

Building on work by Otto Frank in the late 19th century, Ernest Starling described in the early 20th century the ability of the heart to change its force of contraction and therefore stroke volume in response to changes in venous return, which has been called the Frank-Starling law of the heart in honor of these two physiologists. The Frank-Starling effect enables the heart to match cardiac output to venous return on a beat-to-beat basis.1 A major mechanism responsible for the Frank-Starling effect and hence the beat-to-beat autoregulation of cardiac output is the sarcomere length-dependent activation of force development.2
In this issue of Circulation Research, Sequeira and coworkers3 investigate both the Ca and length-dependent regulation of force development in cardiomyocytes harvested from patients with Hypertrophic Cardiomyopathy (HCM), a familial disorder commonly associated with mutations in genes encoding sarcomeric proteins.4 Consistent with previous animal and human studies5, myofilament Ca sensitivity was increased in all 38 HCM patient samples compared to 12 non-failing donor hearts. Notable, the authors observed that length-dependent activation was impaired, which, unlike the Ca sensitivity increase, appeared to be a primary effect of the mutant sarcomeric proteins, because 1), it could not be rescued by increasing protein kinase A (PKA) phosphorylation, and 2), was normalized when mutant troponin proteins were replaced with wild-type protein. These new results suggest that impaired length dependent activation and hence a defective Frank-Starling effect importantly contributes to the pathogenesis of HCM.
Reduced maximal Ca-activated force and increased Ca sensitivity are central findings in human HCM
HCM is a familial disorder commonly associated with mutations in genes encoding sarcomeric proteins. Mutation carriers develop progressive heart hypertrophy and fibrosis, and are at a high risk for sudden death due to ventricular arrhythmia. More than 1000 mutations in >11 genes have been identified that can lead to this disease. The current report3 together with two previous smaller human studies6, 7 indicates that regardless of the disease-causing mutation, human HCM cardiac muscle exhibits reduced maximal Ca-activated force and increased Ca sensitivity of muscle activation. This result raises the possibility that HCM mutations trigger a common cascade of molecular events that result in a similar phenotype (Figure 1).
Figure 1. Pathophysiological framework of HCM.
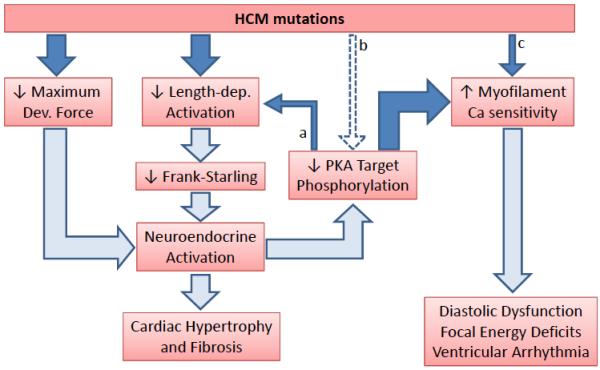
Solid arrows indicate experimental findings reported by Sequeira and coworkers. Open arrows indicate the consequences based on experimental evidence from other studies. (a) Impaired length-dependent activation can also be caused by PKA hypophosphorylation (i.e., of TnI). (b) HCM mutations may directly interfere with phosphorylation of myofilament PKA targets. (c) Only a subset of HCM mutations directly alter myofilament Ca sensitivity.
Based largely on in vitro studies and animal models, myofilament Ca sensitization had previously been associated with HCM (reviewed in8, 9). The study by Sequeira et al.3 suggests that myofilament Ca sensitization indeed is a universal finding in human HCM. However, the authors find that Ca sensitization is often not a primary property conferred by the mutant protein itself. Rather, the increased Ca sensitivity was largely due to an apparent hypophosphorylation of PKA targets in the myofilaments: Although all HCM groups had higher myofilament Ca sensitivity at baseline, in tissue from mutation carriers in myosin binding protein C (MyBPC), troponin I (TnI), tropomyosin and from HCM patients without sarcomeric mutations, Ca sensitivity was restored to normal levels by treatment with PKA. The two exceptions were one patient with a mutation in TnT and another patient with a βMHC mutation, where the Ca sensitivity increase appeared to be a property directly conferred by the mutant protein. As with any patient study, differences in drug therapy between the HCM and the normal donor group may have contributed to the reduced PKA phosphorylation. Nevertheless, this result suggests that the increased Ca sensitivity of human HCM muscle is mostly the result of post-translational modifications, and potentially helps to maintain systolic function despite the reduction in maximal Ca-activated force caused by the HCM mutation (Figure 1).7 Regardless of the underlying cause, the Ca sensitivity increase is likely of major importance for HCM pathophysiology, with experimental evidence indicating that regardless of how myofilaments are Ca sensitized, an increase in myofilament Ca sensitivity can contribute to diastolic dysfunction,10 focal energy deficits11 and arrhythmogenesis (Figure 1).12
Impaired length-dependent activation – a trigger of HCM pathogenesis?
The authors discovered a new common finding in human HCM muscle – impaired length-dependent activation, which, in contrast to increased Ca sensitivity, remained after PKA treatment for all sarcomeric missense mutations studied. Furthermore, replacement of mutant with wild-type troponin corrected the impaired length dependence in the two mutations that could be studied. Hence, impaired length-dependent activation may be a direct effect of many mutations that cause HCM in humans (Figure 1). What is the significance of this finding for the pathogenesis of HCM? Since a functional length-dependent activation is responsible for the Frank-Starling effect, it is intriguing to speculate that HCM mutations generate an intermittent inability to match cardiac ejection to dynamic changes in ventricular filling volume (i.e., during the respiratory cycle or during exercise). The resulting increase in wall stress due to intermittent impaired ventricular emptying could trigger neuroendocrine activation and a hypertrophic gene program analogous to acquired heart disease with increased wall stress and an impaired Frank-Starling effect such as post-MI cardiomyopathy. While not tested directly in the report by Sequeira, activation of neuroendocrine signaling could be the main culprit for the decrease in PKA phosphorylation of myofilament target proteins found universally in HCM patients (Figure 1). In addition, some of missense HCM mutation (i.e., in TnT or TnI) may directly interfere with PKA’s ability to phosphorylate TnI (Figure 1, dashed arrow b). PKA phosphorylation of TnI not only regulates myofilament Ca sensitivity, but also is important for physiological length-dependent activation of cardiac muscle.13 Hence, PKA hypophosphorylation can independently contribute to impaired length-dependent activation, as was demonstrated by the authors for two groups of HCM patients (HCM due to truncation mutations of MyBPC and genotype-negative HCM). In those patients, this can generate a vicious cycle that could be central to the pathogenesis in HCM, as shown in Figure 1.
In addition to the impaired length-dependent activation, which is measured experimentally by the increase in Ca sensitivity (ΔpCa50) induced by increased sarcomere length (Sequeira et al, Figure 2), all sarcomeric missense mutation (but not MyBPC truncation mutants and genotype-negative HCM) also impaired the length-dependent increased in maximum force (Sequeira et al, Table 2). The latter is measured at fully-activating [Ca] and reflects the intrinsic force generating ability of the muscle. While there can be many reasons for the reduced length dependence of maximum force (e.g., loss of myofibrils), the net result is an even further compromised length-tension relationship and hence impaired Frank Starling effect.
Open questions
A number of questions remain. Given that genetic background might affect the functional effect of the mutations and many of the individuals studied by Sequeira et al. were family members, the results of the study may not be quite generalizable to the general HCM population. By necessity, all human myocardial samples came from patients with significant cardiac hypertrophy and/or heart failure. Hence, it is still possible that the altered length-dependent activation is the consequence of pathological remodeling of the sarcomeric apparatus in the diseased myocardium. Nevertheless, the rescue with wild-type troponin protein replacement provides compelling evidence that this is not the case, at least for two specific mutations. Furthermore, passive viscoelastic properties of human HCM cardiomyocytes are similar to those of cardiomyocytes from healthy donor hearts,7 further supporting the hypothesis that altered Ca and length dependent regulation of muscle activation are central to the pathophysiology of human HCM (Figure 1). Not directly investigated in the current study, but likely also important for HCM pathophysiology is the inefficient ATP utilization (increased “tension cost”) caused by many HCM mutations.14 It remains to be seen which of these myofilament properties is the most significant driver of the disease. However, altered myofilament properties affect every myocyte in the heart starting at birth and while some heterogeneity is expected, there are significant questions remaining. For example, what is the reason for the incomplete penetrance of the disease (age dependent, but possibly as low as 41%15)? What is responsible for the focal nature of hypertrophy and fibrosis in HCM hearts? This clearly shows that the presence of a HCM mutation does not inevitably lead to disease, and even in an affected patient some regions of the heart remain normal. Such a phenotype may imply a “second hit” that is necessary to initiate the disease and remains to be identified. A potential culprit is the focal energy deprivation that occurs in HCM mouse models during stress,11 which can directly contribute to reentry arrhythmias,11 but also has the potential to drive heterogeneous hypertrophy and remodeling.16 The next frontier will be to test whether therapeutic interventions aimed at normalizing Ca sensitivity, length-dependent activation or inefficient ATP utilization are beneficial in HCM.
Acknowledgments
SOURCES OF FUNDING
This work was supported in part by the US National Institutes of Health grants HL88635 (to BCK) and HL71670 (to BCK) and by the American Heart Association Scientist Development Grant 10SDG2640109 (to SH).
Footnotes
DISCLOSURES
NONE
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Markwalder J, Starling EH. On the constancy of the systolic output under varying conditions. J Physiol. 1914;48:348–356. doi: 10.1113/jphysiol.1914.sp001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Tombe PP, Mateja RD, Tachampa K, Ait Mou Y, Farman GP, Irving TC. Myofilament length dependent activation. J Mol Cell Cardiol. 2010;48:851–858. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas-Paalberends R, Regan JA, Boontje N, Ten Cate F, Germans T, Carrier L, Sadayappan S, van Slegtenhorst M, Zaremba R, Foster DB, Murphy A, Poggesi C, Dos Remedios CG, Stienen GJ, Ho CY, Michels M, van der Velden J. Perturbed Length-Dependent Activation in Human Hypertrophic Cardiomyopathy with Missense Sarcomeric Gene Mutations. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.111.300436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011;108:743–750. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huke S, Knollmann BC. Increased myofilament Ca2+-sensitivity and arrhythmia susceptibility. J Mol Cell Cardiol. 2010;48:824–833. doi: 10.1016/j.yjmcc.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- 7.Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh S, Avkiran M, Watkins H, Redwood C, Marston SB, Kentish JC. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2010;49:737–745. doi: 10.1016/j.yjmcc.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura A. Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J Hum Genet. 2010;55:81–90. doi: 10.1038/jhg.2009.138. [DOI] [PubMed] [Google Scholar]
- 9.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2012;9:91–100. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- 10.Sirenko SG, Potter JD, Knollmann BC. Differential effect of troponin T mutations on the inotropic responsiveness of mouse hearts--role of myofilament Ca2+ sensitivity increase. J Physiol. 2006;575:201–213. doi: 10.1113/jphysiol.2006.107557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huke SS, Venkataraman R, Faggioni M, Bennuri SC, Hwang HS, Baudenbacher FJ, Knollmann BC. Focal Energy Deprivation Underlies Arrhythmia Susceptibility in Mice with Calcium-Sensitized Myofilaments. Circ Res. 2013 doi: 10.1161/CIRCRESAHA.113.301055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arteaga GM, Palmiter KA, Leiden JM, Solaro RJ. Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J Physiol. 2000;526(Pt 3):541–549. doi: 10.1111/j.1469-7793.2000.t01-1-00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003;19:263–268. doi: 10.1016/S0168-9525(03)00081-7. [DOI] [PubMed] [Google Scholar]
- 15.Michels M, Soliman OI, Phefferkorn J, Hoedemaekers YM, Kofflard MJ, Dooijes D, Majoor-Krakauer D, Ten Cate FJ. Disease penetrance and risk stratification for sudden cardiac death in asymptomatic hypertrophic cardiomyopathy mutation carriers. Eur Heart J. 2009;30:2593–2598. doi: 10.1093/eurheartj/ehp306. [DOI] [PubMed] [Google Scholar]
- 16.Yamanishi K, Fujita M, Araie E, Ohno A, Sasayama S, Okada E. Regional myocardial hypertrophy induced by repeated brief coronary occlusion in conscious dogs. Am J Cardiovasc Pathol. 1990;3:305–310. [PubMed] [Google Scholar]