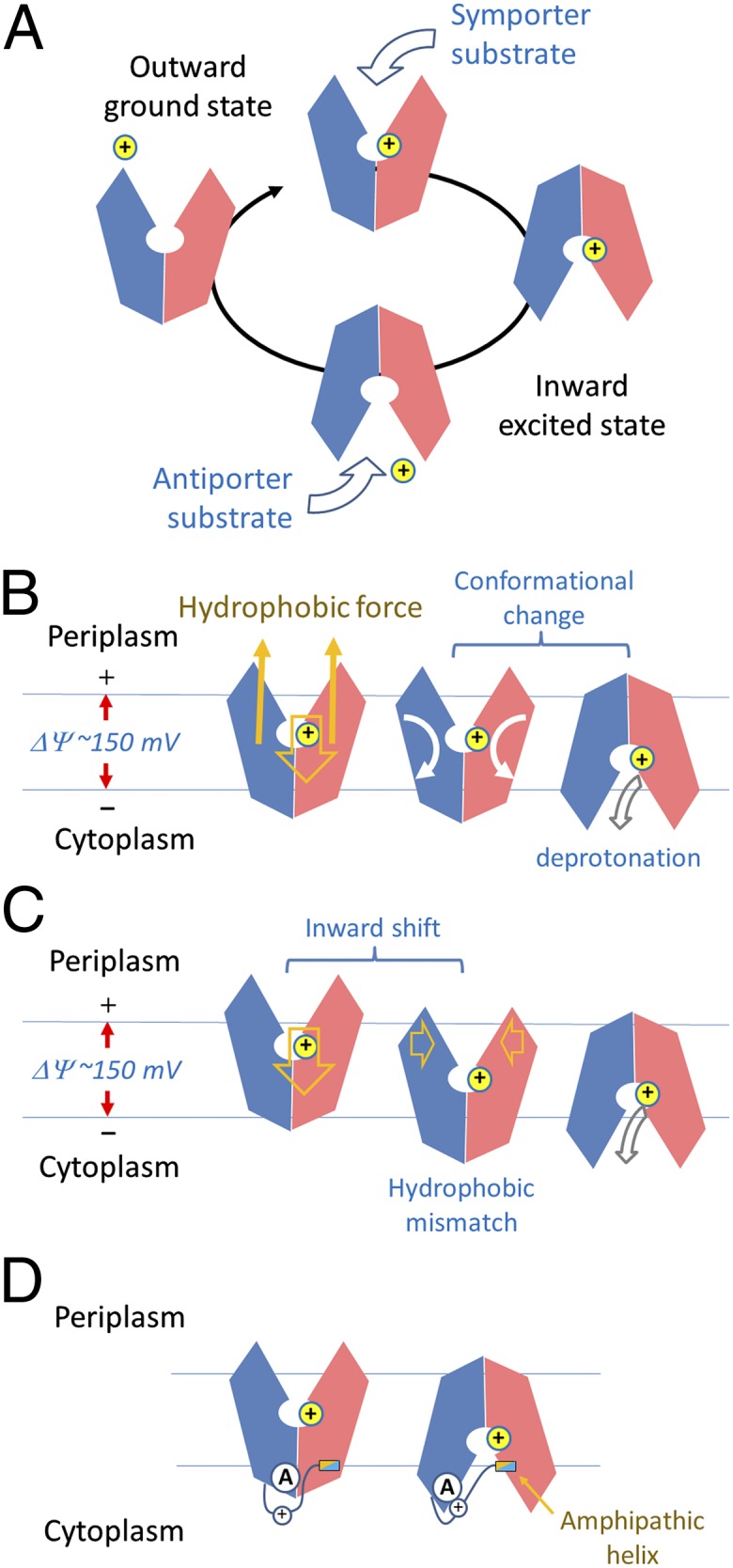
Schematic diagram of MFS conformational changes. (A) Changes in a functional cycle of MFS. (B) Changes driven by protonation. Directions of movements are shown in arrows. (C) Alternative mechanism of proton-driven conformational changes. (D) Effects of the interdomain linker on motif A.
Although crystal structures of several MFS proteins have been elucidated, conserved structural elements that could provide the basis for a common transition mechanism remain to be identified. These crystal structures are found in inward, outward, or intermediate states, reflecting the low-energy conformations of the detergent-solubilized proteins under the corresponding crystallization conditions. Here we divide the mechanistic question into two parts, relating to the outward-to-inward transition and the inward-to-outward transition, respectively, and attempt to address them by using H+-driven, electrogenic MFS transporters as a paradigm, with emphasis on new structural information derived from the YajR crystal structure, combined with existing data from studies on MFS structures and functions.
Transition from the Ground State to Excited State.
In the following, we make an argument that both protonation and a cross-membrane electrochemical potential are required for the outward-to-inward transition. In nearly all available crystal structures of proton-driven MFS transporters, proton titratable amino acid residues can be identified in the central cavity. In YajR, such residues are likely to be the His225-Glu320 pair, where both amino acid residues reside close to each other (at a distance of 3.7 Å between the closest atom pairs of their side chains) and are the only two residues in theory titratable inside the cavity. Generally, a protonated residue may function in two ways: (i) Protonation may create or eliminate some electrostatic interactions, such as a hydrogen bond or a salt-bridge bond. This type of interactions usually contributes approximately 4 kJ/mol (∼1.6 RT) to protein stability (21), and may result in a local conformational change to lower the transition state energy barrier (22). (ii) Proton binding creates a structural point on which the physiological, negative-inside, electrostatic potential (ΔΨ) of the cytoplasmic membrane applies an electrostatic force to the protonated MFS. By transporting one proton across the membrane, the membrane potential [approximately 150 mV (23)] produces approximately 15 kJ/mol (i.e., FΔΨ, or ∼6 RT) energy. The corresponding force, combining with a hydrophobic interaction force that holds the TM core in the membrane, would produce torques that rotate the two domains individually in opposite directions and result in a rocking conformational change between the two domains (Fig. 3B). The most hydrophobic TM helices (i.e., the supporting helices TMs 3, 6, 9, and 12 in Fig. 1B) are located at the two ends of the longest dimension of the TM core, favoring the rotations of the two domains relative to the membrane as well as to each other. Furthermore, the banana-shaped helices that form the interdomain interface (i.e., the rockering helices TMs 2, 5, 8, and 11 in Fig. 1B) provide the pivot points for the rotation. Roughly speaking, for each domain the hydrophobic force is applied at its mass center, and consists of a strength of half of the electric force applied to the proton. If each domain rotates 20° relative to the membrane, the total energy produced is estimated to be approximately One-sixth of FΔΨ (∼2.5 kJ/mol or 1 RT). In addition, the electrostatic force applied on the proton by ΔΨ promotes an inward shift of the protein (Fig. 3C). For example, if the inward movement could transiently reach one-sixth of the thickness (approximately 5 Å) of the lipid bilayer, the energy produced would be approximately 2.5 kJ/mol (∼1 RT). As a result, the movement might drive the opening side of the outward-facing structure more into the membrane from the periplasmic side while forcing the close side to protrude out of the membrane from the cytoplasmic side. Because the central cavity of a MFS protein is in general more hydrophilic than the outside surface in the TM region, and because the cavity wall possesses two clefts on the opening side (Fig. 1B), such an inward shift of the protonated MFS must be energetically unstable, resulting in a so-called hydrophobic mismatch (24). Therefore, an outward-to-inward conformation change can be triggered at this transition state, and the energetically more favorable inward conformation is taken up. In this inward conformation, the closed periplasmic side becomes more hydrophobic, whereas the now open cytoplasmic side becomes more hydrophilic. From an alternative point of view, because the protonation site is present in the middle of the TM core, it is located on the positive side of the so-called focused electric field of the membrane potential in the outward conformation, but becomes located on the negative side in the inward conformation. Thus, the total energy of the overall system is reduced by approximately 15 kJ/mol upon outward-to-inward conformational change. Such a mechanism would not necessarily require the existence of position-specific, conserved, structural motifs, but would demand a hydrophobicity change of the overall structure, for which the rocker-switch architecture of the MFS protein is perfectly suited. Undoubtedly, there are many other energetic factors involved in the rocking conformational change (25). However, most of them would cancel each other out because of the symmetry of the conformational change, and the net result would be the outward-to-inward conformational change of the MFS protein.
Similar concepts of functional requirement of a membrane potential (and thus intact cell membrane) have been widely accepted for membrane proteins that are involved in energy metabolism. For example, in an ATP synthase complex, proton movement driven by the electrochemical potential results in the rotation of the central piece of the complex (approximately 500 kDa in size); in this process, four protons are consumed to synthesize one ATP molecule (i.e., storing 30 kJ/mol energy with an efficiency of 50−75%) (26). Furthermore, voltage-gating ion channels change the conformation of their charge-carrying TM helices upon voltage inversion (27). The requirement of a membrane potential for MFS transporting activity has also been shown experimentally, (e.g., in active transport of lactose by LacY) (28, 29). Numerous mutagenesis experiments demonstrate that protonatible amino acid residues inside the central cavity are essential for H+-driven MFS transporters (12, 30, 31). Interestingly, however, MdfA, a well-studied MFS protein, tolerates displacements of an essential acidic amino acid residue to various locations in the substrate translocation, strongly indicating that it is the protonation per se but not detailed geometry of such a residue is important for the transport activity (32). Taken together, we hypothesize that the interaction between the protonation site and the membrane potential provides (partial) energy source for the conformational transition of the MFS protein from the outward (ground) state to the inward (excited) state.
Transition from the Excited to Ground States.
For the inward-to-outward conformational change of a H+-driven MFS protein, there are at least two contributing factors: deprotonation and distribution of basic residues. Although the protonation of an MFS protein favors the inward (excited) state, a deprotonation occurs in the inward conformation because of the alkaline condition of the cytosol (23), resulting in eliminating of the inward force applied on the protein by the negative-inside membrane potential. In addition, the differential energy associated with protonation and deprotonation of one amino acid residue on the two sides of the membrane with a ΔpH of ∼0.6 (corresponding to approximately 2.3 RTΔpH or 3.5 kJ/mol per proton), combined with a fraction of the ΔΨ-associated energy, would promote substrate release (symporter) or substrate loading (antiporter), by modifying the substrate-binding site in the inward conformation.
Charge distribution of membrane proteins usually follows the “positive inside rule” (18). For example, in the case of YajR, numerous basic residues are located on the cytoplasmic side, and many of them are distributed in the circumference of the protein, presumably interacting with the polar head groups of lipid molecules in the membrane (SI Appendix, Fig. S3). These basic residues would be pushed away from the negatively charged inner leaflet of the lipid bilayer during the above-mentioned inward movement, and thus a portion of the energy generated by proton translation could be stored in form of electrostatic energy. This energy would provide an electrostatic force for the protein to carry out the second conformational transition back to the outward (ground) state after deprotonation, using a mechanism similar to the first outward-to-inward transition. Such a mechanism of conformational change would also not require position-specific interactions, and thus no conserved sequence motif would be necessary for implementing such a mechanism.
According to this hypothesis, for a proton-driven symporter, substrate-loading in the outward state is ideally to be before protonation but not the opposite, to prevent wasteful proton leak. This corollary is consistent with previous works showing that loading of the substrate lactose to LacY is independent of the H+ electrochemical gradient (20, 22). Similarly, the substrate releasing in the inward conformation should be before the deprotonation, to prevent the substrate from being sent back impurposely. In other words, the transporter would stay in its inward conformation until the substrate is released to the cytosol followed by deprotonation, which would explain why LacY can accumulate lactose against a 100-fold concentration gradient (20). Therefore, the binding order of substrate and proton in both loading and releasing processes is likely to be important for effective transport cycles.
Motif A and Regulation of Transition.
Whereas both above mentioned-mechanisms of conformational transitions do not require interactions between specific amino acid residues, many MFS transporters do contain a conserved motif, which was termed motif A (also called an MFS-specific motif or a signature motif), located in the L2-3 region on the cytoplasmic side (19). Although its high degree of conservation implies an important function, motif A must be in its functional state during structure analysis in order for us to understand its function accurately. So far, however, this particular structure has not been available. The YajR crystal structure provides an excellent showcase for the functions of motif A in action (Fig. 2A). In this outward-facing structure, motif A in L2-3 harbors, apart from other features, three essential structural elements: (i) a Gly residue at the position +1 essential for interdomain helix packing (Fig. 2 and SI Appendix, Table S2); (ii) an interdomain charge-helix dipole interaction between Asp73(+5) and the N-terminal end of the helix TM11; and (iii) a charge-relay system with an acidic amino acid residue from TM4 [i.e., Asp73(+5)-Arg77(+9)-Asp126TM4]. Acompanied by other conserved, short side chain residues form TM11, Gly69(+1) is presumably involved in the dynamic interdomain helix packing during conformational change (Fig. 2A), and our data show that a G69W mutation disturbs the outward conformation (Figs. 1A and 2B). Consistently, it is shown in LacY that the +1 position of motif A are critical for its transport activities (33, 34). Moreover, the charge-helix dipole interaction was observed to stabilize the YajR structure in the outward conformation by locking the domain interface on the cytoplasmic side, whereas the charge-relay system may provide a mechanism to regulate the strength of the charge-helix dipole interaction.
A charge-helix dipole interaction on its own is estimated to provide approximately 4 kJ/mol stabilization energy in a soluble protein (35). In YajR, a charge-reverse point mutation, D73R, at the +5 position decreased the melting temperature by nearly 20 °C (Fig. 1A) and increased occupancy of the inward conformation (Fig. 2B). These results support the stabilization role of motif A in the charge-dipole interaction. Many mutations at the +5 position of motif A in a number of MFS proteins were found to be detrimental to the transport function (33, 34, 36, 37). For example, an Asp-to-Asn variant of Tn10 completely lost its activity (36). Even a conservative modification from Asp to Glu diminished the transport activity (36, 38), suggesting that a precisely maintained geometry is crucial for effective interdomain interactions, and thus essential for transport activity. More interestingly, it has been shown that the probability of opening the periplasmic pathway is markedly reduced in Asp(+5) mutations of LacY (39), consistent with a role of motif A including Asp(+5) in stabilizing the outward conformation.
In support of a functional role of the charge-relay system, Arg77 and Asp126 of YajR are conserved among many MFS proteins (19), and point mutations at these positions decrease the thermal stability of YajR. Point mutations of the TetA(P) efflux protein from Clostridium perfringens at the position Arg71 (equivalent to Arg77YajR) or Glu117 (Asp126YajR) are reported to disrupt its transport activity (40). Five acidic amino acids residues in the Lactococcus lactis MFS H+/drug antiporter LmrP have been shown to be essential for its transport activity (31). Among these residues, according to our current model, Asp68 corresponds to the central acidic residue of motif A, Asp73YajR, Glu327 to Glu320YajR of the titratable pair in the central cavity, and Asp128 to the charge-relay acidic residue Asp126YajR. Charge-relay systems are widely used in enzymes. For example, an Asp-Arg-Asp charge-relay system has been reported in an epoxide hydrolase and serves as the catalytic center; and mutations at any one of the three amino acids residues, even an Arg-to-Lys mutation at the middle position, result in loss of the enzyme activity (41). In a charge-relay system, electron movement is delocalized, and thus signals initiated by environmental changes can be sensed by one part of the system, before being transferred to another.
How does motif A on the cytoplasmic surface sense and response to protonation inside the central cavity? In nearly all known crystal structures of proton-driven MFS proteins that contain motif A in L2-3, the charge-relay system is always accompanied by the interdomain peptide. These linker peptides often contain a number of polar residues (e.g., His192 in YajR), as well as an amphipathic α-helix (α6–7) (SI Appendix, Fig. S2), which is likely to bind to the membrane surface. If this amphipathic α-helix maintains membrane association during the inward shift of the overall protein structure, the interdomain peptide will be stretched, resulting in a linker movement that probably interrupts the charge-relay system in motif A (Fig. 3D). Alternatively, some (basic) residues from the linker may maintain interactions with lipid molecules in the membrane during the inward shift of the TM core, thus inducing a conformational change of the linker. In support of such possible scenarios, the central acidic residue Asp68 of motif A in the inward conformation of LacY interacts with His205 in the interdomain linker. A pH-titration experiment on Asp128 of LmrP from L. lactis (equivalent to Asp126YajR) suggests that the charge-relay system of motif A undergoes a change from buried to solvent-exposed environments during the outward-to-inward transition (31). Moreover, in the two MFS structures of PepTSo/2XUT and PepTSt/4APS, an interdomain, reentrant helix pair is inserted between the two domains of the TM core relative to the 12-TM MFS proteins and is likely to move independently from the rest of the TM core (11, 13). Thus, this reentrant helix pair may function in a way similar to the above-mentioned amphipathic α-helix in other known MFS structures. Therefore, we hypothesize that the inward movement of the TM core of the MFS protein, occurring relative to the membrane, triggers the release of the charge-dipole, interdomain lock of motif A in L2-3, thus facilitating the outward-to-inward transition.
Because of the internal symmetry of the MFS protein, motif A-like structural elements (A-like motifs) may occur in four places, namely L2-3, L5-6, L8-9, and L11-12 loop regions. Indeed, analysis of known MFS protein structures indicates that A-like motifs can be found in all four positions (referred to hereafter as motifs AL2-3, AL5-6, AL8-9, and AL11-12, respectively), and some proteins contain more than one of these motifs (SI Appendix, Table S3). For examples, YajR contains motifs AL2-3 and AL8-9 both on the cytoplasmic side; and motif AL11-12 of FucP/3O7Q on the periplasmic side is conserved in homologous l-fucose transporters (12). In addition, our analysis of amino acid sequences of 77 MFS proteins from E. coli shows a similar pattern (SI Appendix, Fig. S5 and Table S4). With the available 3D structures of MFS proteins and homolog sequences, a general trend emerges as that A-like motifs occur more often on the cytoplasmic, rather than periplasmic side, and motif AL2-3 is the most conserved one. Furthermore, in known 3D structures, more A-like motifs on the cytoplasmic side (i.e., AL2-3 and AL8-9) contain the charge-relay system, whereas none of those on the periplasmic side do. Because the cytoplasmic side A-like motifs are often covered either by the interdomain linker (and a linker-coupled structural element in XylE/4GC0) or by a putative regulatory structural element, these motifs are likely to regulate the outward-to-inward conformational change. Of particular interests to our YajR structure, the C-terminal, small YAM domain may sense environmental changes, such as pH and halogen ion variation (SI Appendix, Fig. S6), providing a unique mechanism to regulate the interdomain lock of motif AL8-9. In agreement with this possibility, truncation of the YAM domain slightly perturbs the thermal stability of the YajR protein (Fig. 1A). On the other hand, A-like motifs on the periplasmic side could be regulated allosterically by a ligand. Consistently, a substrate-induced, inward-to-outward conformational change has been reported in the symporter LacY (42), suggesting a possibility that a regulation of conformational change occurs outside of the central cavity and from the periplasmic side (e.g., via its AL11-12 motif). Interestingly, point mutations in LacY that rescue a defective mutation at the +5 position of motif AL2-3 on the cytosol side are found to be clustered around the motif AL11-12 on the periplasmic side (43), implying that balanced stabilities between the outward and inward conformations are important for the transport function. Similar structure-functional relationships are found in eukaryotic MFS proteins. For example, point mutations in either motif AL2-3 or AL8-9 result in loss of transport activity in a nitrate transporter from Aspergillus nidulans (44). Related to human diseases, in variants of the glucose transporter GLUT1 (a homolog of bacterial XylE) from human patients of GLUT1 Deficiency Syndrome, mutations have been identified in positions Gly91 [equivalent to Gly76(+8)YajR] and Arg93 [Lys78(+10)YajR] of motif AL2-3, Glu146 (Asp126YajR) of the charge-relay system associated with motif AL2-3, and Arg333 (Lys276YajR) of the charge-relay system of motif AL8-9. Futhermore, other point mutations for basic amino acid residues in the interdomain linker region (e.g., Arg212, Arg218, and Lys256) of GLUT1 have been reported being found in patient genes (45). These observations indicate the importance of the corresponding structural elements in GLUT1 functions. Therefore, a wide collection of existing data support our hypothesis that A-like motifs function as molecular switches, which regulate the conformational changes of MFS proteins.
Conclusion.
YajR proteins of Gram-negative bacteria contain the typical 12-TM helix core of MFS transporters, as well as a C-terminal ferredoxin-like YAM domain. The crystal structure of its outward conformation illustrates functions of the highly conserved motif A of MFS transporters, thus enabling us to postulate some common mechanisms of the conformational changes between the outward and inward states of H+-potential driven, electrogenic, MFS transporters. For this class of MFS proteins, an inward shift of the protonated protein powered by the negative-inside membrane potential facilitates the outward-to-inward conformational change, and the distribution of basic amino acid residues on the cytoplasmic side of the protein may partially provide the energy for the protein to return to its ground state. Moreover, a charge-helix dipole interaction between the two domains of the TM core can be regulated by the charge-relay system of motif A. We believe that similar mechanisms will apply to most, if not all MFS transporters.
Methods
The full-length YajR gene was cloned from the E. coli BL21 genome. Recombinant YajR protein was expressed in E. coli, extracted from the cell membrane with dodecyl-β-d-maltopyranoside, and purified in the presence of nonyl-β-d-glucopyranoside. The protein sample was crystallized using hanging-drop vapor-diffusion methods. The crystal form belongs to the P6122 space group. There is one YajR molecule per asymmetric unit of 70% solvent content (VM = 4.0 Å3/Da). Initial phases of the structural factors were determined using a mercury-based MAD method (17), and the structure model was refined to 3.15 Å resolution. Fitting of the amino acid sequence to the peptide-chain model was assisted with heavy atom markers from multiple Hg-derivative crystals (including one from a WT crystal and three from isomorphous crystals of Cys-mutations, namely A90C, A149C, and A180C) as well as a Se-Met derivative crystal (SI Appendix, Figs. S1A and S7). More information on materials and methods can be found in SI Appendix.
Supplementary Material
Acknowledgments
We thank the staff of the Protein Research Core Facility at the Institute of Biophysics, Chinese Academy of Sciences for their excellent technical assistance, and staff members of the Shanghai Synchrotron Radiation Facility (China) and SPring-8 (Japan) synchrotron facilities. This work was supported by the Ministry of Science and Technology of China “973” Project Grants 2009CB918803 and 2011CB910301 (to X.C.Z.), and National Science Foundation of China Grants 31130028 and 31225011 (to J.L.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3WDO).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1308127110/-/DCSupplemental.
References
- 1.Marger MD, Saier MH., Jr A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem Sci. 1993;18(1):13–20. doi: 10.1016/0968-0004(93)90081-w. [DOI] [PubMed] [Google Scholar]
- 2.Saier MH, Jr, et al. The major facilitator superfamily. J Mol Microbiol Biotechnol. 1999;1(2):257–279. [PubMed] [Google Scholar]
- 3.Brown S, Chang JL, Sadée W, Babbitt PC. A semiautomated approach to gene discovery through expressed sequence tag data mining: Discovery of new human transporter genes. AAPS PharmSci. 2003;5(1):E1. doi: 10.1208/ps050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 6.Abramson J, et al. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301(5633):610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 7.Zheng H, Wisedchaisri G, Gonen T. Crystal structure of a nitrate/nitrite exchanger. Nature. 2013;497(7451):647–651. doi: 10.1038/nature12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan H, et al. Structure and mechanism of a nitrate transporter. Cell Rep. 2013;3(3):716–723. doi: 10.1016/j.celrep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen BP, et al. Crystal structure of a eukaryotic phosphate transporter. Nature. 2013;496(7446):533–536. doi: 10.1038/nature12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490(7420):361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 11.Newstead S, et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011;30(2):417–426. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dang S, et al. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467(7316):734–738. doi: 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- 13.Solcan N, et al. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012;31(16):3411–3421. doi: 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183(20):5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cummings MD, Farnum MA, Nelen MI. Universal screening methods and applications of ThermoFluor. J Biomol Screen. 2006;11(7):854–863. doi: 10.1177/1087057106292746. [DOI] [PubMed] [Google Scholar]
- 17.Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science. 1991;254(5028):51–58. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 18.von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225(2):487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 19.Paulsen IT, Brown MH, Skurray RA. Proton-dependent multidrug efflux systems. Microbiol Rev. 1996;60(4):575–608. doi: 10.1128/mr.60.4.575-608.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan L, Kaback HR. Binding affinity of lactose permease is not altered by the H+ electrochemical gradient. Proc Natl Acad Sci USA. 2004;101(33):12148–12152. doi: 10.1073/pnas.0404936101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alber T, et al. Contributions of hydrogen bonds of Thr 157 to the thermodynamic stability of phage T4 lysozyme. Nature. 1987;330(6143):41–46. doi: 10.1038/330041a0. [DOI] [PubMed] [Google Scholar]
- 22.Smirnova I, Kasho V, Sugihara J, Vázquez-Ibar JL, Kaback HR. Role of protons in sugar binding to LacY. Proc Natl Acad Sci USA. 2012;109(42):16835–16840. doi: 10.1073/pnas.1214890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zilberstein D, Schuldiner S, Padan E. Proton electrochemical gradient in Escherichia coli cells and its relation to active transport of lactose. Biochemistry. 1979;18(4):669–673. doi: 10.1021/bi00571a018. [DOI] [PubMed] [Google Scholar]
- 24.Mouritsen OG, Bloom M. Mattress model of lipid-protein interactions in membranes. Biophys J. 1984;46(2):141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White SH, Wimley WC. Membrane protein folding and stability: Physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 26.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402(6759):263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 27.Tombola F, Pathak MM, Isacoff EY. How far will you go to sense voltage? Neuron. 2005;48(5):719–725. doi: 10.1016/j.neuron.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 28.Viitanen P, Garcia ML, Kaback HR. Purified reconstituted lac carrier protein from Escherichia coli is fully functional. Proc Natl Acad Sci USA. 1984;81(6):1629–1633. doi: 10.1073/pnas.81.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan L, Sahin-Tóth M, Kálai T, Hideg K, Kaback HR. Probing the mechanism of a membrane transport protein with affinity inactivators. J Biol Chem. 2003;278(12):10641–10648. doi: 10.1074/jbc.M211355200. [DOI] [PubMed] [Google Scholar]
- 30.Smirnova IN, Kasho V, Kaback HR. Protonation and sugar binding to LacY. Proc Natl Acad Sci USA. 2008;105(26):8896–8901. doi: 10.1073/pnas.0803577105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gbaguidi B, Hakizimana P, Vandenbussche G, Ruysschaert JM. Conformational changes in a bacterial multidrug transporter are phosphatidylethanolamine-dependent. Cell Mol Life Sci. 2007;64(12):1571–1582. doi: 10.1007/s00018-007-7031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigal N, Fluman N, Siemion S, Bibi E. The secondary multidrug/proton antiporter MdfA tolerates displacements of an essential negatively charged side chain. J Biol Chem. 2009;284(11):6966–6971. doi: 10.1074/jbc.M808877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frillingos S, Sun J, Gonzalez A, Kaback HR. Cysteine-scanning mutagenesis of helix II and flanking hydrophilic domains in the lactose permease of Escherichia coli. Biochemistry. 1997;36(1):269–273. doi: 10.1021/bi9618629. [DOI] [PubMed] [Google Scholar]
- 34.Jessen-Marshall AE, Paul NJ, Brooker RJ. The conserved motif, GXXX(D/E)(R/K)XG[X](R/K)(R/K), in hydrophilic loop 2/3 of the lactose permease. J Biol Chem. 1995;270(27):16251–16257. doi: 10.1074/jbc.270.27.16251. [DOI] [PubMed] [Google Scholar]
- 35.Nicholson H, Becktel WJ, Matthews BW. Enhanced protein thermostability from designed mutations that interact with alpha-helix dipoles. Nature. 1988;336(6200):651–656. doi: 10.1038/336651a0. [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi A, Ono N, Akasaka T, Noumi T, Sawai T. Metal-tetracycline/H+ antiporter of Escherichia coli encoded by a transposon, Tn10. The role of the conserved dipeptide, Ser65-Asp66, in tetracycline transport. J Biol Chem. 1990;265(26):15525–15530. [PubMed] [Google Scholar]
- 37.Suárez-Germà C, et al. Phosphatidylethanolamine-lactose permease interaction: A comparative study based on FRET. J Phys Chem B. 2012;116(48):14023–14028. doi: 10.1021/jp309726v. [DOI] [PubMed] [Google Scholar]
- 38.Jessen-Marshall AE, Brooker RJ. Evidence that transmembrane segment 2 of the lactose permease is part of a conformationally sensitive interface between the two halves of the protein. J Biol Chem. 1996;271(3):1400–1404. doi: 10.1074/jbc.271.3.1400. [DOI] [PubMed] [Google Scholar]
- 39.Liu Z, Madej MG, Kaback HR. Helix dynamics in LacY: Helices II and IV. J Mol Biol. 2010;396(3):617–626. doi: 10.1016/j.jmb.2009.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bannam TL, et al. The Clostridium perfringens TetA(P) efflux protein contains a functional variant of the Motif A region found in major facilitator superfamily transport proteins. Microbiology. 2004;150(Pt 1):127–134. doi: 10.1099/mic.0.26614-0. [DOI] [PubMed] [Google Scholar]
- 41.Arand M, et al. Structure of Rhodococcus erythropolis limonene-1,2-epoxide hydrolase reveals a novel active site. EMBO J. 2003;22(11):2583–2592. doi: 10.1093/emboj/cdg275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smirnova I, et al. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci USA. 2007;104(42):16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cain SM, Matzke EA, Brooker RJ. The conserved motif in hydrophilic loop 2/3 and loop 8/9 of the lactose permease of Escherichia coli. Analysis of suppressor mutations. J Membr Biol. 2000;176(2):159–168. doi: 10.1007/s00232001085. [DOI] [PubMed] [Google Scholar]
- 44.Kinghorn JR, et al. Missense mutations that inactivate the Aspergillus nidulans nrtA gene encoding a high-affinity nitrate transporter. Genetics. 2005;169(3):1369–1377. doi: 10.1534/genetics.104.036590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pascual JM, et al. Structural signatures and membrane helix 4 in GLUT1: Inferences from human blood-brain glucose transport mutants. J Biol Chem. 2008;283(24):16732–16742. doi: 10.1074/jbc.M801403200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.