

Significance
Torso-like (Tsl) is the sole Drosophila member of the membrane attack complex/perforin-like protein superfamily, generally known for pore-forming function and immune defence roles. Tsl, however, has a well-characterized developmental role in controlling activation of the receptor tyrosine kinase Torso (Tor) to achieve patterning of the termini of the early embryo. Here we report that the second known role of Tor, as the receptor for the hormone that induces metamorphosis, does not require Tsl. Instead, we find that Tsl controls developmental timing and growth independently of Tor. We conclude that Tsl plays a broader than expected role during development and is not merely a specialized cue for Tor signaling.
Keywords: MACPF, growth rate, ecdysis, heterochrony
Abstract
Activation of the Drosophila receptor tyrosine kinase Torso (Tor) only at the termini of the embryo is achieved by the localized expression of the maternal gene Torso-like (Tsl). Tor has a second function in the prothoracic gland as the receptor for prothoracicotropic hormone (PTTH) that initiates metamorphosis. Consistent with the function of Tor in this tissue, Tsl also localizes to the prothoracic gland and influences developmental timing. Despite these commonalities, in our studies of Tsl we unexpectedly found that tsl and tor have opposing effects on body size; tsl null mutants are smaller than normal, rather than larger as would be expected if the PTTH/Tor pathway was disrupted. We further found that whereas both genes regulate developmental timing, tsl does so independently of tor. Although tsl null mutants exhibit a similar length delay in time to pupariation to tor mutants, in tsl:tor double mutants this delay is strikingly enhanced. Thus, loss of tsl is additive rather than epistatic to loss of tor. We also find that phenotypes generated by ectopic PTTH expression are independent of tsl. Finally, we show that a modified form of tsl that can rescue developmental timing cannot rescue terminal patterning, indicating that Tsl can function via distinct mechanisms in different contexts. We conclude that Tsl is not just a specialized cue for Torso signaling but also acts independently of PTTH/Tor in the control of body size and the timing of developmental progression. These data highlight surprisingly diverse developmental functions for this sole Drosophila member of the perforin-like superfamily.
Terminal patterning in the Drosophila embryo involves secretion of the membrane attack complex/perforin-like (MACPF) protein Torso-like (Tsl) from specialized follicle cells at the anterior and posterior ends of the oocyte into the perivitelline space (1–3). Following its secretion, Tsl remains at the embryo poles through association with the vitelline membrane (4–6). Through a poorly understood mechanism that involves the eggshell proteins fs(1)Nasrat, fs(1)Polehole, and Closca, Tsl likely permits localized activation of the cysteine knot-like growth factor Trunk (Trk), possibly by proteolytic cleavage (7, 8). Activated Trk then binds to Tor and activates signaling at the embryo poles (9).
Tsl is the only MACPF-like protein that can be identified in the Drosophila genome (10). The majority of MACPF proteins characterized to date play roles in pore formation in mammalian immunity (including perforin itself and Complement C9) or in bacterial pathogenesis (11, 12). Currently, it is unclear how Tsl functions at the embryo poles, and a simple pore forming function is difficult to reconcile with a central role in activation of the Tor-signaling pathway.
Tor has a second major developmental role, in the prothoracic gland (PG), where it functions as the receptor for prothoracicotropic hormone (PTTH), a brain-derived neuropeptide hormone required for initiation of metamorphosis (13). Recently, it was shown that tsl is also expressed in the PG and that RNAi knockdown of tsl results in a significant (48 h) developmental delay (14). Given that PTTH and Trk belong to the same superfamily of cysteine knot-like growth factors (13), it seemed likely that Tsl plays a role in Tor activation in the PG. Here we confirm that loss of tsl leads to a delay in development. Remarkably, however, we discover that Tsl regulates body size and developmental timing independently of Tor. Our results show that Tsl has diverse functions, some of which are independent of Torso, and is thus not just a specialized cue for Torso signaling.
Results
Torso-Like Null Mutants Have a Developmental Delay.
Previous studies have shown that RNAi knockdown of torso (tor) or tsl specifically in the PG results in an extended developmental period and delayed metamorphosis (13, 14). To begin dissecting the role of Tsl in the PG, and to understand its functional relationship with Tor, we first tested whether loss of tsl in our hands caused a developmental delay phenotype. As available stocks carrying tsl mutant alleles are likely hypomorphic and/or have additional genetic mutations, we used ends-out gene replacement to generate a tsl null mutant allele (tslΔ) in which the entire coding region is removed (Fig. S1 A and B). tslΔ homozygotes are viable and females are sterile, laying embryos that fail to specify terminal cell fate (Fig. S1C).
Developmental timing analyses of tslΔ homozygotes revealed a significant delay of 26 h to reach pupariation compared with heterozygous controls (Fig. 1A, P < 0.001). This length of delay is less than but consistent with that observed by Grillo et al. (14) using RNAi. To ensure that the tslΔ delay was solely due to loss of tsl activity, we showed that it could be rescued by ubiquitous expression of a UAS-tsl transgene in the tslΔ background (Fig. 1A, P < 0.001). In further support, we found that transheterozygotes carrying the tslΔ allele and a strongly hypomorphic allele of tsl, tsl5, also exhibited a developmental delay (Fig. S2).
Fig. 1.

tsl functions in the PG independently of tor to regulate developmental timing. (A) tslΔ homozygotes pupariate ∼1 d later than controls. Weak ubiquitous expression (da-Gal4) of a UAS-tsl transgene results in a significant reduction (∼10 h, P < 0.001) in developmental delay compared with tslΔ. (B) torXR1 and tslΔ larvae exhibit a similar delay to pupariation compared with controls and a similar delayed transition from the L2 to the L3 stage. (C) Larvae deficient for both tor and tsl display a greatly extended delay in time to pupariation (83 h) compared with controls (P < 0.001). h AEL, hours after egg lay. Error bars represent ±1 SEM for all graphs. ***P < 0.001, **P < 0.01 from two-tailed t tests in all cases. n = 6 or greater for all means with no fewer than 52 individuals tested for each genotype.
The tor null allele torXR1 was previously shown to have a delay to pupariation of ∼30 h (13); however, as developmental timing depends on many extrinsic conditions (e.g., temperature, media type), it was important to compare it to the tsl mutant under our laboratory conditions. When we compared homozygotes for torXR1 and for tslΔ, we found that they exhibited a similarly lengthened developmental period (P = 0.065, Fig. 1B). Furthermore, both tslΔ and torXR1 homozygotes displayed a similar (P = 0.760) L2–L3 transition delay of 7 h (P = 0.006 for torXR1; not significant for tslΔ).
Torso-Like Acts Independently of Torso in Developmental Timing and Body Size.
We next asked whether tsl exerts its effects on developmental timing by acting through the tor pathway or whether tsl has a role separate from that of tor in developmental timing. To address this, we tested torXR1; tslΔ double mutants to assess whether the delay caused by loss of tsl was epistatic or additive to loss of tor. We found that the double mutants displayed a drastically extended larval period, pupating 83 h after the controls (P < 0.001, Fig. 1C), and significantly later than either torXR1 or tslΔ alone (both P < 0.001). Thus, tsl and tor show additive effects on developmental timing, indicating different roles.
In further support of tsl having a developmental role that is independent of tor, we also observed a distinct phenotypic difference between tsl and tor mutant adults. Loss of tor or ablation of PTTH-producing neurons results in larger-than-normal adults due to prolonged duration of feeding as larvae (13, 15). Intriguingly, despite the developmental delay observed for tsl null mutants, adults are not larger as would be expected if the PTTH/Tor pathway had been perturbed. Instead, tslΔ homozygotes are 28% smaller than controls (P < 0.001, Fig. 2 A and C, Inset). torXR1; tslΔ double-mutant flies are also smaller than normal (P < 0.001) and similar in size to tslΔ alone (P = 0.076), indicating that for this phenotype tsl is epistatic to tor. Furthermore, tslΔ larvae are notably smaller than heterozygous controls when the controls have reached the wandering stage (Fig. 2B). Indeed, tslΔ larvae measured throughout their entire third instar stage revealed a severe growth rate defect (ANCOVA F1,11 = 8.55, P = 0.014, Fig. S3), likely initiated in the preceding larval stages.
Fig. 2.
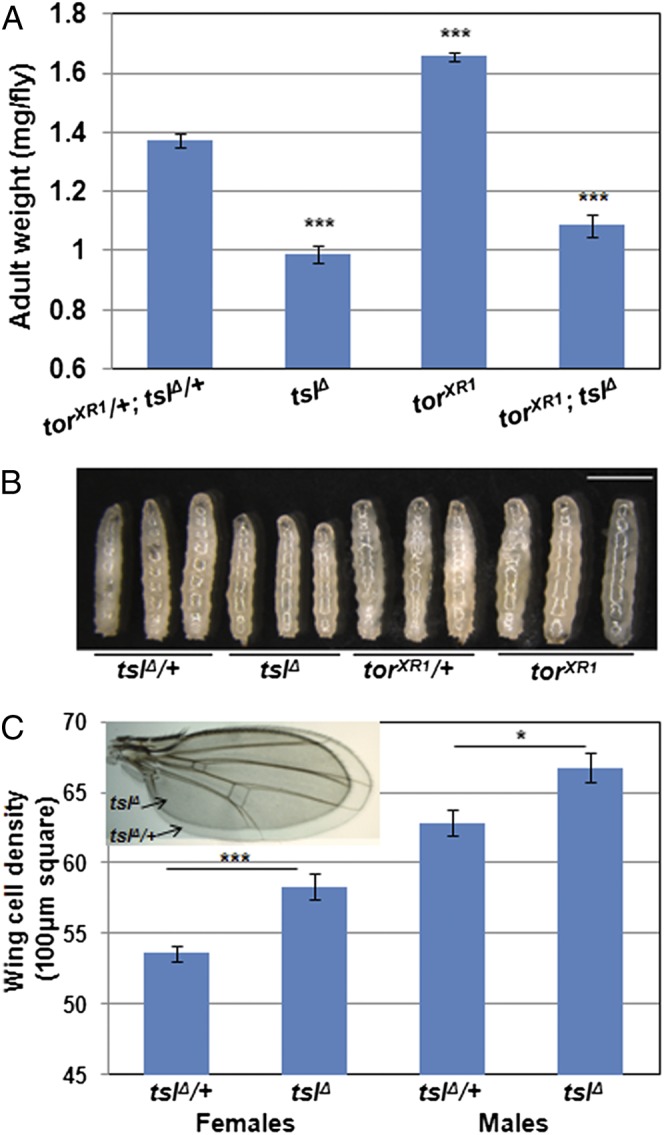
tsl and tor have opposing effects on body size. (A) tslΔ adult flies weigh 28% less than controls (P < 0.001) and 40% less than torXR1 individuals. Flies mutant for both genes weigh the same as flies lacking tsl alone (21% reduction compared with control, P = 0.08 compared with tslΔ). Note that only females were tested here due to insufficient recovery of adult males mutant for both genes; however, effects of tor and tsl on male weight were highly consistent with the female data. n = 5 or greater for each genotype with more than 17 flies weighed per genotype. (B) Third-instar larvae at 127 h AEL. At this stage, control larvae have begun wandering but tsl and tor mutants remain feeding. tslΔ larvae are notably reduced in size at this point, suggesting that tsl is acting earlier than tor to regulate growth (Scale bar, 2 mm.) (C) Wing-cell density is significantly higher in tslΔ flies compared with controls, indicating that cell size is reduced in tslΔ wings [tslΔ wings are smaller than the control (Inset)]. n = 10 for all wing-cell density measurements for each genotype. Error bars represent ±1 SEM for all graphs. ***P < 0.001, *P < 0.05 from two-tailed t tests in all cases.
To determine whether the reduced size is due to loss of cells or to smaller cells, we counted wing-hair density in adults. These results showed that the decreased body size of tslΔ is due to a reduction in cell size (females, P < 0.001; males, P = 0.011; Fig. 2C). This contrasts with results obtained from manipulation of PTTH, which affects cell number rather than size (15). Taken together, these data indicate that the tor and tsl mutant delays are likely to have separate mechanistic bases and suggest an earlier and tor-independent role for tsl in growth regulation.
To try and determine if the PG is the site of action of tsl in control of our observed developmental timing and body size defects, we expressed a tsl transgene specifically in the PG using phm-Gal4. This was unable to rescue the developmental delay or small size of tslΔ mutants (Fig. S4 A and B). We also did not observe a significant delay or small size phenotype when we knocked down tsl in the PG using RNAi (Fig. S4 C and D). However, driving RNAi constructs with the strong ubiquitous driver Actin-Gal4 also did not produce the delay or body-size difference (Fig. S4 E and F); thus in our hands the RNAi constructs are unable to achieve sufficient knockdown to produce tsl loss-of-function phenotypes. From these results we cannot conclude if the observed phenotypes are due to tsl expression in the PG or a different tissue.
Torso-Like Is Not Required for Ectopic PTTH Function.
In terminal patterning, Tsl is thought to mediate conversion of Trk into a form capable of activating Tor signaling (7, 8, 16). Similarities between the Trk and PTTH sequences, including in the relative positions of putative cleavage sites, have led to the suggestion that PTTH might also require Tsl function for its activity (13, 14). As our results for tor;tsl double mutants indicated otherwise, we explored this further. We reasoned that, if Tsl is acting independently of PTTH/Tor signaling in controlling developmental timing and body size, then any effects of PTTH overexpression might not be suppressed by removal of Tsl. To this end, we drove expression of a UAS-ptth transgene directly in the PG using phm-Gal4. This resulted in a marked advancement in the time of pupariation by 19 h compared with the control, consistent with previous observations (15) (P < 0.001, Fig. 3A). Removal of tsl did not restore normal developmental timing (P < 0.001). Furthermore, the reduction in adult size caused by ectopic PTTH expression was enhanced, rather than suppressed, by removal of tsl (females, P = 0.003; males, P < 0.001; Fig. 3B).
Fig. 3.

Ectopic PTTH activity is independent of tsl. (A) Overexpression of PTTH in the prothoracic gland (phm-Gal4) results in a decreased time to pupariation (19 h less than the control, P < 0.001; 35 h less than tslΔ, P < 0.001). Removal of tsl under these conditions marginally slowed pupariation (P = 0.034) but not to close to normal or tslΔ developmental timing (P = 0.034). Means are calculated from n = 10 with a minimum of 108 individuals tested per genotype. (B) Overexpression of PTTH using phm-GAL4 gives a strong reduction in adult weight in males that is further reduced by loss of tsl (all means significantly different from each other; P < 0.001). Females followed a similar but nonsignificant trend due to overexpression of PTTH having little affect on adult weight. n = 7 or greater with at least 28 flies weighed per genotype. (C) Overexpression of Trk in the germ line (nos-Gal4) has no effect on terminal patterning because Trk requires Tsl activity, which is present only at the poles. PTTH overexpression, however, results in a grossly disrupted embryonic cuticle due to ubiquitous Tor signaling throughout the whole embryo, which is also shown by the greatly expanded tailless expression. This is unaffected by removal of tsl. Anterior is to the left and maternal genotypes are as indicated. Images are representatives of more than 60 embryos in each case. Error bars represent ±1 SEM for all graphs. ***P < 0.001, **P < 0.01, *P < 0.05 from two-tailed t tests in all cases.
In addition, because PTTH has been shown to be able to act in place of Trk if it is artificially expressed in the early embryo (13), we asked whether maternal Tsl is required for this action of PTTH. We drove expression of PTTH in the germ line (Gal4:VP16-nos.UTR) and observed ubiquitous over-activation of the Tor pathway (Fig. 4C) as indicated by near ubiquitous tailless (tll) transcription (normally restricted to precise domains at the poles of the embryo). This phenotype suggests that, unlike Trk, PTTH is able to activate Tor in the embryo without a requirement for Tsl, which here is expressed only at the embryo poles. We note that in a previous study overexpression of PTTH in the germ line did not result in Tor over-activation (13); however, this study used a construct in the pUAST vector, and we alternatively used the pUASp vector, which expresses far more strongly in the germ line (17).
Fig. 4.

A tagged form of Tsl functions in developmental timing but not in terminal patterning. The developmental delay (A) and reduced size (B) of tslΔ homozygotes was rescued by the HA:tsl construct (P < 0.001 for delay, P < 0.01 for female size, and P < 0.05 for males all compared with tslΔ). HA:tsl does not rescue the tsl maternal null terminal phenotype (C). Note that structures posterior to abdominal segment 8, including the filzkorper (arrowhead), are absent in larval cuticles, and posterior tll expression is absent in early embryos from HA:tsl, tslΔ mothers. Pupariation times are means representative of n = 8 or greater with at least 65 individuals scored for each genotype. Means for adult weight are calculated from n = 6 or greater with no fewer the 24 flies weighed for each genotype. Error bars represent ±1 SEM for all graphs. ***P < 0.001, **P < 0.01, *P < 0.05 from two-tailed t tests in all cases.
Tagged Form of Tsl Rescues the Developmental Delay and Small Size but Not Terminal Patterning.
Finally, in further support of Tsl having different modes of action in patterning and developmental timing, we observed that a Tsl genomic rescue construct that carries ∼3 kb of promoter and the tsl coding sequence tagged with three tandem hemagglutinin (HA) epitopes at the N terminus (HA:tsl) (4) can completely rescue the developmental delay and reduced size of the tslΔ mutation (Fig. 4 A and B), but intriguingly does not rescue the terminal patterning defect (Fig. 4C). The lack of rescue of the terminal patterning defect is not due to incorrect localization of the tagged Tsl protein, as we and others have confirmed that the HA:Tsl protein is indeed secreted into the perivitelline space (4). We therefore reason that the HA tag is disrupting the mechanism of action of Tsl in the early embryo but not in the developing larva, which again is indicative of different protein activities in different tissues.
Discussion
Our observations define a role for torso-like in the regulation of developmental timing and body size that is independent of the Tor pathway. The recent discoveries that (i) tor encodes the receptor for PTTH (13) and that (ii) tsl is also expressed in the PG (14) prompted us to investigate whether Tsl plays a role in the activation of PTTH similar to the one it plays for Trk in the embryo. Although tsl is similar to tor in terms of expression in the PG and in one aspect of its larval development mutant phenotype (13, 14, and herein), here we show that these similarities are coincidental rather than indicative of cooperative function.
Importantly, the data presented here show that the genes required for activation of Tor in the early embryo are not used in its activation in the PG. We and others (13, 15) have shown that ectopic expression of PTTH in cells that do not normally produce it, in the early embryo and the PG, results in active PTTH that is capable of activating Tor. These data suggest that these cells produce all proteins necessary for activation of PTTH. In contrast, ectopic Trk driven in the PG (where it is not normally produced) has no effect on developmental timing or adult size despite expression of Tsl in this tissue (14). Taken together, we conclude that the activation requirements of Trk and PTTH are quite different. Specifically, tsl expression is unnecessary for PTTH activity and insufficient for ectopic Trk activity.
Why would Tor be activated differently in the embryo and in the PG? We reason that the answer to this question possibly lies with differences in the two ligands. During early embryogenesis Trk is secreted in an inactive form that requires Tsl and other terminal class genes for activation (7–9). In this situation Tor activation requires spatial constraint that is achieved by restricted Tsl expression controlling spatially localized Trk activation. In contrast, because the PG is directly innervated by PTTH-producing neurons that synapse within the gland (15, 18), spatial control of Tor activation might not be necessary in this context. We hypothesize that PTTH may simply be secreted from these neurons in a form that does not require further local activation and thus does not require Tsl.
How might Tsl act in controlling body size and development timing? The tsl mutant phenotypes presented here resemble those observed when insulin signaling is reduced (19, 20). Specifically, overexpression of a dominant negative form of the insulin-like receptor (InR) causes reduced size and delayed development (21). Additionally, larvae carrying heat-sensitive alleles of InR also display developmental delays and effects of size (22). However, despite these phenotypic similarities, experiments in our laboratory have not yielded evidence for interactions between tsl and the InR pathway.
Another possibility is that tsl regulates developmental timing and growth earlier in development or in a tissue distinct from the PG. The growth rate defect that we observe in the tsl mutants may indicate that it acts earlier in development than tor. It must also be noted that our data do not implicate the PG as the source of timing and growth defects observed in tsl null mutants. Consistent with the latter possibility, we have been unable to rescue the tsl null phenotypes by specifically expressing tsl in the PG using phm-Gal4. In addition, we were unable to replicate the delay observed by Grillo et al. (14) using RNAi knockdown of tsl in the PG. As technical difficulties and experimental variations between laboratories can underlie such differences, however, the PG remains a candidate tissue given its crucial role in regulation of developmental transitions in response to nutritional inputs (23, 24). Further experiments to manipulate Tsl function in different tissues and at different times during development will be required to determine the specific tissues and pathways underlying these phenotypes. Taken together, however, our current results reveal the surprising finding that the function of Tsl in its maternal patterning role is mechanistically distinct from its zygotic role in the developing larva.
Materials and Methods
Drosophila Stocks.
The following stocks were used: w1118 (BL5905); hs-FLP hs-I-SceI (BL9634) ; ey-FLP (BL5580); da-Gal4 (BL12429); phm-Gal4 (gift from Leonie Quinn, University of Melbourne, Melbourne, originally from Michael O’Connor, University of Minnesota, Minneapolis); Gal4::VP16-nos.UTR (BL7293); torXR1, a protein null allele of torso (25); HA:tsl (gift from Jordi Casanova, IRB Barcelona, Barcelona); tsl5, a strong EMS-induced loss-of-function mutation (Y148N); UAS-tslRNAi (v14430); and ZH-96E attP (BL24487) for ΦC31 integrase-mediated germ-line transformation.
Transgenics and Generation of tsl Null Mutants.
A genomic fragment containing the coding sequence of ptth including introns and a C-terminal single HA tag was synthesized and cloned (Genscript) into pUASP to make UASP-ptth-HA. UASP-trk was similarly constructed using the full-length trk cDNA. Purified plasmid DNA was injected into w1118 embryos (BestGene Inc.) using standard P-element transformation methods (26) to yield transgenic flies. For UAS-tsl, the coding sequence of tsl was first cloned from cDNA (F-AGA TCT ATG CTG AGC GAT GCG GTC GTG CCT, R-CTC GAG CTA TCG GGT GGG ATG ACT CTG CGG) into the pGEM-T Easy vector (Promega), then sequenced, and then subcloned into pUASTattB. Transgenics were made via ΦC31 integrase-mediated transformation (27, 28) (UAS-tsl) using the ZH-96E attP landing site. For all transgenes, several independent transgenic lines were established and tested.
The tsl null mutant was generated using ends-out gene replacement (29). First, the donor plasmid pW25-tsl was built by cloning 4.2 kbp of upstream (F-AGC AGG CGC GCC TGC AAT CAG CTG TCT TGA CC, R-TAC CCG TAC GCC TTT TAA CCC ACA GCC AAA) and 2.0 kbp of downstream (F-AGC AGC GGC CGC CAT CAA GAA GGT GGC CCT TA, R-TAC CGG TAC CTC CGA CTT GCA TTT CAG CTA) sequence flanking the tsl-coding region into the appropriate sites in the ends-out transformation vector pW25. Purified pW25-tsl was injected into w1118 embryos (BestGene Inc.). The donor targeting element was mobilized by crossing to hs-FLP and hs-I-SceI and applying a heat shock for 80 min at 37 °C daily for days 3–5 of development. Mosaic- and white-eyed virgins were crossed to ey-FLP, and stable lines were established from w+ progeny (∼1/6 vials yielded a w+ individual). Lines were characterized for targeting events via PCR (Fig. S2, A-GCA ATC TGT GAG GTT CAT CTG TAA GGT CAA GAC AG, B-CGG GTA GAG CCG TTT ACC CGT TTC GAT TAC CGG TTC, C-GCA ACT GAA GGC GGA CAT TGA CGC TAT CGA CCT ATT CAG, D-CAG CTG GTA CTC CGG CAA TTG TTG TGC CC). Of the 22 w+ lines that we generated, 4 underwent gene replacement. Tsl transcriptional activity was assessed in the mutants by RT-PCR from ovary-derived cDNA using primers specific for tsl (detail above) and a control gene CyclinK (F-GAG CAT CCT TAC ACC TTT CTC CT, R-TAA TCT CCG GCT CCC ACT G).
RNA in Situ Hybridizations.
A digoxigenin-labeled (Roche) probe against tll was hybridized with 0–4 h old embryos using standard protocols (30) before imaging with differential interference contrast optics.
Cuticle Preparations.
Adults were allowed to lie on media containing apple juice supplemented with yeast paste for 24 h before being removed. Embryos developed for a further 24 h before dechorionation in 50% (vol/vol) bleach and mounting on slides in a mixture of 1:1 (vol/vol) Hoyer’s solution:lactic acid. Slides were incubated overnight at 65 °C and imaged using dark-field optics (Leica).
Phenotypic Measurements.
All stocks were maintained at 25 °C on standard media for all experiments. First-instar larvae were sorted by GFP (on balancer chromosome) 24 h following a 3-h lay into 6–10 groups of 10 or 20 individuals (depending upon experiment) per genotype. Larvae were placed into vials containing standard fly media and scored every 8 h for the time taken to reach pupariation. L2–L3 transition was scored by inspecting larval spiracle and mouthhook morphology every 3 h on three food plates, each containing 20 per genotype. Adults were sorted several days following eclosion and weighed in groups on a microbalance (Mettler Toledo). Wing-cell density was calculated from wing-hair counts within a rectangle (147 × 198 µm) averaged across two zones per wing for 10 wings per genotype. Growth rates were determined by weighing four groups of five larvae for each time point until pupariation. Linear regression was used to calculate growth rates and ANCOVA to assess whether rates were different (Prism 6.0b).
Supplementary Material
Acknowledgments
We thank James Heaney and the Australian Drosophila Biomedical Research Facility (OzDros) for technical support; Jordi Casanova for providing the N-terminally tagged Tsl construct; Marc Freeman, Peter Dearden, and Richard Burke for discussions; and two anonymous reviewers for their valuable feedback. This work was supported by the National Health and Medical Research Council of Australia (NHMRC), the Australian Research Council (ARC), and an Interdisciplinary Research Grant from Monash University. J.C.W. is an ARC Federation Fellow and an honorary NHMRC Principal Research Fellow.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309780110/-/DCSupplemental.
References
- 1.Stevens LM, Frohnhöfer H-G, Klingler M, Nüsslein-Volhard C. Localized requirement for torso-like expression in follicle cells for development of terminal anlagen of the Drosophila embryo. Nature. 1990;346(6285):660–663. doi: 10.1038/346660a0. [DOI] [PubMed] [Google Scholar]
- 2.Savant-Bhonsale S, Montell DJ. torso-like encodes the localized determinant of Drosophila terminal pattern formation. Genes Dev. 1993;7(12B):2548–2555. doi: 10.1101/gad.7.12b.2548. [DOI] [PubMed] [Google Scholar]
- 3.Martin J-R, Raibaud A, Ollo R. Terminal pattern elements in Drosophila embryo induced by the torso-like protein. Nature. 1994;367(6465):741–745. doi: 10.1038/367741a0. [DOI] [PubMed] [Google Scholar]
- 4.Jiménez G, González-Reyes A, Casanova J. Cell surface proteins Nasrat and Polehole stabilize the Torso-like extracellular determinant in Drosophila oogenesis. Genes Dev. 2002;16(8):913–918. doi: 10.1101/gad.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stevens LM, Beuchle D, Jurcsak J, Tong X, Stein D. The Drosophila embryonic patterning determinant torsolike is a component of the eggshell. Curr Biol. 2003;13(12):1058–1063. doi: 10.1016/s0960-9822(03)00379-8. [DOI] [PubMed] [Google Scholar]
- 6.Ventura G, Furriols M, Martín N, Barbosa V, Casanova J. closca, a new gene required for both Torso RTK activation and vitelline membrane integrity. Germline proteins contribute to Drosophila eggshell composition. Dev Biol. 2010;344(1):224–232. doi: 10.1016/j.ydbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Casanova J, Furriols M, McCormick C-A, Struhl G. Similarities between trunk and spätzle, putative extracellular ligands specifying body pattern in Drosophila. Genes Dev. 1995;9(20):2539–2544. doi: 10.1101/gad.9.20.2539. [DOI] [PubMed] [Google Scholar]
- 8.Casali A, Casanova J. The spatial control of Torso RTK activation: A C-terminal fragment of the Trunk protein acts as a signal for Torso receptor in the Drosophila embryo. Development. 2001;128(9):1709–1715. doi: 10.1242/dev.128.9.1709. [DOI] [PubMed] [Google Scholar]
- 9.Sprenger F, Nüsslein-Volhard C. Torso receptor activity is regulated by a diffusible ligand produced at the extracellular terminal regions of the Drosophila egg. Cell. 1992;71(6):987–1001. doi: 10.1016/0092-8674(92)90394-r. [DOI] [PubMed] [Google Scholar]
- 10.Rosado CJ, et al. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317(5844):1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 11.Hadders MA, Beringer DX, Gros P. Structure of C8α-MACPF reveals mechanism of membrane attack in complement immune defense. Science. 2007;317(5844):1552–1554. doi: 10.1126/science.1147103. [DOI] [PubMed] [Google Scholar]
- 12.Law RHP, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468(7322):447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 13.Rewitz KF, Yamanaka N, Gilbert LI, O’Connor MB. The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science. 2009;326(5958):1403–1405. doi: 10.1126/science.1176450. [DOI] [PubMed] [Google Scholar]
- 14.Grillo M, Furriols M, de Miguel C, Franch-Marro X, Casanova J. Conserved and divergent elements in Torso RTK activation in Drosophila development. Sci Rep. 2012;2:762. doi: 10.1038/srep00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBrayer Z, et al. Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Dev Cell. 2007;13(6):857–871. doi: 10.1016/j.devcel.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furriols M, Casali A, Casanova J. Dissecting the mechanism of torso receptor activation. Mech Dev. 1998;70(1–2):111–118. doi: 10.1016/s0925-4773(97)00181-0. [DOI] [PubMed] [Google Scholar]
- 17.Rørth P. Gal4 in the Drosophila female germline. Mech Dev. 1998;78(1–2):113–118. doi: 10.1016/s0925-4773(98)00157-9. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh A, McBrayer Z, O’Connor MB. The Drosophila gap gene giant regulates ecdysone production through specification of the PTTH-producing neurons. Dev Biol. 2010;347(2):271–278. doi: 10.1016/j.ydbio.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nijhout HF. The control of body size in insects. Dev Biol. 2003;261(1):1–9. doi: 10.1016/s0012-1606(03)00276-8. [DOI] [PubMed] [Google Scholar]
- 20.Edgar BA. How flies get their size: Genetics meets physiology. Nat Rev Genet. 2006;7(12):907–916. doi: 10.1038/nrg1989. [DOI] [PubMed] [Google Scholar]
- 21.Slack C, Giannakou ME, Foley A, Goss M, Partridge L. dFOXO-independent effects of reduced insulin-like signaling in Drosophila. Aging Cell. 2011;10(5):735–748. doi: 10.1111/j.1474-9726.2011.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shingleton AW, Das J, Vinicius L, Stern DL. The temporal requirements for insulin signaling during development in Drosophila. PLoS Biol. 2005;3(9):e289. doi: 10.1371/journal.pbio.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Layalle S, Arquier N, Léopold P. The TOR pathway couples nutrition and developmental timing in Drosophila. Dev Cell. 2008;15(4):568–577. doi: 10.1016/j.devcel.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Mirth CK, Truman JW, Riddiford LM. The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr Biol. 2005;15(20):1796–1807. doi: 10.1016/j.cub.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Sprenger F, Stevens LM, Nüsslein-Volhard C. The Drosophila gene torso encodes a putative receptor tyrosine kinase. Nature. 1989;338(6215):478–483. doi: 10.1038/338478a0. [DOI] [PubMed] [Google Scholar]
- 26.Rubin GM, Spradling AC. Vectors for P element-mediated gene transfer in Drosophila. Nucleic Acids Res. 1983;11(18):6341–6351. doi: 10.1093/nar/11.18.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166(4):1775–1782. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA. 2007;104(9):3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gong WJ, Golic KG. Ends-out, or replacement, gene targeting in Drosophila. Proc Natl Acad Sci USA. 2003;100(5):2556–2561. doi: 10.1073/pnas.0535280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomancak P, et al. (2002) Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol 3(12): research0088. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.