

Single mutation turns an inedible symbiotic bacterium into an amoeba’s food source
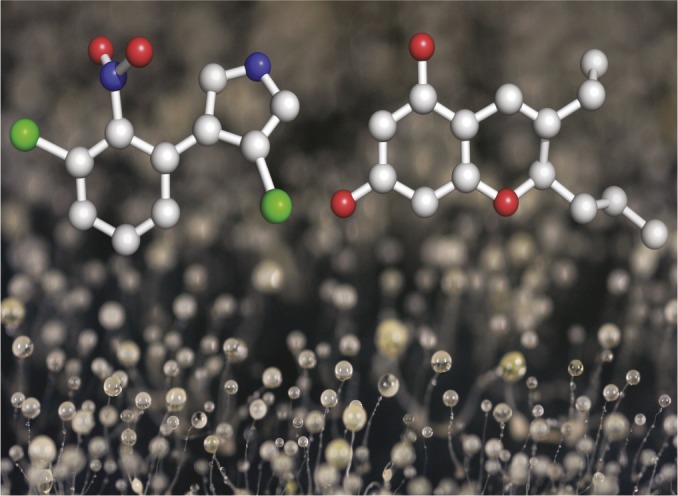
D. discoideum fruiting bodies shown with models of beneficial molecules produced by farmer-associated bacterial symbiont [pyrrolnitrin (Left) and a chromene (Right)]. Image courtesy of Scott Solomon, Rice University, Houston, TX.
“Farmer” clones of the social amoeba, Dictyostelium discoideum, carry bacteria with them, but only half the bacteria they carry serve as food sources. Pierre Stallforth et al. (pp. 14528–14533) investigated the association between D. discoideum and two strains of the bacterium Pseudomonas fluorescens, one that serves as a food source for the amoeba and one that does not. The authors found that the nonfood strain of P. fluorescens produced two diffusible small molecules: pyrrolnitrin, a known antifungal agent, and a previously undescribed molecule, 3-ethyl-2-propyl-2H-chromene-5,7-diol (chromene). Treatment with either pyrrolnitrin or chromene increased spore production in a farmer strain of D. discoideum and suppressed spore formation in a nonfarmer strain; pyrrolnitrin also likely inhibited microbial pathogens. Using genome sequence and phylogenetic analyses, the authors identified a disabling point mutation in a regulatory protein, GacA, of the food strain of P. fluorescens. Mimicking the mutation by deleting the gacA gene in the nonfood strain turned it into a food source for the amoeba and changed its secondary metabolite profile to match that of the food strain. The findings suggest that a single mutational step is sufficient to convert a symbiotic bacterium from an inedible producer of beneficial small molecules into a food source for D. discoideum, according to the authors. — S.R.
Suboptimal doses of antiretroviral drug induce aberrant HIV-1 integration
Integration of HIV-1 into the host genome is critical for viral replication. Integration is a two-step process that is mediated by a viral enzyme called integrase (IN), which first removes two nucleotides from each end of the viral DNA and then catalyzes strand transfer reactions that insert each end of the viral DNA into the target DNA. Janani Varadarajan et al. (pp. 14747–14752) hypothesized that suboptimal concentrations of IN inhibitors, which are widely used as anti-HIV drugs, might block only one of the two strand transfer reactions required to insert the viral genome into host DNA, leading to aberrant integration reactions catalyzed by the cellular DNA repair machinery. To test this hypothesis, the authors analyzed the structures of viral DNAs recovered from HIV-infected cells that had been treated with different doses of raltegravir (RAL), an IN inhibitor. The authors found that all of the integrated viral DNAs isolated from untreated cells were normal. By contrast, approximately 10–15% of the integrated viral DNAs isolated from cells treated with a suboptimal dose of RAL were abnormal and were accompanied by rearrangements of the host genome. The authors suggest that aberrant integrations might occur in HIV-1–infected patients who do not adhere to their RAL treatment regimen, potentially resulting in an increased risk of malignancy. — N.Z.
Spontaneous prion formation in a mouse model of Creutzfeldt–Jakob disease
Prions—misfolded proteins that can hijack living cells, reproduce in them, and gradually destroy brain tissue in humans, cattle, and other mammals—cause remarkably different clinical symptoms depending on the type of mutation in the prion protein, PrP. Walker Jackson et al. (pp. 14759–14764) compared three mouse lines, each expressing the same PrP protein with a single amino acid difference, to study the discrete genetic differences between two human forms of spongiform encephalitis: Creutzfeldt–Jakob disease (CJD) and fatal familial insomnia (FFI). Two previously created mouse lines—a wild-type line containing an intact form of PrP and a model of FFI containing an aspartate-to-asparagine substitution—were compared with a third model containing a glutamate-to-lysine mutation, known to cause inherited CJD in humans. All three lines possessed a molecular barrier to infectious prion agents in the external environment. The authors report that mice expressing either modified form of PrP spontaneously created prions that could be transmitted among mice to induce the hallmark symptoms of either CJD or FFI. According to the authors, altering a single amino acid in one gene in an otherwise normal genome can cause remarkably different neurodegenerative diseases and trigger the formation of two different transmissible prions. — A.G.
Copper impairs the clearance of amyloid-β from mouse brain
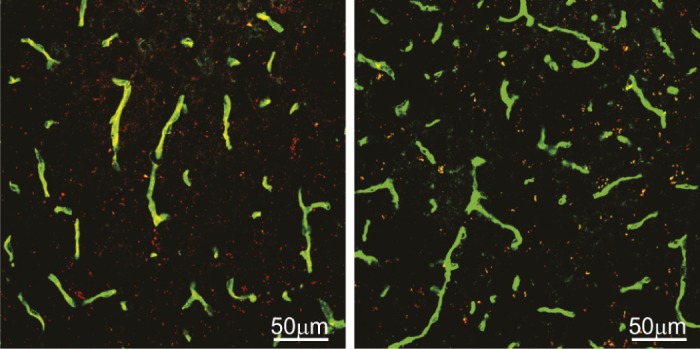
Levels of LRP1 (red) in brain blood vessels (green) is reduced (yellow) in mice dosed with low levels of copper (within the EPA limits for drinking water, Right) compared to vehicle treated mice (Left).
Excessive levels of copper (Cu) in the brain are associated with Alzheimer’s Disease (AD) pathogenesis; amyloid-β (Aβ) accumulates in the brain of laboratory animals dosed with low levels of Cu, and a Cu chaperone has shown promise in phase II clinical trials for AD. Itender Singh et al. (pp. 14771–14776) investigated the molecular mechanism by which Cu leads to Aβ accumulation in the mouse brain and human brain endothelial cells. Compared with young mice, aging mice displayed higher levels of Cu in brain capillaries, accumulation of Aβ, and reduced levels of LRP1, an endothelial protein that removes Aβ from the brain by transporting it across the blood–brain barrier. The authors replicated these observations in young mice by adding low levels of Cu to their drinking water. When Cu was added to cultured human brain endothelial cells, it enhanced binding of the cellular prion protein PrPc to LRP1 and increased proteasome-dependent degradation of LRP1. In a mouse model of AD, administration of Cu depressed LRP1 levels as in normal mice, increased the production of Aβ and proinflammatory cytokines, and impaired cognitive function. According to the authors, the results suggest that Cu regulates LRP1-mediated clearance of Aβ across the blood–brain barrier, and that Cu-chelating agents may reduce Aβ accumulation and improve cognition in AD. — C.B.
Alcohol shifts behavioral control from the brain’s cortex to the striatum
Alcohol and drugs of abuse are known to lead to reduced executive control over behavior and to compulsive drug seeking. Researchers hypothesize that the behavioral change involves a corresponding shift in the brain area controlling behavior: from the prefrontal cortex (PFC) to the dorsal striatum (DLS). While previous research shows that alcohol impairs PFC function, the role of alcohol in shifting behavioral control to the DLS remains unestablished. Lauren DePoy et al. (pp. 14783–14788) used a mouse model to examine the effects of chronic alcohol consumption on the morphology and signaling of the DLS and on various forms of operant learning known to be controlled in the region. The authors found that mice exposed to chronic intermittent alcohol exhibited an expansion of dendritic material in the DLS and a downregulation of DLS CB1 receptor signaling and plasticity. In addition, alcohol facilitated performance on the learning tasks. Together, these findings suggest that alcohol upregulates DLS function and, coupled with prior evidence, support the idea that the DLS may take over behavioral control from the PFC. The authors suggest that the shift to striatal control over behavior may be a critical step in the progression of alcoholism. — B.A.
Failure to clear ATP blocks neural responses to taste stimuli
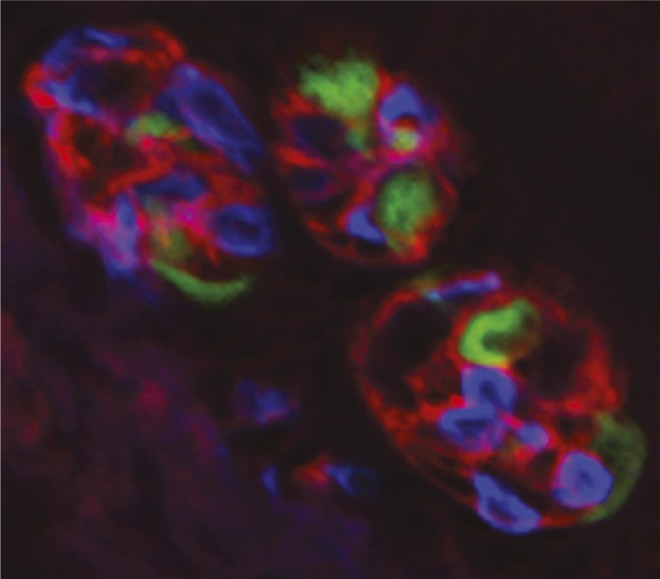
NTPDase2 expressing cells (red) envelop other cell types (blue and green) in three taste buds, facilitating ATP removal from extra-cellular space. Image courtesy of Diane Bartel (University of Colorado School of Medicine, Aurora, CO).
Unique among the special senses, taste relies on ATP as a neurotransmitter to activate sensory nerve fibers. Released in response to taste stimuli, ATP activates P2X2 and P2X3 purinergic receptor subunits on taste nerves and is quickly degraded by the enzyme NTPDase2, thereby preventing extracellular accumulation of the neurotransmitter. To investigate the role of NTPDase2 in the function of taste buds, Aurelie Vandenbeuch et al. (pp. 14789–14794) conducted trials with gene-targeted Entpd2-null mice, which lack the enzyme. Using molecular, histochemical, and anatomical metrics, the authors determined that Entpd2 deletion causes an accumulation of ATP in extracellular spaces that likely desensitizes receptors on nerve fibers and attenuates the transmission of information from taste buds to the taste nerves. The authors also propose that pharmaceutical agents currently being developed, such as drugs that target purinergic receptors and enzymes to treat chronic pain and other disorders, may inadvertently disrupt taste function. The findings suggest that the genetic deletion of NTPDase2, decreases neural responses to taste stimuli, according to the authors. — T.J.