

Abstract
Polytopic membrane proteins are inserted cotranslationally into target membranes by ribosome–translocon complexes. It is, however, unclear when during the insertion process specific interactions between the transmembrane helices start to form. Here, we use a recently developed in vivo technique to measure pulling forces acting on transmembrane helices during their cotranslational insertion into the inner membrane of Escherichia coli to study the earliest steps of tertiary folding of five polytopic membrane proteins. We find that interactions between residues in a C-terminally located transmembrane helix and in more N-terminally located helices can be detected already at the point when the C-terminal helix partitions from the translocon into the membrane. Our findings pinpoint the earliest steps of tertiary structure formation and open up possibilities to study the cotranslational folding of polytopic membrane proteins.
Keywords: protein folding, membrane insertion, helix-helix interactions
When and how do polytopic helix-bundle membrane proteins fold? In vitro studies have shown that purified membrane proteins can fold from a denatured state (1–3). Coexpression or coreconstitution of artificially severed membrane proteins can also yield functional, properly folded protein (4–6). Posttranslational folding of (some) membrane proteins is thus clearly possible; however, nearly all polytopic membrane proteins are integrated into the membrane cotranslationally (7). Given that membrane insertion is a relatively slow process that is essentially limited by the rate of translation, it seems likely that much of the folding process might be cotranslational in vivo. Because of a lack of good assays, this idea has been difficult to test experimentally. The best support for it thus far has been obtained by end-point studies that compare the efficiency of membrane insertion of marginally hydrophobic transmembrane helices (TMHs) in their natural sequence context and when interacting residues in the protein have been removed by mutagenesis (8–13), but such studies cannot identify the precise point during the translation-insertion–folding process when interactions between TMHs first develop.
Bacterial inner membrane proteins and eukaryotic membrane proteins found in the plasma membrane or in organelles along the exo- and endocytic pathways are cotranslationally inserted into the inner membrane (in bacteria) or the endoplasmic reticulum (in eukaryotes) via evolutionarily related Sec-type translocons: the SecYEG translocon in bacteria and the Sec61 translocon in eukaryotes (14, 15). As they exit the ribosome, the TMHs in a nascent membrane protein appear to move sequentially into the membrane through a lateral gate in the wall of the translocon channel (16, 17). The TMHs are thought to sample the environment immediately outside the lateral gate and insert into the membrane with efficiencies that depend on their hydrophobicities (18).
An obvious consequence of a sequential, cotranslational insertion mechanism is that more C-terminally located TMHs become exposed not only to the lipid environment outside the lateral gate, but also to all previously inserted TMHs in the same polypeptide chain. In principle, a C-terminally located TMH could therefore interact with upstream TMHs, facilitating its transition into the membrane. Recent studies have already hinted that distant parts of a polypeptide chain can influence the membrane insertion of a given TMH (8, 19, 20).
Here, we set out to determine the earliest point at which it is possible to detect interactions between a C-terminal TMH and more N-terminally located TMHs during cotranslational protein insertion into the inner membrane of Escherichia coli. We use a recently established in vivo assay (21) to indirectly measure the pulling force exerted on a C-terminal TMH during its cotranslational insertion into the membrane, either in the presence or absence of its natural upstream TMHs. We find that interactions between a C-terminal TMH and upstream TMHs can be detected as an increase in the pulling force already at the point at which the C-terminal TMH partitions from the translocon into the membrane and thereby can guide the formation of the tertiary structure as well as membrane insertion at the same time. Our results help to define the earliest steps along the folding pathway for bacterial inner membrane proteins and to demonstrate the sensitivity of the in vivo force-measurement technique.
Results
Translational Arrest Peptides as in Vivo Force Sensors.
We have recently shown that a SecYEG-mediated, cotranslational insertion of a TMH into the inner membrane of E. coli cells generates a pulling force on the nascent chain that can be detected using a translational arrest peptide (AP) engineered into the nascent chain as a force sensor (21).
In our previous study, pulling forces were measured using constructs with the 17-residue-long AP FSTPVWISQAQGIRAGP from E. coli SecM (22) inserted near the C terminus of the well-characterized E. coli inner membrane protein LepB (Fig. 1A). LepB has two TMHs near the N terminus and a large periplasmic domain. A sufficiently hydrophobic “test” TMH inserted into the periplasmic domain will experience a pulling force (F) when integrating from the translocon into the membrane; F is transmitted via the nascent chain to the AP (Fig. 1B). In the absence of F, the ribosome stalls when the final Pro codon in the AP enters the A site, generating a truncated form of the protein (23). In contrast, if there is sufficient tension in the nascent chain at this point, stalling is prevented and full-length protein is produced. The relative amount of full-length vs. truncated protein is proportional to the hydrophobicity of the test TMH at the point when the ribosome reaches the critical Pro codon (21, 24). The SecM AP can thus be used as an in vivo force sensor, with the fraction of full-length protein (fFL) produced serving as a proxy for the physical force exerted on the test TMH at the precise moment when the ribosome reaches the end of the AP.
Fig. 1.

The pulling force assay. (A) Design of the LepB-derived constructs. (Upper) Construct used to measure pulling forces on the isolated C-terminal TMH. Two N-terminal TMHs of composition 8L/11A (black) are followed by a 151-residue-long spacer segment from LepB. The C-terminal test TMH(i+1) (yellow) is followed by a linker of fixed length, the SecM arrest peptide (AP; brown), and a 23-residue long C-terminal tail. (Lower) Construct used to measure pulling forces on the test TMH in the context of the natural protein. Because all five tested proteins possess a cytoplasmic N terminus and all test TMHs are even-numbered (C), only one 8L/11A TMH is included to maintain the Nout-Cin orientation of the test TMH. The N-terminal part of the test protein [TMH1-TMH(i); blue] and the C-terminal test TMH(i+1) is followed by a linker of fixed length, the SecM arrest peptide, and a 75-residue C-terminal tail. (B) For L = 37 residues, F on the nascent chain is maximal and the TMH spans the translocon (21). The cartoons show LepB constructs with an isolated test TMH (Left) and with the test TMH in its natural sequence context (Right). (C) The free energy of membrane insertion of each TMH in CaiT, BtuC, NhaA, EmrD, and GlpT was predicted using the delta-G predictor (18). The C-terminal test TMHs are highlighted (yellow circle). Structures are color-coded from N terminus (blue) to C terminus (red).
When the test TMH is inserted into LepB at a length (L) = 37 residues upstream of the critical Pro codon in the AP (Fig. 1A), the pulling force on the nascent chain is maximal (21). This corresponds to a situation in which the test TMH has fully entered the SecYEG translocon and is presumably partitioning into the membrane (Fig. 1B).
We reasoned that we might be able to adapt this system to detect cotranslational interactions between a test TMH in a natural protein and its upstream TMHs by comparing two types of LepB constructs: one in which the test TMH is introduced by itself at L = 37 residues before the C-terminal end of the AP and one in which the part of the natural protein that is N-terminal to the test TMH is also included in the construct (Fig. 1 A and B). Our expectation was that interactions between the test TMH and its upstream TMHs, if they add to the free energy of membrane insertion of the test TMH, should give rise to a higher pulling force in the latter type of construct.
The two N-terminal TMHs of LepB were replaced by one (in constructs with multiple TMHs) or two (in constructs with a single test TMH), 19-residue-long hydrophobic segments of composition 8L/11A. The high hydrophobicity of these segments ensures efficient membrane targeting/insertion of all constructs, and the simple sequence composition was chosen to minimize the risk of interfering interactions between these TMHs and the test TMH or its upstream partners. Compared with the LepB construct used in our earlier study (21), the C-terminal tail following the AP in constructs with multiple TMHs preceding the test TMH was lengthened to 75 residues so that the arrested and full-length forms of the protein could be cleanly separated by SDS/PAGE.
For this study, we chose five polytopic membrane proteins from E. coli with solved crystal structures: l-carnitine/γ-butyrobetaine antiporter CaiT [Protein Data Bank (PDB) ID code 2WSX] (25), sodium/proton antiporter NhaA (PDB ID code 1ZCD) (26), multidrug transporter EmrD (PDB ID code 2GFP) (27), vitamin B12 permease BtuC (PDB ID code 1L7V) (28), and glycerol-3-phosphate transporter GlpT (PDB ID code 1PW4) (29). All five proteins have their N terminus facing the cytoplasm and hence are correctly orientated in the inner membrane when fused after the first 8L/11A TMH in LepB (Fig. 1B). The apparent free energy of membrane insertion (ΔGapp) of each of the TMHs in the five proteins was predicted using the delta-G predictor (18). As can be seen in Fig. 1C, some of the TMHs have positive predicted ΔGapp values, indicating an inefficient insertion into the membrane and a low pulling force in the absence of stabilizing interactions. We picked one C-terminally located TMH with a positive predicted ΔGapp value from each protein (yellow dots) and measured the pulling force on the TMH, both in the presence and absence of its natural upstream TMHs.
The Presence of N-Terminal TMHs Increases the Pulling Force on C-Terminal TMHs.
In the absence of N-terminal TMHs, the pulling force acting on the C-terminally located test TMHs at L = 37 residues was low in all cases, as is evident from the low fFL values (Fig. 2B, yellow bars; see Fig. S1 for typical gels). On the other hand, fFL increased significantly in every case when the N-terminal TMHs were present (blue bars). Thus, the presence of the N-terminal TMHs leads to an increase in the pulling force on the test TMH where it partitions from the translocon into the membrane in all five proteins.
Fig. 2.

C-terminally located TMHs experience a stronger pulling force in the presence of N-terminal TMHs. (A) Sequences of the C-terminal test TMHs chosen for analysis, with the residues that are mutated to alanine in the TMH(i+1;mut) and TMH1-TMH(i+1;mut) constructs highlighted in red. Flanking residues from the LepB construct are in gray. (B) Force measurements on C-terminal test TMHs in five E. coli inner membrane proteins. The fFL for constructs including only the C-terminal TMHs [TMH(i+1)] are shown in yellow; results for the same TMHs in the presence of their respective N-terminal TMHs [TMH1-TMH(i+1)] are shown in blue. Orange and green bars show the corresponding measurements for constructs in which polar residues in TMH(i+1) have been mutated to alanine (see A). (C) For TMH8 of BtuC, the residues marked in A were individually mutated to alanine and fFL was determined both in the presence and absence of upstream TMHs (color coding as in B). SEs are shown (number of independent measurements ≥3) and P values with respect to the nonmutated (wt) and mutated (mut) constructs are indicated. The dashed lines show fFL for the wild-type TMH8 in the presence and absence of the N-terminal TMHs.
In an attempt to break interactions between the C- and N-terminal TMHs, we mutated selected polar or charged residues in each of the test TMHs to alanine (Fig. 2A). The chosen residues are buried inside the protein, as seen in the crystal structures, and interact either with backbone atoms or amino acid side chains in the N-terminal TMHs. Because these mutations make the C-terminal test TMHs more hydrophobic, they should significantly increase fFL for the constructs that do not include the N-terminal TMHs.
As expected, the mutations led to an increase in fFL for the constructs that lack N-terminal TMHs (Fig. 2B; compare yellow and orange bars). Strikingly, however, the mutations led to a decrease in fFL in all constructs where the N-terminal TMHs are present (blue vs. green bars); the presence/absence of the N-terminal TMHs made little difference for the mutated test TMHs (orange vs. green bars). The increase in fFL seen for the nonmutated C-terminal test TMHs in the presence of the N-terminal TMHs (yellow vs. blue bars) thus reflects specific interactions between the TMHs.
For TMH8 of BtuC, the three target residues were also individually mutated to alanine (Fig. 2C). All three mutations led to an increase in fFL in constructs lacking the N-terminal TMHs (yellow vs. orange bars). For the P261A mutant, fFL was significantly higher in the construct that includes the N-terminal TMHs than in their absence (orange vs. green bars), indicating that TMH8 still interacts with the N-terminal TMHs and that P261 is not a major contributor to TMH interactions at this early point of BtuC folding. In contrast, in the presence of the N-terminal TMHs, fFL was significantly lower in the G254A mutant compared with wild-type TMH8 (green vs. blue bars); the presence/absence of the N-terminal TMHs made little difference (green vs. orange bars), suggesting that this mutation weakens the interaction between TMH8 and the N-terminal TMHs. In the fully folded structure, G254 is located in a distorted part of TMH8 and we speculate that mutation to alanine might induce a more regular helical structure that cannot interact well with surrounding TMHs. For the H262A mutant, finally, fFL was close to maximal even in the absence of the N-terminal TMHs (orange bar); we therefore cannot say whether the upstream TMHs affect the pulling force in this case (orange vs. green bar).
Mutations in N-Terminal TMHs Can Reduce the Pulling Force on C-Terminal TMHs.
If interactions of hydrophilic residues in C-terminal TMHs with more N-terminal parts of the protein contribute to the pulling force, the pulling force should also be sensitive to mutations in the N-terminal TMHs. Judging from the X-ray structures, most polar interactions between the test and N-terminal TMHs are between the mutated residues marked in Fig. 2A and the peptide backbone in N-terminal TMHs, making mutagenesis difficult. Nevertheless, for GlpT and BtuC, distinct side-chain interactions can be seen in the structures. We therefore mutated the interacting residues in the N-terminal parts of these two proteins to alanine but kept the C-terminal TMHs unchanged.
In the crystal structure of GlpT, E299 in the C-terminal TMH8 is located close to possible interaction partners K46 and H165 (30), both of which are buried deep in the protein interior (Fig. 3A). To test if interactions between E299 and either one of these residues occur during membrane insertion of TMH8, the mutations K46A and H165A were introduced separately and together in the N-terminal part of the protein and the pulling force on the test TMH8 was measured. We observed a decrease in fFL for both mutations, indicating that these residues actively influence the insertion of TMH8. When both residues were simultaneously changed to alanine, a more marked decrease in fFL was observed, corresponding roughly to the sum of the changes in fFL observed for the single mutations.
Fig. 3.
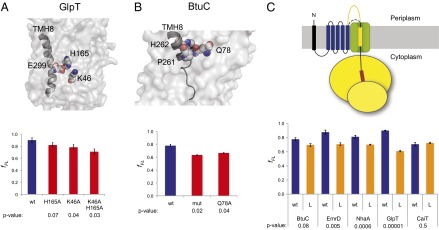
Interacting residues in N-terminal TMHs and the length of the loop preceding the C-terminal TMH affect the pulling force exerted on C-terminal TMHs. (A) Surface representation of the GlpT crystal structure, with E299 in TMH8 and interacting residues K46 and H165 shown as Corey-Pauling-Koltun (CPK) models. The red bars show the fFL for mutants H165A, K46A, and K46A+H165A. wt is the nonmutated GlpT TMH1-TMH(i+1) construct (blue bar, Fig. 2B). (B) Same as in A, but for BtuC. Residues P261 and H262 in TMH8, together with the interacting residue Q78, are shown as CPK models. (C) fFL for TMH1-TMH(i+1) constructs in which the loop preceding the C-terminal test TMH(i+1) was lengthened by 85 residues (L and orange bars). wt indicates results for the nonmutated constructs (blue bars, Fig. 2B). SEs are shown (number of independent measurements ≥3) and P values with respect to the nonmutated (wt) constructs are indicated.
In the C-terminal TMH8 of BtuC, P261 and H262 both appear to interact with Q78 in the N-terminal part of the protein (Fig. 3B). Mutating Q78 to alanine led to a decrease in fFL of a similar magnitude as seen when G254, P261, and H262 in TMH8 were simultaneously mutated to Ala (Fig. 2).
As a control, we also mutated one polar residue located far from the C-terminal test TMHs in each of the five proteins. As expected, none of these mutations affected fFL (Fig. S2).
These results further strengthen the conclusion that specific interactions between residues in C- and N-terminally located TMHs form early during membrane insertion and possibly align TMHs for tertiary structure formation.
Increased Separation Between C- and N-Terminal TMHs Reduces the Pulling Force on C-Terminal TMHs.
It is conceivable that the separation between the C-terminal test TMH and the N-terminal TMHs could influence the pulling force. To address this possibility, we introduced an additional 85 residues from the periplasmic domain of LepB into the loop between the test TMH and the N-terminal TMHs. A significant reduction in fFL was observed for EmrD, NhaA, and GlpT (Fig. 3C). A marginal decrease was seen for BtuC, and no effect was apparent for CaiT. Thus, long intervening loops can reduce the efficiency with which contacts between the C-terminal TMH and the N-terminal TMHs are formed, at least in some cases.
Hydrophobic Interactions of Lipid-Exposed Side Chains Promote the Membrane Insertion of C-Terminal TMHs.
Finally, we probed the contribution of lipid-exposed residues to the insertion of C-terminal TMHs in the presence of the N-terminal TMHs. Single residues with a lipid-exposed hydrophobic side chain were mutated to alanine (Fig. 4). If these hydrophobic side chains contribute to the membrane insertion of the test TMHs, mutating them to alanine should result in decreased pulling force. Indeed, fFL decreased in all cases, indicating that not only hydrophilic interactions with N-terminal protein parts, but also hydrophobic interactions with the surrounding lipid contribute to the pulling force.
Fig. 4.

Surface-exposed hydrophobic residues in C-terminally located TMHs contribute to the pulling force. The indicated surface-exposed hydrophobic residues (orange) in the test TMHs (yellow) of the structures (Upper) were mutated to alanine. Dashed lines indicate the approximate location of the lipid bilayer. The bar graph shows the fFL for the nonmutated (blue bars) and mutated (orange bars) TMH1-TMH(i+1) constructs. SEs are shown (number of independent measurements ≥3).
Discussion
In E. coli, most helix-bundle inner membrane proteins are cotranslationally inserted into the membrane via the SecYEG translocon. Individual TMHs are believed to be released from the translocon into the membrane in a sequential fashion (17). There is, however, a wide gap in our knowledge concerning the steps that lead from the insertion of individual TMHs to the final folded state of polytopic membrane proteins. At some point, individual TMHs have to engage in interactions to form the tertiary structure, and many proteins further incorporate cofactors and/or form oligomers (31).
When does the tertiary structure of a polytopic membrane protein begin to form and what are the sequence determinants? In this study, we have tried to define the earliest point at which a TMH engages in specific interactions with more N-terminally located TMHs. Using a recently developed assay in which the SecM translational arrest peptide is used as a force sensor, we have measured pulling forces on C-terminally located TMHs where they partition from the translocon into the membrane for five different E. coli membrane proteins of known structure. The assay does not measure forces directly, but given the typical membrane insertion free energies of TMHs [ΔGapp ∼ 1 kcal/mol (8)] and the typical distances (d) over which a TMH is expected to move during transition from the translocon into the membrane (d ∼ 10 Å), we expect forces on the order of F = ΔGapp/d ∼ 10 pN (for a detailed physical model of the force-measurement assay, see ref. 32).
For all five proteins, we find that interactions between residues buried in the interface between the chosen C-terminal test TMH and N-terminal TMHs contribute to the overall pulling force exerted on the C-terminal TMH (Fig. 2). The pulling force is sensitive to mutations of interacting residues both in the C-terminal and the N-terminal TMHs (Figs. 2 and 3) and is weakened when the loop that connects the C-terminal TMH to the N-terminal TMHs is lengthened (Fig. 3). Finally, reducing the hydrophobicity of lipid-exposed residues in the C-terminal TMHs reduces the pulling force (Fig. 4).
These observations suggest a folding pathway for polytopic membrane proteins in which at least the early steps are cotranslational. More C-terminally located TMHs can “sense” the presence of N-terminal TMHs already at the point when they are exiting the translocon, and interactions between polar residues in the C-terminal TMH and in N-terminal TMHs help promote membrane integration. Thus, tertiary interactions can already form in or in the immediate vicinity of the translocon. When the loop separating the C-terminal TMH from the upstream parts of the protein is lengthened, the tertiary interactions are weakened, possibly because the N-terminal TMHs are no longer held in close proximity to the translocon when the C-terminal TMH emerges from the ribosome. This may be one reason why loops between TMHs in polytopic membrane proteins tend to be short (33) and multidomain polytopic membrane proteins are rare (34).
Translational arrest peptides provide exquisitely sensitive in vivo sensors to detect forces acting on a nascent polypeptide chain during translation (21). As shown here, using such sensors, it is possible to pick up forces generated by interactions between TMHs during translocon-mediated membrane insertion, opening up possibilities to study the early folding steps that guide tertiary structure formation in polytopic membrane proteins.
Materials and Methods
Cloning and Mutagenesis.
The previously described pING plasmid encoding a variant of leader peptidase (LepB), with an introduced test segment of composition 6L/13A followed by the E. coli SecM arrest-peptide (20; Fig. S1), was altered to fit the requirements of this study. Synthetic oligonucleotides were obtained from MWG, coding for either one or two 8L/11A TMHs with restriction sites NcoI and EagI. These segments were ligated into respectively digested pING plasmid and ligations were confirmed by sequencing. To differentiate between the full-length and the arrested form on SDS gels, 55 residues from EGFP (residues 21–76; GenBank database accession number ACO05019.1) were introduced into the C terminus of constructs carrying the test segments with all N-terminal TMHs, resulting in a 75-aa-residue-long C terminus. Restriction sites AflII and XmaI were present in both pING plasmids [amino acid sequence: WPTGLRLS (“LS” = cTTAAGt AflII)—PGILEDER (“PG” = cCCCGGGa XmaI)] and were introduced into the EGFP fragment amplified by PCR. Ligation was carried out with subsequent sequencing to confirm successful ligation.
The respective parts of the polytopic membrane proteins from E. coli were amplified from the E. coli genome by PCR. Each forward and reverse oligonucleotide contained a KpnI and SpeI restriction site and encoded GGPG or GPGG flanks, respectively. The amplified fragments were PCR-purified, digested with the respective restriction enzymes, and ligated into KpnI/SpeI-predigested pING plasmid in the same location as the H segments used in ref. 20 [i.e., between the amino acid segments GIRLSETS (“TS” = ACTAGT SpeI) and VPGQQNAT (“VP” = GGTACCa KpnI)]. Successful cloning was confirmed by sequencing. Single TMHs were introduced by ligating preannealed and phosphorylated oligonucleotides into KpnI/SpeI-digested pING plasmid. Successful ligation was confirmed by sequencing. Mutagenesis of all constructs was done using partial overlapping oligonucleotides carrying the respective sequence exchanges (35) and was confirmed by sequencing.
The loop region between the C-terminal TMH and the N-terminal part was extended by introducing 85 amino acids from LepB (residues 92-176; GenBank accession no. AAA24064.1) by PCR. Successful insertion was confirmed by sequencing.
Protein Expression, Pulse-Labeling, Immunoprecipitation, and Detection.
E. coli MC1061 cells were transformed with the respective pING plasmids. Cells were grown overnight at 37 °C in M9 minimal medium, containing 19 amino acids (1 µg/mL) but not methionine. The medium was further supplemented with 0.4% (wt/vol) fructose to repress chimeric protein expression, 100 µg/mL thiamine, 0.1 mM CaCl2, 2 mM MgSO4 and 100 µg/mL ampicillin. Cells were diluted 1:10 and grown to an OD600 of 0.3. A total of 500 µL of cells were then shaken at 37 °C and expression was induced with 0.2% (wt/vol) arabinose for 5 min. Chimeric proteins were radiolabeled with [35S]-methionine for 2 min and the reaction was stopped by the addition of 250 µL of ice cold 50% trichloroacetic acid. After samples were incubated for 60 min on ice, they were centrifuged for 5 min and the precipitate was washed with ice cold acetone. After recentrifugation, the samples were dried at 98 °C and the precipitate was solubilized by the addition of SDS containing buffer [10 mM Tris⋅Cl pH 7.5, 2% (wt/vol) SDS] at 98 °C by three cycles of vortexing and heating.
Insoluble material was removed by centrifugation. The supernatant was diluted below the critical micellar concentration of SDS and used for immunoprecipitation using Pansorbin (Calbiochem) and LepB antisera (rabbit). After incubation at 4 °C overnight, samples were centrifuged and the immunoprecipitated samples were washed with buffer containing 10 mM Tris⋅Cl pH 7.5, 150 mM NaCl, 2 mM EDTA, and 0.2% triton X-100. After recentrifugation, the immunoprecipitated sample was solubilized in SDS sample buffer and used for SDS/PAGE. Labeled protein bands were detected by exposing film plates to the dried gel and by scanning in a Fuji FLA-3000 phosphorimager. Intensity profiles were obtained by using Fujifilm ImageGauge software and quantifications were done with EasyQuant software. All structures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.3; Schrödinger, LLC.) and ∆G predictions were obtained with the delta-G predictor (http://dgpred.cbr.su.se) (18).
Supplementary Material
Acknowledgments
We thank Nurzian Ismail, Rickard Hedman, and Nina Schiller for helpful comments. This work was supported by grants from the European Research Council (ERC-2008-AdG 232648), the Swedish Foundation for Strategic Research, the Swedish Research Council, and by a research fellowship (CY 74/1-1) from the Deutsche Forschungsgemeinschaft (to F.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306787110/-/DCSupplemental.
References
- 1.Allen SJ, Curran AR, Templer RH, Meijberg W, Booth PJ. Controlling the folding efficiency of an integral membrane protein. J Mol Biol. 2004;342(4):1293–1304. doi: 10.1016/j.jmb.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 2.Gorzelle BM, et al. Reconstitutive refolding of diacylglycerol kinase, an integral membrane protein. Biochemistry. 1999;38(49):16373–16382. doi: 10.1021/bi991292n. [DOI] [PubMed] [Google Scholar]
- 3.Anbazhagan V, Cymer F, Schneider D. Unfolding a transmembrane helix dimer: A FRET study in mixed micelles. Arch Biochem Biophys. 2010;495(2):159–164. doi: 10.1016/j.abb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Ridge KD, Lee SSJ, Yao LL. In vivo assembly of rhodopsin from expressed polypeptide fragments. Proc Natl Acad Sci USA. 1995;92(8):3204–3208. doi: 10.1073/pnas.92.8.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debnath D, Nielsen KL, Otzen DE. In vitro association of fragments of a beta-sheet membrane protein. Biophys Chem. 2010;148(1-3):112–120. doi: 10.1016/j.bpc.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Bibi E, Kaback HR. In vivo expression of the lacY gene in two segments leads to functional lac permease. Proc Natl Acad Sci USA. 1990;87(11):4325–4329. doi: 10.1073/pnas.87.11.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid DW, Nicchitta CV. Primary role for endoplasmic reticulum-bound ribosomes in cellular translation identified by ribosome profiling. J Biol Chem. 2012;287(8):5518–5527. doi: 10.1074/jbc.M111.312280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedin LE, et al. Membrane insertion of marginally hydrophobic transmembrane helices depends on sequence context. J Mol Biol. 2010;396(1):221–229. doi: 10.1016/j.jmb.2009.11.036. [DOI] [PubMed] [Google Scholar]
- 9.Meindl-Beinker NM, Lundin C, Nilsson I, White SH, von Heijne G. Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep. 2006;7(11):1111–1116. doi: 10.1038/sj.embor.7400818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buck TM, Wagner J, Grund S, Skach WR. A novel tripartite motif involved in aquaporin topogenesis, monomer folding and tetramerization. Nat Struct Mol Biol. 2007;14(8):762–769. doi: 10.1038/nsmb1275. [DOI] [PubMed] [Google Scholar]
- 11.Calamia J, Manoil C. Membrane protein spanning segments as export signals. J Mol Biol. 1992;224(3):539–543. doi: 10.1016/0022-2836(92)90542-r. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, et al. Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc Natl Acad Sci USA. 2007;104(20):8263–8268. doi: 10.1073/pnas.0611007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bañó-Polo M, et al. Charge pair interactions in transmembrane helices and turn propensity of the connecting sequence promote helical hairpin insertion. J Mol Biol. 2013;425(4):830–840. doi: 10.1016/j.jmb.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450(7170):663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 15.White SH, von Heijne G. How translocons select transmembrane helices. Annu Rev Biophys. 2008;37:23–42. doi: 10.1146/annurev.biophys.37.032807.125904. [DOI] [PubMed] [Google Scholar]
- 16.Egea PF, Stroud RM. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc Natl Acad Sci USA. 2010;107(40):17182–17187. doi: 10.1073/pnas.1012556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadlish H, Pitonzo D, Johnson AE, Skach WR. Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat Struct Mol Biol. 2005;12(10):870–878. doi: 10.1038/nsmb994. [DOI] [PubMed] [Google Scholar]
- 18.Hessa T, et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450(7172):1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 19.Chin CN, von Heijne G. Charge pair interactions in a model transmembrane helix in the ER membrane. J Mol Biol. 2000;303(1):1–5. doi: 10.1006/jmbi.2000.4122. [DOI] [PubMed] [Google Scholar]
- 20.Hermansson M, von Heijne G. Inter-helical hydrogen bond formation during membrane protein integration into the ER membrane. J Mol Biol. 2003;334(4):803–809. doi: 10.1016/j.jmb.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Ismail N, Hedman R, Schiller N, von Heijne G. A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat Struct Mol Biol. 2012;19(10):1018–1022. doi: 10.1038/nsmb.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakatogawa H, Ito K. The ribosomal exit tunnel functions as a discriminating gate. Cell. 2002;108(5):629–636. doi: 10.1016/s0092-8674(02)00649-9. [DOI] [PubMed] [Google Scholar]
- 23.Nakatogawa H, Ito K. Secretion monitor, SecM, undergoes self-translation arrest in the cytosol. Mol Cell. 2001;7(1):185–192. doi: 10.1016/s1097-2765(01)00166-6. [DOI] [PubMed] [Google Scholar]
- 24.Butkus ME, Prundeanu LB, Oliver DB. Translocon “pulling” of nascent SecM controls the duration of its translational pause and secretion-responsive secA regulation. J Bacteriol. 2003;185(22):6719–6722. doi: 10.1128/JB.185.22.6719-6722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulze S, Köster S, Geldmacher U, Terwisscha van Scheltinga AC, Kühlbrandt W. Structural basis of Na(+)-independent and cooperative substrate/product antiport in CaiT. Nature. 2010;467(7312):233–236. doi: 10.1038/nature09310. [DOI] [PubMed] [Google Scholar]
- 26.Hunte C, et al. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435(7046):1197–1202. doi: 10.1038/nature03692. [DOI] [PubMed] [Google Scholar]
- 27.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 2002;296(5570):1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 30.Law CJ, et al. Salt-bridge dynamics control substrate-induced conformational change in the membrane transporter GlpT. J Mol Biol. 2008;378(4):828–839. doi: 10.1016/j.jmb.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelman DM, et al. Membrane protein folding: Beyond the two stage model. FEBS Lett. 2003;555(1):122–125. doi: 10.1016/s0014-5793(03)01106-2. [DOI] [PubMed] [Google Scholar]
- 32.Rychkova A, Mukherjee S, Bora RP, Warshel A. Simulating the pulling of stalled elongated peptide from the ribosome by the translocon. Proc Natl Acad Sci USA. 2013;110(25):10195–10200. doi: 10.1073/pnas.1307869110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7(4):1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Gerstein M, Engelman DM. Transmembrane protein domains rarely use covalent domain recombination as an evolutionary mechanism. Proc Natl Acad Sci USA. 2004;101(10):3495–3497. doi: 10.1073/pnas.0307330101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32(14):e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.