

Abstract
Background
Several theories propose alternative explanations for drug addiction.
Objectives
We propose a general theory of transition to addiction that synthesizes knowledge generated in the field of addiction into a unitary explanatory frame.
Major principles of the theory
Transition to addiction results from a sequential three-step interaction between: (1) individual vulnerability; (2) degree/amount of drug exposure. The first step, sporadic recreational drug use is a learning process mediated by overactivation of neurobiological substrates of natural rewards that allows most individuals to perceive drugs as highly rewarding stimuli. The second, intensified, sustained, escalated drug use occurs in some vulnerable individuals who have a hyperactive dopaminergic system and impaired prefrontal cortex function. Sustained and prolonged drug use induces incentive sensitization and an allostatic state that makes drugs strongly wanted and needed. Habit formation can also contribute to stabilizing sustained drug use. The last step, loss of control of drug intake and full addiction, is due to a second vulnerable phenotype. This loss-of-control-prone phenotype is triggered by long-term drug exposure and characterized by long-lasting loss of synaptic plasticity in reward areas in the brain that induce a form of behavioral crystallization resulting in loss of control of drug intake. Because of behavioral crystallization, drugs are now not only wanted and needed but also pathologically mourned when absent.
Conclusions
This general theory demonstrates that drug addiction is a true psychiatric disease caused by a three-step interaction between vulnerable individuals and amount/duration of drug exposure.
Keywords: Drug addiction, Drug abuse, Drug use, Drug dependence, Drugs of abuse, Loss of control, Individual differences, Addiction theory
Introduction
This paper is not an extensive review of the literature on addiction but a position paper largely based on our own work and more generally on contributions of the Bordeaux School of Psychobiology. Here we provide the foundation for what we believe is the first “general” theory of transition to addiction. We qualify this as a general theory because it allows the organization and fitting together of most data produced by researchers in the field of addiction. However, our theory is limited to this transition and does not attempt to address the mechanisms of drug relapse (for recent reviews on this topic, see Kalivas 2009; Pickens et al. 2011; Bossert et al. 2013).
We describe the fundamentals of this general theory of transition to addiction in three sections. In the first section, we discuss the frame of reference we use to define addiction, which allows us to identify three separate steps in this process: (1) recreational, sporadic (ReS) drug use; (2) intensified, sustained, escalated (ISuE) drug use; (3) loss of control (LoC) of drug intake and full addiction. In the second section, we critically assess the roles of drug exposure and individual vulnerabilities in the process of transition to addiction, identifying the respective and essential roles of these two variables. In the third section, we describe the potential biological and psychological bases of the three phases of transition to addiction (see Fig. 1 for a summary of the theory). Finally, in the “Conclusion,” we describe crucial assumptions that can be used to further prove or falsify our theory.
Fig. 1.

Summary of the phases and processes of transition to addiction. Transition to addiction is a progression of three consecutive phases: (1) Recreational, sporadic (ReS) drug use, in which drug intake is moderate and sporadic and still one among many recreational activities of the individual. (2) Intensified, sustained, escalated (ISuE) drug use, in which drug intake intensifies and is now sustained and frequent and becomes the principal recreational activity of the individual; although some decreased societal and personal functioning start appearing, behavior is still largely organized. (3) Loss of control (LoC) of drug use and full addiction that results in disorganization of the addict’s behavior; drug-devoted activities are now the principal occupations of the individual. The three phases are consecutive but independent: entering one phase is necessary but not sufficient to progress toward the next phase, because specific individual vulnerabilities are needed. The first phase (ReS) occurs in most individuals (use prone); drugs overactivate the same substrates of natural rewards and therefore are perceived as extremely salient and likable stimuli. The second phase (ISuE) occurs in a vulnerable subset of individuals (escalation prone) because of quantitative differences in the activity of the brain reward-related system, which increases the motivational effects of the drug, for example, a hyperactive (sensitized) dopaminergic system and an impaired prefrontal cortex. The ISuE phase is then stabilized by additional drug-induced adaptations, inducing an allostatic state that makes drugs not only strongly wanted but also needed in order for the individual to function normally and, in certain cases, by habit formation. The last phase (LoC) leads to full addiction and is due to a second vulnerable phenotype that we term loss of control prone. This phenotype is characterized by a persistent loss of long-term depression of synaptic transmission (LTD) in reward-related brain areas, which can induce a crystallization of behavior around drug-taking, resulting in losing control of drug intake. In the addicted state, the presence of the drug is not only needed to function normally, as at the end of the ISuE phase, but its absence is experienced as an irreplaceable loss and strongly pathologically mourned. When the individual goes from liking drugs to pathologically mourning them when they are not available, the process of transition to addiction is complete
One of the major contributions of our theory is to further support the idea that addiction is a true psychiatric pathology and not an iatrogenic disease resulting from a vice. This vision of addiction is a much-needed evolution of how most societies view and treat addiction. In agreement with what has previously been proposed by Leshner (1997), addiction should not be considered a condition for which the individual is largely responsible and for which he or she should be punished but as a disease that needs treatment.
Section 1: How to define transition to addiction?
Leave preconceptions of the disease aside and focus on the patient
In addressing the issue of defining addiction, the most complicated point is to avoid a circular definition or a partial view of the disease based on personal, cultural, scientific, or theoretical backgrounds. One way of avoiding this is to clarify goals and define priorities. In this context, the goal and the priority of research on addiction and other diseases is to find a treatment. The definition of the disease should then reflect the reality of the final recipient of the research effort, the addict. For this purpose, the definition of addiction should be operational and based on the behavior of those whom our society identifies as drug addicts. We should not say that addiction is this and that; instead, we should start saying that an addict is this and that. As the identification of human addicts is based on specific criteria in diagnostic manuals [principally the Diagnostic and Statistical Manual of Mental Disorders (DSM) and the International Classification of Diseases (ICD), but also see the recent evolution of the Research Domain Criteria Initiative (RDoC)], the behavioral definition of addiction should be based on these diagnostic criteria. The neurobiological basis and potential treatments for addiction should then be tested and evaluated in animal models that are based on criteria used in these manuals. We believe that a definition of addiction that encompasses the addict’s behavior allows the highest chances for us to help find a treatment for that disease.
Transition to addiction in humans
The first observation that arises from reading addiction-related diagnostic manuals is that taking drugs in itself is not a pathological behavior; rather, taking drugs in certain ways and with certain consequences is what defines a drug-related problem and addiction.
Recreational nonpathological drug use
The intake of natural or synthetic substances for their psychoactive properties is a behavior widely represented in humans. Among the most common substances used in Western societies, one could cite coffee, tea, alcoholic beverages, and tobacco, followed by the most widely used illegal drug, cannabis. At least two of these substances, alcoholic beverages and tobacco, are capable of generating a very severe form of addiction that is highly resistant to treatment. Why, then, are alcohol and tobacco cigarettes legal substances? Clearly there are huge economic interests that make it impractical to declare alcohol and cigarettes illegal. However, probably the true reasons that allow this economic drive to win over the risk to develop addiction are twofold:
Experiencing a “high” is one of the major goal-directed activities of the human species. The major consequence of a large number of recreational activities is to strongly alter brain activity. In fact, an enormous effort on the part of human societies is dedicated to enabling these activities. The list is very long, and we cite only a few for which entire industries have been developed and dedicated facilities are built in most cities and towns. This is the case for many sports, either directly practiced or watched. It is the case for music-related activities of playing, listening, and dancing. It is also the case for many forms of visual entertainment that screen features that aim to induce in the audience intense emotions. Last but not least are the gourmet food and sex industries, in which the goal is clearly not one to feed or reproduce but to give intense pleasure. Taking drugs can be reasonably seen as another form of recreational activity in which brain activity is not modified through the sensory system but directly through specific pharmacological compounds. Why humans spend so much time and money and have such a strong motivation to experience a high is certainly a very important question. However, it is not the subject of this paper, and we accept it merely as a fact. The interested reader could refer to the book by Ronald K. Siegel, Intoxication, which discusses this issue at length and posits that intoxication is an innate drive seen across species (Siegel 2005).
Addiction is something believed to be happening to others. For most drugs of abuse, approximately only 15 % of users will progress from recreational use to a substance-related disorder (Anthony et al. 1994; Nutt et al. 2007), with the highest being nicotine (30 %) and the lowest cannabis (10 %) (Lopez-Quintero et al. 2011). The fact that drugs seem to be the pleasure of many and the doom of just a few “others” contributes strongly to the difficulty in controlling and forbidding drug use at the societal level. However, drugs are used by many millions of people, and consequently, millions of individuals develop addiction, making it the disease, among behavioral pathologies with the highest prevalence and, because of the nefarious health consequence of chronic drugs use, one of the most costly health problems of the so-called developed countries (EMCDDA 2011).
Pathological drug intake
Defining symptoms to provide distinction between nonpathological and pathological drug users is a matter of debate. Indeed, drug addiction is the behavioral pathology for which the clinical conception has evolved the most during the last 20 years. The first considerable evolution was between the early 1980s and the mid-1990s, as testified by the profound revisions of the reference DSM. This manual provides a series of behavioral symptoms (called items) that if present in an individual in preestablished numbers and preestablished frequency, allows making the diagnosis of specific pathological behaviors. In the DSM of the early 1980s (APA 1980) (DSM-III), two drug-related effects had to be present to make a diagnosis of drug dependence and addiction (1) Tolerance to drug effects: the effects of the drug decrease over time or one needs increasingly more drug to get the same effect. (2) Appearance of withdrawal symptoms when drug use is discontinued. In the DSM of the late 1980s [DSM-III revised (DSM-III-R)] (APA 1987), the category of dependence was expanded to include some criteria that in the DSM-III were considered symptoms of abuse. For example, the DSM-III-R described dependence as including both physiological symptoms, such as tolerance and withdrawal, and behavioral symptoms, such as impaired control over drug use. In the DSM-IV (APA 1994) and DSM-IV-R (APA 2000) of the 1990s and 2000s, tolerance and withdrawal remain among the diagnostic items but are no longer necessary criteria to diagnose drug dependence. In contrast, behavioral criteria related to loss of control over drug-taking and seeking are expanded, and at least one of these behavioral symptoms must be present. The DSM-IV describes five possible behavioral alterations characterizing loss of control (Table 1) and that can be grouped into three main categories: (1) difficulty limiting drug use and seeking (items 3 and 4); (2) extremely strong motivation to procure the drug (items 5 and 6); (3) maintaining drug use despite awareness of negative consequences (items 7).
Table 1.
Comparison of the diagnostic items of drug use related disorders in DSM IV and DSM 5, sufficient and necessary conditions for a diagnosis of addiction and corresponding major behavioral dimensions usable within an RDoC-like approach to behavioral pathologies
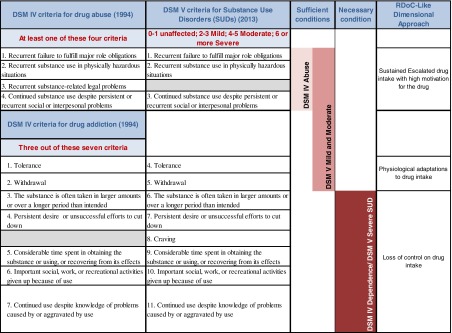
In conclusion, whereas in the DSM-III transition to addiction was defined as the appearance of changes in drug effects (tolerance and withdrawal), in the DSM-IV, it is characterized by changes in the modality of drug-taking: from controlled drug use to loss of control (Table 1).
All these diagnostic manuals once indicated drug abuse as a second category of pathological drug use. In the most recent version of the DSM-IV-R (2000), drug abuse is basically defined by the shift from sporadic drug intake to habitual and sustained drug-taking and is measured mainly by the appearance of drug-related behavioral problems and high motivation for the drug, such as: (1) recurrent substance use resulting in a failure to fulfill major role obligations; (2) recurrent substance use in situations in which it is physically hazardous; (3) recurrent substance-related legal problems; (4) continued substance use despite persistent or recurrent social or interpersonal problems caused or exacerbated by the effects of the substance.
It is important to emphasize that in the DSM-IV, drug abuse and drug dependence are not defined as two stages—mild and severe—of the same disease but two independent diseases carried by orthogonal dimensions. In other words, in the DSM-IV, abuse and dependence are two conceptually independent categories representing a different realm of problematic use. The consolidation of these two categories is probably the major evolution from the DSM-IV (APA 1994) to the DSM-5 (APA 2013). The DSM-5 replaces the two separate DSM-IV (substance-specific) categories of dependence and abuse with a single (substance-specific) category: Substance Use Disorders (SUDs). Criteria for SUDs results from merging the previous lists of dependence and abuse criteria into a single list of 11 criteria (Table 1). In the process, the criterion of recurrent legal problems of drug abuse was dropped and a criterion for craving was added. The DSM-5 also introduces the concept of different severities of SUD that is now graded by the number of criteria met: 0–1, unaffected; 2–3, mild; 4–5, moderate; ≥6, severe. Another important addition is that tolerance and withdrawal do not count if an individual is taking a medicine such as an opioid analgesic under medical supervision. This change will avoid the mislabelling of patients as dependent or addicted when they develop normal physiological dependence while adhering to a prescribed regimen (Compton et al. 2013).
As shown in Table 1, when one analyzes the DSM-5 on the basis of necessary and sufficient conditions to make a diagnosis of mild, moderate, or severe SUD, one can see that the previous criteria of abuse plus tolerance and/or withdrawal are sufficient to make a diagnosis of mild or moderate SUD. As was previously the case for the dependence category in the DSM-IV, at least one criterion of loss of control needs to be present for a diagnosis of severe SUD. In other words, our comparison predicts a substantial overlap between the abuse category of the DSM-IV and the mild and moderate SUD of the DSM-5. This prediction is confirmed by data contained in Table 2 that were recalculated from Table 3 in the report by Peer et al. (2013). It can be seen that, across several drugs, diagnosis of drug abuse in the DSM-IV corresponds in 99 % of cases to a diagnosis of mild or moderate SUD (inferior to severe) with the DSM-5. Similarly, but as could have been expected with less accuracy, a diagnosis of drug dependence with the DSM-IV corresponds in at least 85 % of cases to a diagnosis of severe SUD with the DSM-5—except for cannabis, where it is 67 %. The higher prevalence of SUD of moderate and mild intensity in the case of cannabis could be indicative of a lower potency of this drug to induce full addiction (Table 2).
Table 2.
Correspondence, across different drugs of abuse, between drug abuse and drug dependence in the DSMIV and mild to moderate SUD (<Severe) and severe SUD in the DSM 5
| DSM-IV | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Alcohol n (%) | Cocaine n (%) | Opioid n (%) | Cannabis n (%) | ||||||
| Abuse only | Dependence | Abuse only | Dependence | Abuse only | Dependence | Abuse only | Dependence | ||
| DSM-V | <Severe | 931 (99) | 569 (15) | 171 (99) | 422 (10) | 115 (100) | 168 (7) | 876 (99) | 692 (33) |
| Severe | 11 (1) | 3,238 (85) | 11 (1) | 4,042 (91) | 0 (0) | 2,332 (93) | 5 (1) | 1,395 (67) | |
A very recent evolution in the way in which pathological behavior could be analyzed is the one prompted by the National Institute of Mental Health (NIMH) with the approach named RDoC (Miller 2010). This is an attempt for a more endophenotype-based classification, which could allow developing biological diagnostic markers of psychiatric disease. If one looks at the behavioral symptoms of pathological drug intake across the DSM-IV and DSM-5 using an RDoC-like approach, it appears evident that three dimensions emerge. The first is characterized by an escalated and sustained drug intake with an enhanced motivation for the drug, which principally encompasses the abuse category of the DSM-IV and the mild and moderate SUD of the DSM-5. The second is characterized by physiological modifications of drug effects—tolerance and withdrawal—which are neither necessary nor sufficient conditions for a diagnosis of pathological drug-taking. The third is loss of control of drug intake, which is a necessary condition both for a dependence diagnosis in the DSM-IV and severe SUD in the DSM-5. Based on a synthesis of the three classification approaches—DSM-IV, DSM-5, and RDoC—we propose the following: (1) discard physiological adaptations to drugs (tolerance and withdrawal), as these two phenomena are neither necessary nor sufficient conditions for identifying pathological drug use; (2) use the two major categories of behavioral alterations that are either sufficient or necessary conditions to define drug intake as pathological: (a) escalated sustained drug use; (b) loss of control of drug intake (Table 1).
Transition to addiction in humans is a three-step process
Based on the above considerations, we propose that transition to addiction is composed of three consecutive and independent phases (Fig. 1): consecutive, because entering one phase is a necessary condition to go to the next one; independent, because entering one phase is not a sufficient condition to progress to the next one. The first step is a nonpathological phase of drug-taking in which there is Recreational Sporadic (ReS) drug use. The ReS phase starts when the individual learns to take drugs as one among many of his or her recreational activities. Drug use is then sporadic and occupies a small portion of the behavioral repertoire. This can be considered normal behavior and is present in a large proportion of the human population when both legal and illegal drugs are taken into account. The second step, Intensified Sustained Escalated (ISuE) drug use, starts with an increase in frequency, amount, and motivation to take drugs. Drug-taking intensifies and becomes sustained; additionally, drug-related problems appear but are not sufficient to promote spontaneous abstinence. The ISuE phase is the first moderate pathological state, one in which the individual takes too much drug but behavior is still organized and the individually generally well integrated into society. The third and final step signals the beginning of the most serious pathological state. In this case, the individual largely has Loss Control (LoC) of drug intake and becomes fully addicted. In this phase, drug-taking becomes the individual’s major goal-directed activity, invading most of the space normally occupied by other components of his or her normal behavioral repertoire. Then, social degradation is inevitable, and relapses to addiction—even after prolonged abstinence—is the rule.
It is important to emphasize that during the transition to addiction, drug intake first changes principally quantitatively (from recreational to sustained) and then qualitatively (from sustained to loss of control). In the transition from ReS to ISuE, the individual progresses from taking drugs sporadically and moderately to taking them frequently and in large quantity. In contrast, in the transition from ISuE to LoC, the principal difference is not how much drug is taken but where the individual is able to confine drug-taking. During ISuE, the individual still has a good degree of control, so that it remains largely confined within the boundaries of recreational activities. During LoC, control of drug intake is largely lost, and the individual’s behavioral repertoire is invaded by drug-taking that becomes the major goal-directed behavior. In summary, during transition to addiction, the drug user first switches from normal to moderately pathological, taking too much drug (quantitative change) and from moderately pathological to fully addicted by losing control of drug-taking (qualitative change).
Nonpathological, escalated, and fully addicted drug intake in animals
The voluntary intake of drugs of abuse is a behavior largely conserved throughout phylogeny. Thus, the preference for drug-associated environments or the learning of tasks reinforced by drugs has been found in several species, including worms, honey bees, rodents, and nonhuman primates (Siegel 2005). Voluntary drug intake has been a major subject of experimental research since 1962. This is largely due to the development of the intravenous drug self-administration model that showed that in rodents, drugs can act as positive reinforcers in the operant conditioning paradigm (Weeks 1962). In other words, rodents can learn to make an operant response—press a lever or nose-poke in a hole—to obtain intravenous delivery of an abused drug.
Models of nonpathological recreational sporadic drug use
Pioneering research in experimental psychology and experimental analysis of behaviors (e.g., Skinner 1930, 1950; Dews 1955; Brady 1956; Morse and Skinner 1958; Kelleher 1958; Dews and Morse 1958; Weiss and Laties 1964; Laties and Weiss 1966) led to the development of the intravenous self-administration model in the 1960s and opened the addiction research field to experimental research (Weeks 1962; Thompson and Schuster 1964; Pickens and Thompson 1968; Goldberg et al. 1969). The major and fundamental contribution of these early studies was to demonstrate that most drugs abused by humans can serve as positive reinforcers in laboratory rats and monkeys. However, drug-reinforced behavior is not per se a model of addiction. Depending on the circumstances under which it occurs, this behavior can be a model of nonpathological drug use (ReS), sustained escalated drug use (ISuE), or the fully addicted state (LoC). The study of the self-administration using unit doses above the median effective dose (ED50), fixed ratio schedules, and short access to the drug (1–3 h) for short periods of time (few weeks) more closely models a normal learning behavior than a pathological one. These types of procedures mostly model recreational drug use rather than the later phases of transition to addiction.
Models of intensified-sustained-escalated drug use
The first demonstration in animals that something is different from recreational drug use and is more similar to sustained escalated drug use was provided using unlimited access to drugs in monkeys (Johanson et al. 1976), dogs (Risner and Jones 1976), and rats (Bozarth and Wise 1985). It was in these studies that erratic drug intake and overdosing were first described. These models can certainly be considered the first demonstration that it is possible to observe a transition from moderate drug-taking to sustained and excessive drug use in laboratory animals. However, these early attempts had two problems that probably explain why they did not receive the attention they deserved. The first is that these data paradoxically suggest that transition to addiction in animals and humans are two very different phenomena. For example, in these models, especially with cocaine, animals rapidly overdosed and died. Although the death of the animals is a most impressive demonstration of excessive drug intake, human drug users clearly do not massively overdose and die over their first few contacts with the drug. In fact, although the amount of drug taken at any stage of pathological drug use—and sometimes during recreational use—is often impressive, it is quite an exception that individuals with different degrees of SUD overdose. In other words, the type of pathological drug intake shown in laboratory animals by these early studies is completely different from that observed in humans. In addition, 24 h/day access models are difficult to implement in most research institutes because of ethical and procedural considerations.
At the end of the 1980s, models of ISuE more close to the human condition started to appear. Investigating individual differences, we showed (Piazza et al. 1989) that when low-unit doses of drugs are made available, some rats (high responders, HRs) rapidly escalated their drug intake, reaching and maintaining a high level, whereas others (low responders, LRs) did not. Subsequently, we confirmed and extended this model by showing that when rats have access to a variety of unit drug doses, including high doses, HRs will consistently take a very large quantity of drug, whereas LRs will maintain moderate levels (Piazza et al. 2000). These differences manifest as an upward shift in the self-administration dose–response function in HRs and are associated with a higher motivation for the drug, as measured using a progressive ratio reinforcement schedule.
Approximately at the same time, models of sustained drug intake based on modification of access to drugs regained broad attention (Ahmed and Koob 1998; Tornatzky and Miczek 2000; Roberts et al. 2002). Ahmed and Koob (1998) showed that a phenotype similar to the HR phenotype is also identified by increasing daily access to the drug from 1 to 6 h. Long-access rats would escalate drug intake and, similarly to HRs, would take more drugs over a large range of doses (Mantsch et al. 2004), showing an upward shift in dose–response functions. Several complementary lines of evidence show that an increase in drug intake can be induced by increasing the exposure of an individual to the drug, as, for example, repeatedly treating rats with the drug before the start of the self-administration session (Piazza et al. 1989; Pierre and Vezina 1997; Vezina et al. 1999; Suto et al. 2002, 2003; Ferrario and Robinson 2007).
A very thorough study of conditions that can induce the transition from moderate to sustained drug intake and increase motivation for the drug have also been provided by Roberts et al., who primarily used the progressive ratio reinforcement schedule. These authors show an upward shift in the ratio-response function under certain conditions of drug access: pretreatment with the drug, high-unit dose, and some specific protocols of extended access [for an extended review, see Roberts et al. (2007)]. An important conclusion from their work is that chronic drug intake or extended access is not a necessary or sufficient condition to increase motivation for the drug, because under certain extended access conditions and training with high doses, motivation for the drug can decrease (Roberts et al. 2002).
From the studies reviewed above, we can infer the behavioral phenotypes that can be used in animals as the basis of a model of transition from recreational to sustained drug use. In particular, we propose two phenotypes that show the best face and construct validity:
Acquisition of a high rate of drug self-administration under conditions of low drug-unit doses and restricted access (Piazza et al. 1989). Under these conditions, all rats learn to self-administer drugs during the first few days of training, but only some vulnerable rats (HRs) escalate their drug intake, whereas in most rats (LRs), drug intake decreases. The difference in behavior between HRs and LRs provides good face validity to the model because humans who try drugs can be similarly divided into those who do or do not shift to escalated drug use. One piece of evidence for the construct validity of the model is that the HR phenotype can also be induced by conditions known to favor drug abuse in humans, such as repeated stress (Piazza et al. 1996; Piazza and Le Moal 1996).
Upward shift in self-administration dose–response functions and in the ratio-response functions. Thus, vulnerable rats will show a sustained drug intake over a large range of doses and an enhanced motivation to self-administer the drug. Convergent evidence for the construct validity of this model is that the upward shift was found by studying individual differences (HR vs LR) (Piazza et al. 2000) or by varying conditions of drug access (Suto et al. 2002, 2003), two approaches in which escalation of drug intake at a fixed dose is observed.
In conclusion, an animal that can acquire sustained drug intake when low drug-unit doses are used and that shows an upward shift in dose–response and ratio-response functions shows a behavior similar to that of individuals in the ISuE phase.
Models of loss of control of drug use and full addiction
In the late 1960s, early 1970s (Wikler and Pescor 1967; Wikler et al. 1971), and early to mid-1990s (Wolffgramm 1991; Wolffgramm and Heyne 1991; Wolffgramm et al. 2000), the first attempts were initiated to develop animal models of loss of control of drug use. Rats were given extended (many months) oral access to morphine, alcohol, and other drugs. For unknown reasons, this pioneering research was either largely forgotten or largely ignored by the addiction research field. In the middle of the first decade of the twenty-first century, loss of control models for intravenous self-administration of cocaine were described (Deroche-Gamonet et al. 2004; Vanderschuren and Everitt 2004). Subsequently, a clear distinction between making rats take drugs and making them addicted began to appear in the literature (Sanchis-Segura and Spanagel 2006; Roberts et al. 2007; Ahmed 2012; Vanderschuren and Ahmed 2013).
The existence of a fully addicted state with loss of control in rats was demonstrated in a publication by our group in 2004 (Deroche-Gamonet et al. 2004). The paper provided a multisymptomatic loss of control model that was drawn directly from the diagnostic criteria of the DSM-IV. We found that after prolonged exposure to cocaine, behaviors analogous to those used to identify loss of control in humans also appear in some rats. These rats cannot refrain from seeking cocaine even when they have previously learned that it is no longer available; become extremely motivated for cocaine, making thousands of responses for a few drug injections; and keep responding for cocaine even when its delivery is associated with adverse consequences, such as an electric shock. As observed in humans, rats in which loss of control appeared also showed a high propensity to relapse even after long periods of withdrawal. A paper published back to back (Vanderschuren and Everitt 2004) with ours also supported loss of control of cocaine self-administration by showing that after extended access to the drug (6 h/day), rats continued to press a lever in the presence of a cue previously associated with foot shock. Although there were no contingencies with adverse consequences of responding for the drug in this setup, their data suggest that rats can become increasingly resistant to avoiding lever pressing after longer periods of drug training. Pelloux et al. (2007) extended these observations to show that rats with an extended cocaine history were also more resistant to a punishment contingent with cocaine seeking Critically, these observations confirmed the predominant role of individual vulnerability in loss of control over extended drug intake. Enhanced resistance to punishment was attributable to a subpopulation that showed minimal or no suppression of drug-seeking behavior (Pelloux et al. 2007).
The bingeing model
We do not propose the bingeing model as a model of transition to addiction. The main reason is that our approach is based fundamentally on the behavior of individuals with SUDs and in particular, on diagnostic criteria or derived behavioral dimensions that across the DSM-IV, DSM-5, and RDoC are either a sufficient or a necessary condition for a diagnosis of pathological drug-taking. Based on these requirements, we have not proposed as relevant models of transition to addiction important drug–induced physiological adaptations, such as tolerance or withdrawal, which are diagnostic criteria for SUD but are not necessary or sufficient conditions for a diagnosis of the disease. Thus, tolerance and withdrawal can appear in response to medications that do not generate pathological drug-taking.
We believe that bingeing belongs to a similar category as tolerance and withdrawal. First, bingeing is not explicitly a diagnostic criterion of SUD. Second, bingeing can be observed in drug users even when they are still in the recreational phase, as for example, in the weekend bingeing observed in many recreational drinkers (e.g., college students). As such, bingeing in itself is not a sufficient condition to identify a pathological drug intake. Third, if cocaine-addiction-like behavior in rats can be associated with a deregulation in drug intake, i.e., a burst-like pattern of use (Belin et al. 2009a), close to the one observed in binge access procedures (Tornatzky and Miczek 2000), the development of bingeing is neither a necessary condition to the development of addiction-like behavior (Belin et al. 2011; Kasanetz et al. 2012; Deroche-Gamonet and Piazza, in press) nor a necessary consequence of prolonged drug intake.
In conclusion, we do not say that binge-like deregulation of drug intake is not important, but studying it independent of specific addiction-associated dimensions carries the risk of studying a phenomenon that may not be related to the shift from normal to pathological drug use.
Conclusions: three makes it simpler, not more complicated
Animal models now provide approaches to efficiently study the three steps that constitute the process of transition to addiction: (1) learning non pathological drug intake (ReS phase), studied over the first few weeks of self-administration training with short drug access: this is basically a normal behavior that would be observed in response to any positive reinforcer; (2) development of the ISuE phase in which drug intake increases and becomes sustained over a large range of doses, either because of individual vulnerabilities or extended access to drugs; (3) development of the LoC phase in which loss of control of drug intake leads to a fully addicted state.
This classification has the potential to help simplify and clarify the interpretation of data obtained in addiction research. In fact, what today are often presented as alternative models or approaches to addiction appear now as models of the different phases of the addiction process. In parallel, alternative views of the psychobiological and neurobiological basis of addiction become different substrates of the different phases of the addiction process. Finally, keeping in mind the behavior of addicts and the three steps of transition to addiction also provides a framework to help develop new pertinent models and understanding of addiction.
Section 2: transition to addiction depends on an individual–drug interaction
Theories of addiction
Based on experimental data, two main theoretical frameworks have been proposed to explain transition to addiction: drug-centered and individual-centered theories. These two groups of theories were initially derived from two common clinical observations, respectively: (1) transition to addiction occurs only after prolonged drug use (drug centered); (2) transition to addiction is observed only in a limited number of drug users (individual centered).
Drug-centered theories
Drug-centered theories of addiction are the ones most often followed in experimental research. This family of theories includes all views of addiction for which taking a drug repeatedly is the major cause of addiction. According to these theories, addiction is principally a consequence of drug intake and results from the psychopharmacological changes that follow chronic drug use. Theories regarding this group focused on specific drug-induced changes, such as tolerance (Roberts et al. 2002), sensitization (Wise and Bozarth 1987; Robinson and Berridge 1993, 2000, 2008), withdrawal and allostasis (Koob and Le Moal 2005, 2008a, b) or drug-induced cognitive changes in impulsivity (Jentsch and Taylor 1999), decision making (Bechara 2001, 2005), and conditioning (Robbins et al. 2008a). These views of addiction were reinforced by the very significant neurobiological alterations at molecular, cellular (Nestler 1992; Nestler et al. 1993; Nestler 1994; Nestler and Aghajanian 1997; Nestler 2000), synaptic (Hyman et al. 2006; Kauer and Malenka 2007), and network levels (Wolf 1998; Everitt and Wolf 2002; Wolf 2002, 2010a, b) that follow chronic drug intake. More recently, they have also been strengthened by observations showing that the longer the within- or between-day exposure to a drug, the greater the likelihood of increases in drug intake (Ahmed and Koob 1998) and subsequent appearance of signs of loss of control (Deroche-Gamonet et al. 2004).
Individual-centered theories
Individual-centered theories of addiction are fundamentally inspired by observations in humans, which are well summarized by Charles P. O’Brien, who stated: “Some addicts go for months or years using heroin or cocaine only on weekends before becoming a daily (addicted) user. Others report that they had such an intense positive response that they became addicted with the first dose…” (O’Brien et al. 1986). For this group of theories, drugs are a necessary but not sufficient condition for the development of addiction. According to this theoretical framework, addiction results from a pathological response to the drug that is generated in a few individuals by a vulnerable biological phenotype. Several experimental observations strengthen the hypothesis of the existence of a drug-vulnerable phenotype. Indeed, in rodents, large differences in vulnerability to escalate drug intake have been repeatedly shown either within outbred rat strains or between inbred mice strains. The main differences between theories in this family principally concern different degrees of etiological relevance given to genetic factors (Crabbe et al. 1999; Ellenbroek et al. 2005; Foroud et al. 2010; Meyer et al. 2010; Crabbe et al. 2010), environmental factors (Piazza et al. 1996; Goeders 2002; Bardo et al. 2013), developmental factors (Koehl et al. 2002; Barron et al. 2005; Doremus-Fitzwater et al. 2010), and gene + environment interactions (Cabib et al. 2000; van der Kam et al. 2005; Caspi and Moffitt 2006).
Addiction: drugs or individuals?
Research in laboratory animals during the last 20 years has progressively demonstrated that individual vulnerabilities to drugs exist and are due to underlying biological factors that mediate transition to addiction.
Individual vulnerabilities exist and are a necessary condition to transition to addiction
Beside phenotypic vulnerabilities, one possible origin of individual differences is experimental error. Thus, especially in operant learning, even small differences in experimental settings can induce large differences in learning. For example, a small difference in the contingency between response and delivery of the reinforcement can induce large differences in rates of acquisition of drug self-administration. How can we determine the difference between phenotypic differences and experimental error when, by definition, we cannot eliminate experimental error? How do we rule out something that by definition cannot be eliminated or systematically controlled? Obviously, we cannot.
If experimental error cannot be eliminated, what can be done, however, is to show that the observed differences between individuals do not result from experimental error. Experimental error is by definition not predictable. Thus, behavioral differences arising from an experimental error should not be predictable by variations in another independent phenotype. In other words, if we can predict individual differences in self-administration on the basis of behaviors observed in other procedures, then what is observed is, by definition, not an error. This was shown in our 1989 publication (Piazza et al. 1989) in which we demonstrated that a higher locomotor response to stress predicted individual vulnerability to rapid escalation in drug intake and maintenance of high levels of drug intake. These observations were confirmed shortly after by results from our group and others showing that HR rats had a higher preexisting dopaminergic activity (Piazza et al. 1991b; Hooks et al. 1992; Rougé-Pont et al. 1993) and a higher corticosterone secretion in response to stress (Piazza et al. 1991a) than did LR rats.
These data were replicated and extended by several groups, confirming that individual differences exist and depend on biological differences. However, such data did not address another major issue: would individual differences also exist in the real word? Thus, we and others studied individual differences in drug self-administration using low-unit drug doses (e.g., Piazza et al. 1989; Pierre and Vezina 1997). This experimental condition is indeed very different from the one observed in the real world, where humans can freely titrate their ideal dose. In other words, the model used to identify individual differences in the laboratory showed differences in threshold sensitivities to the drug that could be irrelevant in the human condition. Translated into pharmacological terms, this is, in fact, a very serious issue that needs to be addressed. General pharmacology tells us that individual differences in drug effects could originate from differences either in potency or efficacy, the two variables that classically characterize dose–response curves. So how is all this relevant to individual-based addiction theories?
Changes in potency (changes in sensitivity to the drug) are translated to horizontal shifts in the dose–response function. In this case, a vulnerable individual, compared with a more resistant one, would have a left shift in the dose–response function. Response intensity would be identical in all individuals, with the only difference being the dose at which the maximal response is obtained: lower in vulnerable individuals and higher in resistant ones. Therefore, if the dose is not an issue when the individual can freely choose the ideal dose, as in the real world, then individual differences are not likely to play a key role in determining transition to addiction.
Changes in efficacy, a measure of the maximum possible intensity of drug effect, are indicated by a vertical shift in the dose–response function. In this case, a vulnerable individual will have an upward shift in the dose–response function, and no matter the dose, this individual will self-administer a larger quantity of drug than a nonvulnerable individual. Consequently, even under conditions in which the dose of the drug can be freely titrated, some vulnerable individuals will escalate to drug abuse, whereas resistant individuals will maintain a moderate drug intake.
Vertical, not horizontal, shifts in dose–response functions explain individual differences in drug self-administration. Thus, when a large range of doses was used, individual differences not only persisted but were amplified. Some rats (HRs) would constantly self-administer large amounts of cocaine (vertical upward shift in the dose–response function), whereas others (LRs) took very low amounts (vertical downward shift in the dose–response function) (Piazza et al. 2000). In addition, the reinforcing effects of cocaine, as assessed in a progressive ratio reinforcement schedule, were higher in rats taking more of the drug and lower in rats taking less. Thus, it appears that independent of dose, cocaine has a higher reinforcing efficacy in some individuals, who will rapidly escalate drug intake.
The last but not least major issue to be addressed in order to establish the role of individual vulnerabilities is to show that these vulnerabilities are relevant to full addiction. Thus, as seen earlier, HR and LR are at best models of ISuE drug use but not of the most severe SUD, which is characterized by loss of control. This issue was one of the most complicated to address, in part because true addiction was once largely considered a purely human condition. To address this issue, in rats self-administering cocaine, we operationalized the DSM-IV diagnostic criteria related to loss of control and evaluated these criteria at regular intervals (Deroche-Gamonet et al. 2004). Criteria were: (1) high motivation for the drug (items 5 and 6), measured by a progressive ratio reinforcement schedule; (2) inability to refrain from drug seeking, even if the drug was not available (items 3 and 4), measured by active responses during periods of signaled drug nonavailability; (3) drug use despite negative consequences (item 7), measured by resistance to foot-shock-induced punishment at cocaine self-administration.
After short or intermediate exposures to cocaine (up to 40 days; daily sessions of 2 h/day), no rat showed any of the three criteria of addiction. After this period and up to 3 months of self-administration, the three addiction-like behaviors appeared in a small percentage of rats (∼20 %), whereas the largest proportion of rats maintained good control over drug intake. These individual differences were not dependent on the daily amount of the drug taken by the individual. Thus, rats that did or did not develop addiction-like behavior had an identical intake of cocaine during the entire drug self-administration period (3 months).
These findings lead to three major conclusions: (1) full addiction is not a purely human phenomenon but also exists in laboratory rats; (2) despite the use of a large amount of drug over a prolonged period, most individuals are resistant to addiction; (3) some of the nosographic constructions used to classify psychiatric diseases in humans correspond to unitary behavioral dimensions in laboratory animals. More generally, our studies suggest that it is possible to develop new models of psychiatric diseases that bridge a dimensional and nosographic approach and truly mimic/simulate the human condition.
Not one but at least two independent vulnerabilities
One completely unexpected but important finding of our 2004 publication was that vulnerability to loss of control was completely unrelated to vulnerability to sustained drug use. Thus, locomotor response to stress (HR and LR), anxiety-like behavior, and some measure of impulsivity, which predicted vulnerability to the development of sustained drug intake (Piazza et al. 1989; Bush and Vaccarino 2007; Anker et al. 2009; Marusich and Bardo 2009; Schramm-Sapyta et al. 2011), did not predict or were associated with the development of addiction-like behavior (Deroche-Gamonet et al. 2004; Belin et al. 2008). The distinction between the ISuE-prone and the LoC-prone phenotype was confirmed by Belin et al. (2008), who showed again that although HRs and LRs have an upward shift in the dose–response function (Piazza et al. 2000; Belin et al. 2008), they do not differ in addiction-like behavior (Deroche-Gamonet et al. 2004; Belin et al. 2008). The discovery of this distinction is important because it shows that completing the transition to addiction requires having two independent vulnerability phenotypes. The first, like the one found in HR and LR (Piazza et al. 1989), predisposes individuals to switch from recreational drug use to sustained escalated drug use. The second is responsible for the shift to loss of control in individuals in whom drug use is already sustained. Recent publications (Kasanetz et al. 2010, 2012) support the existence of these two independent phenotypes by showing that vulnerability to loss of control is associated with a different type of biological modification than the vulnerability to develop sustained escalated drug use.
How many vulnerable individuals?
To evaluate the role of individual vulnerabilities in the sequential steps of transition to addiction, it is important to estimate how many individuals actually make those transitions. This is a tricky question to answer unitarily because different dependent variables are measured in the two phases of transition to addiction and the individual vulnerabilities involved are of a very different nature.
How many individuals are vulnerable to develop sustained drug intake?
In animal models of ISuE as well as in humans, analyzed variables are principally quantitative. Drug use in the recreational phase is sporadic and generally moderate, whereas in the escalated phase, it becomes frequent and sustained. This principally quantitative difference between use and abuse has generated complicated discussions about the threshold that should be used in humans to differentiate the two phases; in other words, how much drug is too much?
This difficulty of establishing in humans a clear-cut threshold may be explained in laboratory animal studies that show that individual differences in drug intake intensity follows a normal distribution—or actually, a log-normal distribution that becomes normal after data log transformation. The normal distribution of drug intake intensity means that vulnerability to drug abuse is distributed along a continuum without a clearcut threshold between normal and too much or between resistant and vulnerable. This is why in humans, normal and too much is largely determined by arbitrary social conventions, and sometimes by health consequences that cannot be confidently foreseen. In animal studies, too, arbitrary thresholds are used to separate vulnerable and resistant individuals; depending on the authors, the upper 33rd or the upper 50th percentile has been defined as the vulnerable subset. However, the fact that a difference can be seen using a 50/50 split indicates that the Gaussian distribution is quite flat and that practically an equivalent number of individuals take moderate and high amounts of drugs, respectively.
The normal distribution describing drug intake in animals also explains why several publications show that the development of sustained escalated drug intake is strongly influenced by drug availability (Ahmed and Koob 1998; Roberts et al. 2002). If sporadic recreational drug use and sustained escalated drug use are two ends of a continuum, then a shift from one to the other condition can be obtained by changing independently the vulnerability of the individual or exposure to the drug. This is why in protocols using extended access, as with the one used by Ahmed and Koob (1998), an increase in drug intake can be observed in a number of individuals that is large enough to be studied behaviorally or neurobiologically by simple group comparison without taking into account individual differences.
When using models of sustained drug use, it is then important to set drug availability within a range that best mimics the human condition. In humans, by definition, drug access during the recreational phase is quite restricted, principally because of the large space occupied by competing activities. In many cases, the intensification of drug use is also initially opposed by the increase in drug-related expenses. This is probably at least part of the reason that most human drug users never shift to sustained escalated drug intake. As a consequence, we believe that the best models of human transition to pathological sustained escalated drug use are the ones using restricted access to drugs and in which the development of abuse-like behavior is not observed in a majority of individuals.
Models in which animals are offered the choice between drugs and competing activities are certainly alternatives to restricted-access procedures (Ahmed et al. 2013). Although these models seem to have higher face validity (Kalivas 2005) than restricted-access ones, they are also procedurally much more complicated. The field would certainly benefit from a direct comparison of the two approaches in order to evaluate their respective advantages and drawbacks.
In conclusion, it is difficult to determine an absolute number of individuals who will shift from sporadic recreational drug use to sustained escalated drug use, because this percentage depends on the interaction between individual vulnerability and drug exposure. As a consequence, in a general population, it is likely that the more a drug is easily available, the higher the number of individuals who will shift from recreational use to pathological escalated use.
How many individuals are vulnerable to loss of control and become fully addicted?
The percentage of individuals vulnerable to develop loss of control and become fully addicted can be more precisely calculated, and this phenotype seems resistant to changes in external contingency, such as the extent of daily drug access. These differences between escalation-prone and addiction-prone phenotypes are because the two phenotypes are of very different natures and are measured in very different ways. When vulnerability to addiction is studied using the three addiction-like behaviors to define the phenotype, only a small percentage of rats (17 %) show a complete manifestation of the phenotype (presence of three intense addiction-like behaviors), whereas most individuals maintain perfect control (41 %, no addiction-like behaviors) or good control (27 %, one addiction-like behavior) over drug intake. The different associations between these three behaviors in different individuals are not artifacts of the classification. Thus, in a factorial analysis, they all load on the same factor. This is probably why changing the criteria for intensity that are used to determine whether each behavior is positive or negative does not significantly change the final percentage of addict rats but maintains it in a range (16–20 %) that appears similar to that observed in humans (Belin and Deroche-Gamonet 2012).
The different natures of vulnerability to escalated and sustained drug use and to full addiction are also shown by the apparent insensitivity of the loss of control prone phenotype to changes in drug availability. We tried to find conditions that speed up the process of transition to addiction and thus increase the final number of addict rats for obvious experimental needs. The first thing we attempted (Deroche-Gamonet and Piazza, unpublished results) was to increase daily access to cocaine, increasing it from 2 to 6 h. As discussed earlier, this is a common procedure used to increase the number of individuals that escalate in sustained drug use. However, we were unable to increase the final number of addicted animals or accelerate the addiction process. Probably one reason for this relative insensitivity to changes in drug availability in the loss of control phenotype is because at least one of the addiction-like behaviors is not normally distributed but follows a bimodal distribution, which automatically restricts the excursion that a given phenotype can show in response to contingent factors.
In conclusion, interaction between the degree of drug exposure and individual vulnerability in determining the shift from sporadic to sustained drug use and from sustained drug use to addiction seems to follow different rules. Thus, the percentage of individuals who will escalate in drug intake can be increased by extending the daily duration of drug availability. In contrast, the addiction phenotype seems insensitive to within-day changes in drug availability and more dependent on the duration of drug exposure over days, necessitating at least 2 months of drug self-administration for addiction-like behaviors to appear even in the few vulnerable individuals. It is noteworthy that although intensified drug use has been found using several types of drugs, the shift to the LoC phase has only been studied with cocaine. It is then possible that the difficulty of increasing the number of subjects shifting to the LoC phase by modifying drug exposure, as described above, may not apply to other drugs (Badiani et al. 2011).
Individual vulnerabilities are a necessary condition to develop addiction
In conclusion, research in laboratory animals during the last 20 years demonstrates that individual vulnerabilities to drugs exist and are a necessary condition to first develop sustained drug use and then escalate to loss of control and full addiction. Transition to addiction is not mediated by one but by two distinct vulnerable phenotypes: the first phenotype promotes drug escalation and the shift to the ISuE phase, the second triggers the shift to the LoC phase (Deroche-Gamonet and Piazza 2010). The necessity of having two distinct and independent vulnerable phenotypes to complete the transition to addiction explains why only a small number of individuals exposed to drugs develop the most severe form of the disease.
Implications of drug-centered and individual-centered theories for medical and societal approaches to addiction
Why is it important to reconcile the two families of theories? Why is it important to accept that although taking drugs is a necessary condition for developing an addiction, it is not sufficient alone, and that transition to addiction occurs only in some vulnerable individuals? The reason is simple: The two families of theories generate opposite scientific, medical, and social conceptions of addiction and suggest different ways of dealing with this health problem.
Implications for disease classification
In the framework of drug-centered theories, addiction is approximated to an iatrogenic disease. An iatrogenic disease literally means a disease that is induced by a physician (iatros, Greek for physician), but this definition is more generally used to indicate diseases that result from the acute or chronic intake of prescription/therapeutic drugs. Classically, side effects of therapeutic drugs, those described in the drug labelling, are iatrogenic diseases. Even more generally, the definition of iatrogenic disease can be extended to the side effects of pharmacological compounds that are taken for other desired effects.
In the case of the drug-centered vision of addiction, this classification seems appropriate for two major reasons: (1) Drugs of abuse are pharmacological compounds that are taken because they induce intoxication—a state highly desired by our species—and that have a major side effect, i.e., addiction. (2) Most drugs were initially introduced for their therapeutic properties, as, for example, cocaine as a local anesthetic and morphine as an analgesic. The same situation in reverse applies to cannabis, which is now prescribed in certain conditions for medical purposes.
Under individual-centered theories, addiction should be considered a true psychiatric disease. Thus, most psychiatric diseases are related to stimuli largely innocuous for the general population but that are nevertheless highly pathological for a subset of vulnerable individuals. Examples of vulnerability are people with depression, anxiety disorders, and posttraumatic stress disorder (PTSD). The major causes of reactive depression in France are divorce and moving from one house to another; such events are generally well endured by the general population. Similarly, anxiety disorders can be associated with stimuli, such as spiders, snakes, and elevators, which do not induce pathological reactions in most people, although they are very often experienced as unpleasant. Finally, even the most traumatic experiences are dealt with efficiently by the largest portion of the population, and only some individuals develop PTSD. Consequently, if, as postulated by individual-centered theories, taking drugs is not a sufficient condition for addiction (which occurs only in some vulnerable individuals), then there is no reason to classify addiction differently from other psychiatric diseases.
Implications for research and social policies
From a scientific point of view, drug-centered theories suggest that our major efforts should be to identify and reverse the brain modifications induced by drug exposure. From a social and political standpoint, major efforts should be devoted to preventing exposure to particular drugs, either by repressive measures or preventive actions. Indeed, when a therapeutic drug shows serious side effects, the appropriate response is to stop its commercialization, forbid access to it, and alert the public to its dangers. Conversely, if the individual-centered vision of addiction is correct, research should focus on identifying the biological basis of vulnerability to drugs with the goal of reversing such vulnerability in order to treat addiction. In particular, this type of research should identify phenotypes predisposing to transition to addiction and the drug-induced adaptations that selectively appear in individuals who develop addiction. From a social and political standpoint, as addiction is one among other psychiatric diseases, health and research measures should be promoted. We do not forbid divorce, moving, and wars or eradicate spiders, snakes, and elevators, although such measures would probably substantially decrease the prevalence of depression, anxiety, and PTSD. However, suggesting such courses of actions as the principal approach to fighting these psychiatric diseases would probably not be well received.
It could be argued that divorce, moving, wars, spiders, snakes, elevators, are “natural” behaviors or elements that also play a positive role in the preservation of our species and our environment, whereas drugs are artificial stimuli that do not have such a function.
However, if drugs are unnatural, nonuseful, artificial stimuli, why do we keep two of them legal—alcohol and tobacco, which are the principal causes of health problems? This is because drugs are not always harmful, and their use is not unnatural. One of the most easily identified functions of drugs is their stress-relieving and anxiolytic effects, which certainly have an important role in helping individuals function in most human societies that are largely very demanding, often unjust, and practically never egalitarian. Also, as we have seen, getting high by different means is one of the principal activities in humans. Experiencing a high, therefore, has the features of a strong innate drive that cannot be considered unnatural. Thus, we generally consider “natural” what exists today because of a selection process and “unnatural” what we have artificially created. Certainly, we have not created the drive for getting high; it is one of the most rooted innate drives in the human species.
Adequacy of current social trends
Most developed nations have implicitly endorsed drug-centered theories of addiction: most public money spent on addiction goes to repressive and preventive measures, and most publicly funded addiction research investigates the effects of chronic drug intake on brain physiology. Finally, in most countries, an addict is regarded not as someone who has a psychiatric pathology but as someone who became addicted to a drug because of a lack of will—as someone with a vice. It is important to emphasize that the fate we reserve for addicts is quite different from that of individuals with well-accepted psychiatric diseases, such as depression or schizophrenia. In the latter cases, even individuals performing major crimes go to psychiatric facilities to be treated and not to prison, as most often happens to addicts.
On the basis of the observations summarized in this section, we believe that this societal approach to addiction should change profoundly because addiction seems to have all the characteristics of a typical psychiatric disease. Do we regard a depressed patient as someone who is lazy and does not want to go to work or someone with PTSD as an individual with no guts? Certainly not; and in Europe, at least, depressed and PTSD patients are taken care by publicly funded healthcare systems, with their absence from work financially compensated and their treatment costs refunded. Why? Because we believe that the individual is not to blame for the limitations associated with his or her disease. A similar treatment should then be reserved for addicts. If addiction develops principally in vulnerable individuals, as posited by the individual-centered vision of addiction, addicts are not individuals who have poor judgment and a vice but patients afflicted by a psychiatric disease.
It could be argued that although addiction it is not a vice, it is nevertheless a vice-like disease. First-time users have probabilistic knowledge that recreational use might lead to addiction. In other words, addiction would not be a 100 % true disease because it would arise from a risk-taking behavior. An appropriate way to address this argument is to ask whether conditions are not considered as true diseases when they arise from risk-taking behaviors. The answer to this question is clearly no. A large portion of cases of diseases for which etiological factors are known derive from risk-taking behavior. Many cases of very serious metabolic and cardiovascular diseases and infectious and traumatic diseases could be avoided by modifying behaviors that put us at risk.
The subsequent argument of a vice-like vision of addiction is that most of the above-mentioned diseases deserve to be endured by the afflicted individual because they result from too strong a push from the natural instincts that are fundamental to preserving the individual and the species, such as food and sex. Following this argument, we should then start looking with suspicion and not consider as true diseases many other conditions. Just to make a few examples, we should not consider as true diseases the multiple muscular and articular problems that result from several high-risk sports and body training activities. In fact, it is very complicated to justify the evolutionary usefulness of certain sports, including American football, car races, mountain skiing, sky diving, and even tennis. It would be also difficult to argue on a relative scale that risky sports (and why not fat-enriched food) have a usefulness superior to the stress- and pressure-relieving effects of drugs that help individuals to cope with society and maintain societal peace.
In conclusion, we believe there is no reason we should not consider pathological drug-taking as a true disease. Nonpathological drug-taking is a behavior for which we have a strong innate drive and that has an identifiable social function. Pathological drug-taking is, then, exactly like other psychiatric diseases, being the result of the interaction between a vulnerable phenotype and stimuli that are not pathogenic, but could be considered useful, for the general population. If this view is correct, most human societies are using the wrong approach to fighting addiction, and societal behaviors toward this disease should change profoundly.
Section 3: psychobiological basis of transition to addiction
This section is not an extensive review of the literature but a description of several pertinent examples that support the existence of three concatenated, independent steps in the transition to addiction. For this reason, we apologize in advance to our colleagues who have produced very important work that is not cited here.
Psychobiological basis of recreational, sporadic drug use
Activation of the brain substrate of natural reinforcers mediates the learning of drug use
The first phase of addiction is a learning process based on drugs of abuse activating the same brain substrates that mediate the positive reinforcing effects of natural reinforcers. The study and discovery of this fundamental basic mechanism was the focus of the early days of the addiction research field. Thus, for the most part, our neurobiological knowledge derives from studies using limited-access and behavioral procedures, such as conditioned place preference. As a consequence, most knowledge we have today concerns “normal” recreational drug use rather than pathological drug use. However, many important contributions have been made in this context, in particular, identification of the crucial role of activation of the mesocorticolimbic dopamine system.
Briefly, the general idea that was progressively put forward and is still largely believed is that drugs of abuse increases dopamine release in the nucleus accumbens and, in particular, in the shell of this nucleus, and that this biological change mediates their appetitive effects. Using complementary approaches, the work of the Di Chiara group (Di Chiara and Imperato 1988), the Concordia group (Wise 1978, 1984, 1987; Kalivas and Stewart 1991; Wise 1994), and of the Koob and Le Moal groups (Le Moal et al. 1979; Koob et al. 1989) are fundamental in proving this concept. Later on, the dopamine-activated downstream intracellular signals, such as adenylate cyclase (Self and Nestler 1995), and the mitogen-activated protein kinase (MAPK) pathways (Valjent et al. 2004) have been discovered and characterized.
An important role of dopamine and of its downstream intracellular mechanisms was also extended to conditioned stimuli associated with drug delivery. Such stimuli become important discriminative stimuli of drug availability and, under some training conditions, secondary reinforcers. The idea here is that the increase in dopamine is backward-shifted and is now activated by discriminative stimuli that predict drug availability more than by the drug itself (Wanat et al. 2009). It is noteworthy that exactly the same phenomenon happens with natural reinforcers, showing that these neurobiological mechanisms activated by drugs are part of the normal learning sequence of the behaviors directed at acquiring any kind of appetitive stimulus (Schultz et al. 1997).
Liking drugs is the major substrate of drug use
As the majority of research in the first 30 years of the addiction research field was performed using models of recreational drug use and not of the later phase of transition of addiction, it is not surprising that one of the first interpretations of drug addiction was actually the explanation of why we take drugs recreationally, or in other words, why drugs can act as positive reinforcers. This initial theory can be summarized by saying that we become addicted because we strongly like (Wise and Bozarth 1982) or want (Robinson and Berridge 1993) drugs, and we like or want drugs because they, or cues associated with them (Stewart et al. 1984), activate mesolimbic dopamine. We believe that this theory cannot explain the entire process of transition to addiction. In contrast, we think that this is a sound explanation of why we use drugs recreationally. Certainly, the primary reinforcing effects of the drugs and the perception of positive effects are the major reasons we learn to use drugs of abuse.
Overactivation of dopamine transmission is why drugs are extremely likeable and wanted stimuli
Despite the fact that learning to obtain drugs and food is mediated by identical brain substrates, drugs of abuse are described by most users as more salient and appetitive than natural reinforcers. Why is that? Two reasons can be advanced: (1) increase in dopamine induced by most drugs is significantly higher than that induced by natural reinforcers; (2) increase in dopamine induced by natural reinforcers rapidly habituates; however, this does not occur, or at least occurs more slowly, in response to drugs of abuse (Bassareo and Di Chiara 1997; Di Chiara 1998).
How dopamine makes us like or want drugs is still unclear, and the initial idea that dopamine is the neurotransmitter of pleasure seems increasingly implausible today. In particular, pleasure is defined as the sensation we perceive during the consummatory phase of a primary reinforcer: tasting the food, reaching the climax. However, in our pleasurable relationship with positive reinforcers, the ultimate consummatory phase is just a small component of the entire behavioral sequence leading to them. What we also strongly respond to is the sight of the stimulus and to its predictors. Salience of the stimuli or how attractive they look is then a very important hedonic dimension of our lives. It is fundamental in controlling and motivating behavior even more than the sensation we receive from the final consummatory phase. We believe that this is the role of an increase in dopamine: to light up stimuli, to increase their salience, and to make them strongly pleasant and irresistibly appetitive without the need to actually consume them (Schultz et al. 1997; Robinson and Berridge 2000). This is a tremendous power that justifies why the dopaminergic response to natural reinforcers and their predictors is moderate and habituates quickly and explains why, when this does not happen—as for drugs of abuse—the stimuli stand out and are perceived as strongly appetitive.
Is there a role of individual differences in recreational drug use?
Given the large number of humans that use drugs recreationally, and the observation that most animals learn to self-administer drugs (approximately 90 % of the population in both cases), we believe that the question should be turned around to ask whether there is a psychobiological “resistance” to using a drug recreationally. The answer is probably yes, since, as we said before, there are few laboratory animals that do not learn to self-administer drugs and few humans that do not take any type of drug. One possible explanation for this behavior is that drugs are aversive and not appetitive in these few individuals. This is a likely possibility, because drugs of abuse, particularly cocaine, are complex stimuli that possess both aversive and appetitive properties, with their final effects resulting from the algebraic summation of the two (Ettenberg 2004). Thus, if in some individuals the aversive effects of drugs outweigh their rewarding effects, then they will avoid drugs of abuse (Schechter 1992). Although this seems a likely hypothesis, to our knowledge, the mechanisms of resistance to recreational drug use have not been directly studied. Although recent investigations start elucidating why drugs, and in particular psychostimulants, can be aversive (Wenzel et al. 2011; O’Neill et al. 2013; Jhou et al. 2013), this hypothesis remains mainly speculative and needs direct validation.
Psychobiological basis of intensified, sustained, escalated drug use
For transition from physiology (drug use) to pathophysiology (sustained drug use and loss of control), we must go from independent to relevants variables
Knowledge of many neurobiological substrates involved in addiction that the research field, including ourselves, has identified so far is based on data collected after few weeks of exposure to drugs or from data obtained from noncontingent, limited drug exposure in locomotor sensitization studies (Piazza et al. 1996; Deroche-Gamonet et al. 2003; Kalivas and Volkow 2005; Nestler 2005; Hyman et al. 2006; Koob and Le Moal 2008a, b; Robbins et al. 2008b; Robinson and Berridge 2008; Ambroggi et al. 2009). This time frame is too short to allow the development of addiction-like behavior. As a consequence, most of these neurobiological changes more likely concern the mechanisms of the transition from recreational to sustained drug use than the substrate of loss of control and full addiction.
For some of these studies, a central issue is then to understand which of the proposed drug-induced modifications mediate sustained drug use. Drugs of abuse have a large number of biological effects, but very few of those effects are abuse related. Just comparing drug-treated animals with drug-naive animals does not allow distinguishing abuse-related from abuse-unrelated effects. In addition, most of these studies were done using a small number of rats or mice and without taking into account individual differences. As a limited number of individuals develop sustained drug use and finally addiction, it is possible that the observed changes reflect what happens in the brain of individuals resistant to drugs, which represent the majority of individuals within a population. As a consequence, some of the proposed putative mechanisms of addiction could be either completely unrelated to it or, alternatively, mechanisms of resistance to transition to addiction.
What approach will allow us to identify with a good degree of confidence the biological basis of transition to drug abuse and then to addiction? The answer to this question is simple: we should stop using approaches that are valid for physiological studies and start using approaches that are adapted to pathophysiological investigations. Physiology is discovering what the biological mechanisms (independent variables) are of a given bodily function (dependent variable). In this context, practically all biological factors for which manipulations modifies the target function can be safely defined as a necessary or sufficient condition of that function and is therefore part of its physiology.
Pathophysiological studies go one step further and attempt to identify the variables that induce pathology in a physiological function rather than attempting to identify all independent variables. In real life, pathological states do not develop by a random modification of all variables involved in the physiology of the diseased system. They result from deregulation of a precise subset of independent variables along a precise cascade of events. This specific selection of a subset of independent variables in the development of a disease is probably because pathology is often the result of the failure of an organ or biological system to adapt to a bodily or environmental constraint, i.e., the etiological factor of the disease. Adaptation or failure to do so depends on the adaptive capacity of the system and from what is called its “functional reserve,” i.e., how much that system can be impaired before the function it subserves is disrupted (Harrison 2011). It is then likely that the subsets of independent variables involved in a specific pathological process are the ones with the smaller functional reserve. Alternatively, they could also be the independent variables that play multifunctional roles. Thus, adaptation that protects one function can be disruptive for another. Clearly, these are not exhaustive examples of how a pathological state can occur. They are just few examples aimed at clarifying why the pathophysiology of a disease selects a subset of the variables involved in the physiology of the target system. We propose naming these independent, disease-specific variables “relevant variables” to differentiate them from the more generic independent variables involved in physiology.
How do we go, then, from studying independent variables to identifying relevant ones? The answer was outlined many years ago by Claude Bernard in his book on fundaments of experimental medicine (Bernard 1865). A biological factor or a biological chain of events can be considered a physiopathological mechanism; i.e., an independent variable can be considered a relevant variable when: (1) changes in its activity can be correlated with the predisposition of vulnerable individuals and/or with the appearance of the disease; (2) known etiological or predisposing external conditions modify the activity of such a factor; (3) opposite manipulations of such a factor can induce and reverse the disease.
In other words, to go from physiology to pathophysiology, we need models that allow either identification of spontaneously vulnerable and resistant individuals, or models that use known etiological factors to induce the disease in a subgroup of individuals. This is particularly important when, as in the case of transition to addiction, only a small percentage of individuals exposed to the pathogen (the drug) develop the disease. It is only by comparing individuals exposed to drugs who do or do not develop sustained drug use followed by loss of control that we can distinguish the three major families of variables that can be modified by drug exposure: (1) variables unrelated to transition to addiction, which will be modified in all individuals; (2) variables that protect against developing sustained drug use and loss of control, which will be modified in individuals who do not develop addiction; (3) variables involved in sustained drug use and loss of control, which will be modified in individuals who will develop these pathological behaviors.
For these reasons, in the following paragraph, we discuss only some of the data that we believe can be specifically related to the pathophysiology of transition to sustained drug use and not all psychobiological modifications induced by repeated exposure to drugs.
Sensitized dopamine: a pathophysiological mechanism of transition to intensified drug use
Sensitization has become a complicated word to use in the drug-abuse research field because many different meanings have been attached to it. Originally, sensitization is the opposite of tolerance. Tolerance refers to a decrease in a drug effect over repeated exposure to the drug. Sensitization defines the converse phenomenon: an increase in drug effect over repeated drug exposure. By definition, the motivational effects of drugs increase, or sensitize, during transition to escalated and sustained drug use. If the motivational effects underwent tolerance, individuals would take increasingly less drug and not increasingly more. Although this seems logical, for a long time, the opposite was argued: individuals would take increased amounts of the drug because they felt their effects less. The debate between tolerance and sensitization has long animated the drug-abuse research field.
In the late 1980s and first half of the 1990s, we and others demonstrated that individuals who were vulnerable for esclation to drug use had a spontaneously sensitized dopaminergic system (Piazza et al. 1991b; Hooks et al. 1992) and a very high response to initial drug exposure (Hooks et al. 1991). Over the same period, we also showed that this sensitized response and the associated vulnerability to escalate to sustained drug use could be induced either by repeated stress or repeated drug exposure (Deroche et al. 1995; Piazza et al. 1996; Piazza and Le Moal 1996; Ambroggi et al. 2009). In particular, vulnerable rats had a higher stress-induced increase in dopaminergic transmission in the nucleus accumbens (Rougé-Pont et al. 1993). Later, we showed that over an intermediate period of drug intake, including at least the first stages of the intensified-use phase, the incentive and reinforcing properties of drugs were increased or “sensitized,” not decreased or “undergoing tolerance” (Deroche et al. 1999).
These and related data were used as the basis to the sensitization theory of drug addiction that proposed sensitization of the motivational systems—which makes the individual increasingly want drugs—as a comprehensive explanation of addiction (Robinson and Berridge 1993, 2001). Available data certainly show that sensitization of dopaminergic transmission is an important element in transition to addiction. However, these data more particularly point to an important role for sensitized dopamine in triggering the ISuE use phase, inducing an escalation in drug intake. Later on, an increase in dopamine seems to play a less important role (Ahmed et al. 2003), and other factors contribute to maintaining the ISuE phase and to the final transition to the LoC phase and true addiction. Thus, dopamine sensitization is not an exhaustive explanation of transition to addiction but probably a crucial factor specific for the first step of this process.
Clearly, a sensitized dopamine response is not the only possible mechanism mediating escalated intensified drug use, but it serves as an example to show that the different phases of transition to addiction have distinct biological bases. For example, another well documented mechanism that we believe could play an important role in the ISuE phase is an impairment of the prefrontal cortex, and we refer readers to several good reviews on the subject (Jentsch and Taylor 1999; Kalivas et al. 2005; Volkow et al. 2011).
How the accumbens dopaminergic system get sensitized
Several independent variables can increase activity (sensitize) of the dopaminergic projections to the nucleus accumbens. We describe here variables that have been identified as relevant and can consequently be advocated as part of the pathophysiological mechanism leading to transition to addiction.
One mechanism of transition to intensified drug use that has been rather extensively studied as a pathophysiological mechanism is the interaction between glucocorticoid hormones and the dopaminergic system [for review see Piazza et al. (1996); Piazza and Le Moal (1996, 1997); Marinelli and Piazza (2002)]. In a series of publications, we showed that glucocorticoid tone is one of the most crucial regulators of dopaminergic transmission activity in the accumbens and that these hormones are involved in the pathophysiological mechanisms that induce transition from recreational drug use to escalated sustained drug use (Deroche et al. 1997). Thus, HR rats have a higher production of glucocorticoids (Piazza et al. 1991a), and these hormones were a crucial factor in determining their vulnerability to drug abuse (Rougé-Pont et al. 1998; Deroche-Gamonet et al. 2003). Glucocorticoid hormones increase the activity of the dopaminergic system by acting on one of the two brain receptors, more specifically, the glucocorticoid receptor (GR) expressed by striatal medium spiny neurons that project back to the ventral tegmental area (VTA). Suppressing the GR specifically in these neurons dramatically reduced the activity of the dopaminergic projection to the accumbens and induced a behavioral phenotype identical to the one of abuse-resistant rats; the LR (Ambroggi et al. 2009). On the contrary, administering glucocorticoids repeatedly increases vulnerability to drug self-administration (Deroche et al. 1992).
The pathophysiological role of glucocorticoids was confirmed by another series of studies showing that stress-induced sensitization of dopaminergic transmission (Rougé-Pont et al. 1995) and the subsequent vulnerability to escalate to the ISuE phase is dependent on stress-induced glucocorticoid production (Deroche et al. 1995; Marinelli and Piazza 2002). Thus, it was enough to block glucocorticoid secretion to completely suppress the effects of stress on vulnerability to drug self-administration. Similarly, suppressing the GR in accumbens medium spiny neurons suppressed the sensitization induced by repeated exposure to drugs of abuse (Ambroggi et al. 2009).
In conclusion, an increase in GR activation seems a sufficient and necessary condition to the expression of increased vulnerability to drug abuse shown by certain individuals. It is probably because of this GR overactivation that some individuals increasingly want more of a drug and have a higher vulnerability to develop the ISuE phase.
Impulse and desire: two servants of one master
Desire or impulse? Do we escalate in drug abuse because we increasingly desire more of a drug or because we cannot refrain from taking it? This is basically the debate between supporters of motivational views of addiction (such as the incentive sensitization theory) and those who favor the role of impulse control and behavioral disinhibition (Jentsch and Taylor 1999; Volkow et al. 2011). We believe that available evidence suggests that both phenomena contribute to the shift from recreational to sustained drug use. At the behavioral level, rats vulnerable to escalated sustained drug use show signs of increased motivation to take drugs and a degree of impulsivity, in particular, intolerance for delayed reward (Anker et al. 2009; Marusich and Bardo 2009). Similarly, in human addicts, motivation to seek a drug is certainly increased and a higher incidence of impulsivity was found [for review see (de Wit 2009)]. At the neurobiological level, the increased activity of dopaminergic neurons in the nucleus accumbens is consistent with a higher “desire” to take drugs, whereas impairment of the prefrontal cortex was found in human addicts (Goldstein and Volkow 2011) with a higher impulsivity (Jentsch and Taylor 1999).
In conclusion, concerning the second phase of transition to addiction, pathological motivation and increased impulsivity seem to be two servants of the same master, i.e., transition from sporadic recreational drug use to ISuE drug use. What remain unclear is the relationship between impulsivity and vulnerability to full addiction. We and others found impulsivity to be associated with sustained escalated drug use but not with the propensity to lose control and become fully addicted (Deroche-Gamonet et al. 2004). In contrast, Belin et al. (2008) found that a certain measure of impulsivity predicted LoC. One possible reason for this discrepancy is that different components of impulsivity may be associated with different phases of transition to addiction. Impulsivity is a complex trait composed of multiple components, including behavioral disinhibition, intolerance for delayed reward, and impaired ability to consider the consequences of behaviors (Evenden 1999). Vulnerability to develop sustained drug use seems reliably associated with intolerance to delayed reward (Perry et al. 2005, 2008; Belin et al. 2008; Anker et al. 2009), which differs in HRs and LRs. In contrast, behavioral disinhibition seems to predict the development of LoC (Belin et al. 2008; Winstanley et al. 2010). This result seems quite difficult to reconcile with the finding that extinction (also a measure of behavioral disinhibition) and addiction-like behaviors are not correlated and load on orthogonal factors (Deroche-Gamonet et al. 2004). It is then possible that behavioral disinhibition predicts loss of control but does not mediate it, because it is no longer present when addiction-like behavior appears.
Allostasis: a pathophysiological mechanism that stabilizes sustained drug use
The establishment of an allostatic state has been proposed by Le Moal and Koob as the crucial phenomenon in the addiction process (Koob and Le Moal 1997, 2001, 2005). In this context, the idea that the word allostasis carries is that following extended drug use, reward systems adapt to the daily overexposure of the brain to drugs by shifting the homeostatic set point (allostasis) to adapt to this continuous overstimulation. Because of this shift, the drug state progressively becomes the normal state and the nondrug state is now perceived as a pathological, or at least, as an unpleasant state. In other words, drugs progressively shift from being strongly wanted to also strongly needed.
The major empirical evidence for this theoretical construct is modification of the hedonic set point observed in rats that develop drug abuse. In such rats, the reward system seems to become less sensitive and needs stronger stimulation to achieve the same level of reward, as assessed in the electrical brain-stimulation reward procedure (Ahmed et al. 2002). The fact that this modification is very short lasting after discontinuation of drug self-administration reinforces our idea that this process plays a major role specifically in stabilizing an ongoing state of sustained drug use.
What makes these data relevant in our opinion is that a valid experimental medicine approach was used to obtain them, i.e., comparison of short- and long-access animals, which differ in the development of signs of sustained drug use. Although this approach does not take into account individual differences but maximizes the influence of drug availability, it remains a valid experimental medicine approach. Koob and Le Moal, even before their close collaboration, were pioneers in seeing addiction for what it is: a pathological condition that should be explained by a pathophysiological process and not the use of a “normal” behavioral response to a nonnatural reward.
In addition to a shift in the hedonic set point, other neurobiological and molecular long-term adaptations to chronic drug intake (see for example Nestler 2005; Kalivas 2009; Self and Nestler 1995; Wolf 2010a), are potentially involved in the allostatic state. The only incertitude in this impressive body of work is to clarify which of the described drug-induced neuroadaptations are relevant variables for transition to addiction.
In conclusion, as we said earlier for other available theories of addiction, we do not think that the published evidence indicates that allostasis and a shift in the hedonic set point can explain the entire process of transition to addiction and, in particular, loss of control of drug intake. However, drug-induced psychological and physiological allostatic changes clearly play an important role in maintaining a pathological and sustained drug use.
Habitual behavior
Habitual behavior is an experimental psychology concept that describes how after long periods of training in operant tasks (e.g., pressing a lever for obtaining food) performed in perfectly standardized conditions, the behavior becomes automated in the sense that it is no longer initiated with reference to the goal. Two views of habit learning exist: behaviorist and ideomotor. According to the ideomotor view, although the behavior is automated (i.e., triggered by environmental cues), its remains controlled by its consequences (the value of the reinforcer). In contrast, according to the behaviorist view, the behavior is decreasingly controlled by its consequences (the delivery of the primary reinforcer) and increasingly by environmental contingencies (i.e., the light associated with drug delivery, the position of the lever in the chamber, the physical feature of the chamber, etc.). Because of this, when delivery of the primary reinforcer (food or drug) is interrupted or the primary reinforcer is devalued (by adulteration or punishment), individuals with a habit will keep responding, whereas changes in lever position or physical features of the operant chamber will more effectively disrupt the behavior. In the case of addiction, this implies that the drug becomes increasingly less necessary to sustain and control behavior, which becomes more strictly controlled by the conditions and context in which the drug is taken (drug paraphernalia, administration context, etc).
The potential role of associative learning and habitual behavior in addiction is well summarized in recent reviews by Hogarth et al. (2013). Importantly, the role of habit in addiction has been progressively revised (Everitt and Robbins 2005; Belin et al. 2009b, 2013). Indeed, it now seems unlikely that habit by itself can be responsible for loss of control of drug use (Robinson and Berridge 2003; Everitt and Robbins 2005). More recently, in its behaviorist conception, a role for habit even in earlier phases of transition to addiction has also been questioned (Hogarth et al. 2010).
Drawing the parallel between the behaviorist habit and full addiction was certainly tempting because both imply a form of loss of control. However, controllers and the controlled show opposite relationships between behaviorist habit and full addiction. In addiction, the individual loses control over the primary reinforcer, the drug. In habit, the reinforcer loses control over the behavior of the individual. In other words, during addiction, the drug gains increasing control over behavior, whereas during habit formation, the reinforcer/reward, i.e. the drug, progressively loses that control. The limited usefulness of placebo as a treatment for addiction is exemplary in this context. If drug addiction was a habit, by substituting the drug with a placebo, the addict should scarcely notice it for a reasonable amount of time. However, this is not the case, and even compounds that target the same receptor as the original abused drug but with different pharmacokinetics—as, for example, methadone—are not completely satisfactory in treating addiction. In addition, experimental data in human (Sheeran et al. 2005; Hogarth et al. 2010) and laboratory animals (Olmstead et al. 2001; Root et al. 2009) show that drug-seeking does not become independent of its consequences, as postulated by the behaviorist habit theory.
The first phase of the addiction process—recreational drug use—cannot be explained by a habit, as the behavior is sporadic and controlled by the appetitive effect of the drug (the primary reinforcer). Escalation to sustained drug use is also unlikely to be explained by the formation of habit. In this intermediate phase of addiction, the drug still plays an important role; the higher the impact of the primary reinforcer, the higher the chances of escalation to sustained drug use. When sustained drug intake is in place, a pathological habit can contribute to its stabilization. However, this can happen only in the very specific conditions of standardized and habitual patterns of drug consumption necessary for habit development. In the context of the ideomotor view of automated behavior (Aarts and Dijksterhuis 2000; Aarts et al. 2008), habit could contribute to the impulsive-like drug-seeking observed during sustained drug intake (Sheeran et al. 2005; Hogarth et al. 2010). Behaviorist habit could also play a role in stabilizing sustained drug intake. Thus, once sustained drug use is in place, the development of an allostatic state and the shift of the hedonic set point appear. Now the individual takes drugs not to feel high but to feel right. This implies a devaluation of the reinforcing effects of the drug despite intense use, which is consistent with habit behavior but also with an increased motivational drive created by the appearance of a discomfort when the drug is absent. In any case, habit behavior can influence intensified drug intake only if the pattern of drug use is standardized enough to allow for its development—a condition that is probably very infrequent in real-life addiction.
Conclusions: desire, impulse, and need—the three keys to transition to sustained drug use
In conclusion, development of sustained drug use (the ISuE phase) can be mediated by sensitized dopaminergic transmission in the accumbens (as described here) and by decreased functionality of the prefrontal cortex (Jentsch and Taylor 1999; Goldstein and Volkow 2011). These changes can be either spontaneously present or induced by stress and drug exposure. Sensitized dopamine transmission and impaired prefrontal cortex converge to make drugs highly appetitive and difficult to resist. In other words, these processes increase the desire and impulse for the drug and facilitate the development of an escalation in drug intake and sustained drug use. The establishment of sustained drug intake will then induce, as a form of additional adaptation, an allostatic state associated with a downregulated reward system that will progressively bring the nondrug state out of the comfort zone. The drug now is not only the object of strong impulses and desire but is also strongly needed–the transition to an escalated, sustained drug use is now complete. Finally, under certain conditions of highly standardized pattern of drug use, the establishment of habit behavior can also contribute to the maintenance of sustained drug intake.
Psychobiology of loss of control and full addiction
Impaired synaptic plasticity is associated with the development of loss of control and full addiction
Our knowledge concerning the pathophysiological mechanisms of the loss of control-prone phenotype is very limited. Indeed, the only biological modification yet specifically associated with loss of control of drug intake is a loss of synaptic plasticity (Kasanetz et al. 2010, 2012). It is important to emphasize here that the implication of synaptic plasticity to addiction was not proposed by us but by several of our colleagues who showed that, depending on the protocol and the conditions used, repeated drug intake would induce impaired synaptic plasticity (White and Kalivas 1998; Kalivas 2005; Kauer and Malenka 2007; Thomas et al. 2008; Russo et al. 2010; Lüscher and Malenka 2011). Our contribution was to show that impairment in long-term depression of synaptic transmission (LTD) in the brain is not just another drug-induced change but to date the only one specifically associated with cocaine-addiction-like behavior.
Briefly, we have shown that after a short period of self-administration (around 7 days), there are no impairments in synaptic plasticity (in the form of LTD) in the cortex or the nucleus accumbens. However, after 18 days, before the appearance of addiction-like behavior, a loss of N-mathyl-D-aspartate (NMDA)-dependent LTD appears in the accumbens of all individuals self-administering cocaine. On the contrary, no modifications of synaptic plasticity are observed in the prefrontal cortex. Later on, after 60 days of self-administration, a normal NMDA-dependent LTD is recovered in the accumbens of rats that maintain a controlled drug intake. In contrast, in rats that have developed addiction-like behavior, LTD in the accumbens appears to be permanently lost (at least up to 3 months). This stable impairment in accumbens LTD is accompanied by the appearance of an impaired metabotropic glutamate receptor 2/3-mediated LTD (mGluR2/3-LTD) that is observed selectively in the cortex of addicted rats.
In conclusion, rats that develop addiction-like behaviors have impaired NMDA and mGluR2/3-dependent LTD in the nucleus accumbens and prefrontal cortex, respectively, whereas both types of LTD are functional in nonaddicted rats. An exception is the endocannabinoid-mediated long-term synaptic depression (eCB-LTD) that after 60 days of self-administration is impaired both in addicted and nonaddicted rats. Thus, eCB-LTD is an example of drug-induced neuroadaptations that are unrelated to addiction that we previously discussed.
These data change the frame of reference in which vulnerability and resistance to addiction should be conceptualized. One way to view resistance and vulnerability to addiction is to say that some vulnerable individuals have a pathological response to addictive drugs, whereas resistant individuals are insensitive to the addiction-promoting effects of drugs of abuse. Our data show that this is not the case. Most drug-induced impairments in synaptic plasticity are initially observed in all individuals, but the majority of individuals, the resistant ones, are able to adapt and recover from most of them. Thus, resistance to drugs is not a passive immunity or insensitivity to the deleterious effects of drugs; this resistance is an active, biological, adaptive process—an active resilience. Conversely, the addict does not develop a response to drugs that is completely different from that of resistant individuals but is principally an individual who cannot adapt to the changes in the brain induced by the drug. As a consequence, vulnerability to addiction can be conceptualized as involving a degree of “anaplasticity,” the inability to recover a lost function, rather than a unique sensitivity to the drug’s deleterious effects.
Behavioral crystallization: a “missing” hypothesis of the psychological process underlying loss of control and full addiction
We propose here an evidence-based hypothesis of the psychological process underlying loss of control and full addiction. This hypothesis is based on the first and only neurobiological impairment specifically associated with loss of control in a rat model: i.e., a persistent loss of synaptic plasticity in the nucleus accumbens and medial prefontal cortex. We propose to the reader to follow two subsequent steps: (1) conceptualize what behavioral deficit could follow a loss in synaptic plasticity; (2) assess whether such a behavioral deficit could be consistent with the behavior observed in individuals showing the severe form of SUD characterized by loss of control of drug intake.
It is largely believed, and we agree with this hypothesis, that synaptic plasticity, as studied by long-term potentiation (LTP) and LTD, represents the ability of the brain to strengthen or depress neuronal circuits in order to maintain adaptive behavioral responses to changes in environmental contingencies (Goto et al. 2010; Neiman and Loewenstein 2013). In other words, one can say that the synapses of the brain circuit mediating a current behavioral sequence are “fully potentiated.” However, if an environmental event makes a current behavioral sequence maladaptive, your brain is able to depress the synaptic strength in that circuit and potentiate synaptic strength in a new circuit, thus mediating a new and more adaptive behavioral sequence. In other words, the balance between potentiation (LTP) and depression (LTD) can shift from one behavior to another and maintain a flexible and adaptive behavior.
Let us imagine that, as observed in rats showing addiction-like behaviors, the brain loses the ability to perform LTD, especially in regions such as the nucleus accumbens and prefrontal cortex, which are important in selecting appropriate goal-directed behaviors. The consequences would be quite catastrophic: behavior would remain crystallized around one behavioral goal and would not be able to shift to another. There would be great perseverance to achieve the crystallized goal no matter the costs or consequences. This would be externally manifested as a considerable loss of control of drug use that will become very difficult to change. However, this is not a state of compulsive pathological motivation for the drug but a “prison” from which the behavior, crystallized around one unique goal, taking drugs, is not very likely to escape.
There is a metaphoric example that we like to provide to students to explain the concept of behavioral crystallization. Imagine that the brain is a tank full of water and behavioral goals are cylinders of very different shapes. You behave in a certain way when one of the behavioral cylinders is immersed in the water tank. When the brain functions normally, you can easily remove a given cylinder, submerge a different one, and change behavior. However, if the water freezes, i.e., crystallizes (loss of synaptic plasticity), it will hold tight to the cylinder that was submerged at the time (taking drugs), and you will then be stuck in the prison of this behavioral goal. With some help, effort, and scraping (painful first period of any detoxification therapy), you will be able to extract the active cylinder from the crystallized water. However, it will be impossible to fill the remaining hole with any other behavioral cylinder. Your behavior is now crystallized, you can learn to live with this hole but it will be too strong a temptation, given the occasion, to fill it up again with the only cylinder that fits: taking drugs (quick relapse to drug addiction even after prolonged abstinence). Clearly, the water could melt again and the behavior regain plasticity. However, this seems to happen in a very restricted number of individuals; rather, addiction becomes a “frozen” chronic relapsing disease in 90 % of cases.
In conclusion, because of behavioral crystallization, drugs are not only wanted and needed but also irremediably missed when they are not present. The absence of drugs is felt as the irreplaceable loss of something very dear and precious, like a pathological mourning that cannot be overcome. When we start pathologically mourning the “missing” drug and can no longer escape from its embrace, there and then the transition to addiction is complete.
Changing places: from the VTA to the accumbens to the cortex—a likely road to full addiction
One interesting hypothesis put forward recently is that during transition to addiction, modifications in synaptic plasticity migrate following a ventrodorsal gradient (Lüscher and Malenka 2011). Changes in plasticity would occur quickly in the VTA, migrate then to the accumbens, and later on to the prefrontal cortex. This process of the transfer of learned information between structures of the brain is a well-known concept that has been described for motor learning (Salmon and Butters 1995; Karni et al. 1995; Jueptner et al. 1997; Wu et al. 2008) and for spatial or declarative memory processes in both rodents and humans (Bontempi et al. 1999; Frankland and Bontempi 2005; Winocur et al. 2010; Helie et al. 2010).
Regarding memory formation, it is recognized that spatial information about a given environment is acquired through the hippocampus and then moves to the cortex for long-term storage (Winocur et al. 2010). Rule-based categorization in humans appears to move from subcortical to cortical regions automatically (Helie et al. 2010). The classic view of motor learning postulates that novel behaviors are dependent on the cortex, whereas automatic behaviors are primarily mediated by subcortical structures, e.g., the striatum. Although this view is now questioned (Ashby et al. 2010) and the role of the striatum in automatic motor tasks (such as playing a well-learned musical piece) and habitual behavior has been challenged, it remains true that plasticity phenomena within (Karni et al. 1995) and between brain structures occur (Wu et al. 2008) when automatic behaviors develop.
In the case of addiction, evidence suggest that the regions controlling drug use progress from the VTA to the accumbens and finally to the cortex. Thus, the accumbens seems completely spared during the first week of self-administration, during which the VTA should be affected. Modifications in the accumbens appear only later on but before the appearance of addiction-like behaviors. The last region to show a change in synaptic plasticity (which appears only in addicted rats) is the prefrontal cortex (Kasanetz et al. 2010, 2012).
It has also been suggested, based on neurochemical and pharmacological evidence (Veeneman et al. 2012; Willuhn et al. 2012), that the response to drugs of abuse and cues associated with them moves from the nucleus accumbens to the dorsal striatum (Belin and Everitt 2008; Belin-Rauscent et al. 2012). In the original framework that describes addiction as a form of habit-like behavior (Tiffany 1990), this shift is seen as the fundamental trigger of a stimulus–response habit (Belin et al. 2013).
In conclusion, whatever the exact structures involved and the exact timing of progression, the observations summarized above indicate that depending on the phase of transition to addiction, different structures mediate responses to drugs. These data strongly support the idea posited by our theory that the process of addiction is mediated by independent but concatenated phases.
Conclusions
Why this is a general theory
The reader will notice that we call this not just a theory but a general theory—the first of its kind in the field of addiction research. What is the difference? Theories are usually stand-alone explanations of a phenomenon; they are backed by a series of arguments and by careful refutation of other pre-existing theories. In contrast, general theories attempt to provide a framework that explains and integrates all existing relevant knowledge and previous existing theories.
We believe that ours is a general theory of transition to addiction because it consolidates the knowledge generated by the drug-addiction research field over 40 years of research. Different competing explanations now appear as important pieces of a complex process that is not one or the other but all of them. The only processes that are discarded or not discussed here are those for which the relevance to addiction remain to be proven. The framework in which to solve this incertitude (going from independent to relevant variables) is also provided here.
Major fundamental concepts and predictions of the general theory
When positing a theory, it is important to define its exact postulates and predictions and what should be demonstrated to invalidate it. For this reason, the three following paragraphs describe the fundamental concepts of our theory and which deviation from them would or would not invalidate it.
Transition to addiction depends on an interaction between individual vulnerabilities and drug exposure
This statement is supported by the observations that transition to addiction is the result of sequential interactions between: (1) degree of vulnerability of an individual, because addiction appears only in a few individuals, even when all individuals were exposed to similar quantities of drugs; (2) degree of exposure to drugs, because addiction—even in the few individuals—appears only after extended access to drugs.
Variation in the degree of these interactions cannot be seen as a fundamental fallacy of our theory. On the other hand, if it is demonstrated that one of these two variables is not necessary in the development of pathological drug use in conditions that can be approximated to the ones occuring in human societies, then our theory will be falsified.
Transition to addiction is a process that develops along at least three steps
This statement is supported by the observations that different types of individual vulnerabilities and different psychobiological processes are involved in the three behavioral phases of the process of transition to addiction. The division into three steps proposed here and the underlying biological basis seem most appropriate on the basis of current knowledge. Consequently, new evidence could increase the number of steps and add to or revise the psychobiological substrates. This should not to be seen as a major revision of the theory. On the other hand, if it is demonstrated that transition to addiction develops in less than three steps, then our theory will be falsified.
Transition to addiction is a true psychiatric disease
This statement is based on the observation that transition to addiction is a multistep pathological process that, similarly to other psychiatric diseases, depends on the interaction between a vulnerable individual and a stimulus—drugs of abuse—which is not pathogenic in the general population.
The discovery of new types of vulnerabilities, of new compounds susceptible to inducing transition to addiction, or revision of the percentage of individuals shifting from drug use to addiction, cannot be seen as invalidating of our theory. On the other hand, the demonstration that in most conditions drug exposure is both necessary and sufficient to induce addiction, will invalidate or falsify our theory.
Farewell
Pleasure, desire, need, and mourning: could these four words summarize addiction, one of the most serious psychiatric diseases of our society? Could it be so simple? The evidence accumulated by the addiction research field over the last 40 years suggests that this is likely the case. Probably tomorrow, this simple sequence of four internal states will be laughed at or more likely forgotten. This is the fate of all evidence-based theories and a fate that we gladly accept. We prefer the shaky ground of evidence to the more solid one of theoretical preconceptions. Science is certainly much better served by an evolving “what it is” than from a durable “what it should be.” Crystallization, even of ideas, is not good.
Acknowledgments
We are immeasurably thankful to Drs. David Epstein and Yavin Shaham for their insightful and knowlegeable comments and contributions to this manuscript. PVP & VDG are supported by ANR (Neurorelaps, TIMMS), EquipEx OptoPath (ANR-10-EQPX-08), and Labex BRAIN grants, by INSERM, the Conseil Regional d’Aquitaine and the University of Bordeaux 2.
Contributor Information
Pier Vincenzo Piazza, Email: pier-vincenzo.piazza@inserm.fr.
Véronique Deroche-Gamonet, Email: veronique.deroche@inserm.fr.
References
- Aarts H, Dijksterhuis A. Habits as knowledge structures: automaticity in goal-directed behavior. J Pers Soc Psychol. 2000;78:53–63. doi: 10.1037//0022-3514.78.1.53. [DOI] [PubMed] [Google Scholar]
- Aarts H, Custers R, Marien H. Preparing and motivating behavior outside of awareness. Science. 2008;319:1639. doi: 10.1126/science.1150432. [DOI] [PubMed] [Google Scholar]
- Ahmed SH. The science of making drug-addicted animals. Neuroscience. 2012;211:107–125. doi: 10.1016/j.neuroscience.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Kenny PJ, Koob GF, Markou A. Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci. 2002;5:625–626. doi: 10.1038/nn872. [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Lin D, Koob GF, Parsons LH. Escalation of cocaine self-administration does not depend on altered cocaine-induced nucleus accumbens dopamine levels. J Neurochem. 2003;86:102–113. doi: 10.1046/j.1471-4159.2003.01833.x. [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Lenoir M, Guillem K. Neurobiology of addiction versus drug use driven by lack of choice. Curr Opin Neurobiol. 2013 doi: 10.1016/j.conb.2013.01.028. [DOI] [PubMed] [Google Scholar]
- Ambroggi F, Turiault M, Milet A, Deroche-Gamonet V, Parnaudeau S, Balado E, Barik J, van der Veen R, Maroteaux G, Lemberger T, Schütz G, Lazar M, Marinelli M, Piazza PV, Tronche F. Stress and addiction: glucocorticoid receptor in dopaminoceptive neurons facilitates cocaine seeking. Nat Neurosci. 2009;12:247–249. doi: 10.1038/nn.2282. [DOI] [PubMed] [Google Scholar]
- Anker JJ, Perry JL, Gliddon LA, Carroll ME. Impulsivity predicts the escalation of cocaine self-administration in rats. Pharmacol Biochem Behav. 2009;93:343–348. doi: 10.1016/j.pbb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony JC, Warner LA, Kessler RC (1994) Comparative epidemiology of dependence on tobacco, alcohol, controlled substances, and inhalants: basic findings from the National Comorbidity Survey. 2:244–268
- APA (1980) DSM-III Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association
- APA (1987) DSM-III-R Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association
- APA (1994) DSM-IV Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association
- APA (2000) DSM-IV-R Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association
- APA (2013) DSM-5 Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Association
- Ashby FG, Turner BO, Horvitz JC. Cortical and basal ganglia contributions to habit learning and automaticity. Trends Cogn Sci. 2010;14:208–215. doi: 10.1016/j.tics.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Belin D, Epstein D, Calu D, Shaham Y. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011;12:685–700. doi: 10.1038/nrn3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo MT, Neisewander JL, Kelly TH. Individual differences and social influences on the neurobehavioral pharmacology of abused drugs. Pharmacol Rev. 2013;65:255–290. doi: 10.1124/pr.111.005124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron S, White A, Swartzwelder HS, Bell RL, Rodd ZA, Slawecki CJ, Ehlers CL, Levin ED, Rezvani AH, Spear LP. Adolescent vulnerabilities to chronic alcohol or nicotine exposure: findings from rodent models. Alcohol Clin Exp Res. 2005;29:1720–1725. doi: 10.1097/01.alc.0000179220.79356.e5. [DOI] [PubMed] [Google Scholar]
- Bassareo V, Di Chiara G. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci. 1997;17:851–861. doi: 10.1523/JNEUROSCI.17-02-00851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A. Neurobiology of decision-making: risk and reward. Semin Clin Neuropsychiatry. 2001;6:205–216. doi: 10.1053/scnp.2001.22927. [DOI] [PubMed] [Google Scholar]
- Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8:1458–1463. doi: 10.1038/nn1584. [DOI] [PubMed] [Google Scholar]
- Belin D, Deroche-Gamonet V (2012) Responses to novelty and vulnerability to cocaine addiction: contribution of a multi-symptomatic animal model. Cold Spring Harb Perspect Med 2(11). doi:10.1101/cshperspect.a011940 [DOI] [PMC free article] [PubMed]
- Belin D, Everitt BJ. Cocaine seeking habits depend upon dopamine-dependent serial connectivity linking the ventral with the dorsal striatum. Neuron. 2008;57:432–441. doi: 10.1016/j.neuron.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Belin D, Mar AC, Dalley JW, Robbins TW, Everitt BJ. High impulsivity predicts the switch to compulsive cocaine-taking. Science. 2008;320:1352–1355. doi: 10.1126/science.1158136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin D, Balado E, Piazza PV, Deroche-Gamonet V. Pattern of intake and drug craving predict the development of cocaine addiction-like behavior in rats. Biol Psychiatry. 2009;65:863–868. doi: 10.1016/j.biopsych.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: relevance for the understanding of addiction. Behav Brain Res. 2009;199:89–102. doi: 10.1016/j.bbr.2008.09.027. [DOI] [PubMed] [Google Scholar]
- Belin D, Berson N, Balado E, Piazza PV, Deroche-Gamonet V. High-novelty-preference rats are predisposed to compulsive cocaine self-administration. Neuropsychopharmacology. 2011;36:569–579. doi: 10.1038/npp.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin D, Belin-Rauscent A, Murray JE, Everitt BJ. Addiction: failure of control over maladaptive incentive habits. Curr Opin Neurobiol. 2013 doi: 10.1016/j.conb.2013.01.025. [DOI] [PubMed] [Google Scholar]
- Belin-Rauscent A, Everitt BJ, Belin D. Intrastriatal shifts mediate the transition from drug-seeking actions to habits. Biol Psychiatry. 2012;72:343–345. doi: 10.1016/j.biopsych.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Bernard C (1865) Introduction à l’étude de la médecine expérimentale
- Bontempi B, Laurent-Demir C, Destrade C, Jaffard R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature. 1999;400:671–675. doi: 10.1038/23270. [DOI] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, Shaham Y (2013) The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl) [DOI] [PMC free article] [PubMed]
- Bozarth MA, Wise RA. Toxicity associated with long-term intravenous heroin and cocaine self-administration in the rat. Jama. 1985;254:81–83. [PubMed] [Google Scholar]
- Brady JV. A comparative approach to the evaluation of drug effects upon affective behavior. Ann N Y Acad Sci. 1956;64:632–643. doi: 10.1111/j.1749-6632.1956.tb36837.x. [DOI] [PubMed] [Google Scholar]
- Bush DEA, Vaccarino FJ. Individual differences in elevated plus-maze exploration predicted progressive-ratio cocaine self-administration break points in Wistar rats. Psychopharmacology (Berl) 2007;194:211–219. doi: 10.1007/s00213-007-0835-7. [DOI] [PubMed] [Google Scholar]
- Cabib S, Orsini C, Le Moal M, Piazza PV. Abolition and reversal of strain differences in behavioral responses to drugs of abuse after a brief experience. Science. 2000;289:463–465. doi: 10.1126/science.289.5478.463. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583–590. doi: 10.1038/nrn1925. [DOI] [PubMed] [Google Scholar]
- Compton WM, Dawson DA, Goldstein RB, Grant BF (2013) Crosswalk between DSM-IV dependence and DSM-5 substance use disorders for opioids, cannabis, cocaine and alcohol. Drug Alcohol Depend [DOI] [PMC free article] [PubMed]
- Crabbe JC, Phillips TJ, Buck KJ, Cunningham CL, Belknap JK. Identifying genes for alcohol and drug sensitivity: recent progress and future directions. Trends Neurosci. 1999;22:173–179. doi: 10.1016/s0166-2236(99)01393-4. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Belknap JK. The complexity of alcohol drinking: studies in rodent genetic models. Behav Genet. 2010;40:737–750. doi: 10.1007/s10519-010-9371-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit H. Impulsivity as a determinant and consequence of drug use: a review of underlying processes. Addict Biol. 2009;14:22–31. doi: 10.1111/j.1369-1600.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche V, Piazza PV, Maccari S, Le Moal M, Simon H. Repeated corticosterone administration sensitizes the locomotor response to amphetamine. Brain Res. 1992;584:309–313. doi: 10.1016/0006-8993(92)90911-r. [DOI] [PubMed] [Google Scholar]
- Deroche V, Marinelli M, Maccari S, Le Moal M, Simon H, Piazza PV. Stress-induced sensitization and glucocorticoids. I. Sensitization of dopamine-dependent locomotor effects of amphetamine and morphine depends on stress-induced corticosterone secretion. J Neurosci. 1995;15:7181–7188. doi: 10.1523/JNEUROSCI.15-11-07181.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche V, Marinelli M, Le Moal M, Piazza PV. Glucocorticoids and behavioral effects of psychostimulants. II: cocaine intravenous self-administration and reinstatement depend on glucocorticoid levels. J Pharmacol Exp Ther. 1997;281:1401–1407. [PubMed] [Google Scholar]
- Deroche V, Le Moal M, Piazza PV. Cocaine self-administration increases the incentive motivational properties of the drug in rats. Eur J Neurosci. 1999;11:2731–2736. doi: 10.1046/j.1460-9568.1999.00696.x. [DOI] [PubMed] [Google Scholar]
- Deroche-Gamonet V, Piazza PV. Transition to addiction. In: Koob GF, Le Moal M, Thompson EB, editors. Encycl. Behav. Neurosci. Oxford: Academic; 2010. pp. 422–428. [Google Scholar]
- Deroche-Gamonet V, Piazza PV (in press) Psychobiology of cocaine addiction: contribution of a multi-symptomatic animal model of loss of control. Neuropharmacology [DOI] [PubMed]
- Deroche-Gamonet V, Sillaber I, Aouizerate B, Izawa R, Jaber M, Ghozland S, Kellendonk C, Le Moal M, Spanagel R, Schütz G, Tronche F, Piazza PV. The glucocorticoid receptor as a potential target to reduce cocaine abuse. J Neurosci. 2003;23:4785–4790. doi: 10.1523/JNEUROSCI.23-11-04785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science. 2004;305:1014–1017. doi: 10.1126/science.1099020. [DOI] [PubMed] [Google Scholar]
- Dews PB. Studies on behavior. I. Differential sensitivity to pentobarbital of pecking performance in pigeons depending on the schedule of reward. J Pharmacol Exp Ther. 1955;113:393–401. [PubMed] [Google Scholar]
- Dews PB, Morse WH. Some observations on an operant in human subjects and its modification by dextro amphetamine. J Exp Anal Behav. 1958;1:359–364. doi: 10.1901/jeab.1958.1-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol Oxf Engl. 1998;12:54–67. doi: 10.1177/026988119801200108. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doremus-Fitzwater TL, Varlinskaya EI, Spear LP. Motivational systems in adolescence: possible implications for age differences in substance abuse and other risk-taking behaviors. Brain Cogn. 2010;72:114–123. doi: 10.1016/j.bandc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek BA, van der Kam EL, van der Elst MCJ, Cools AR. Individual differences in drug dependence in rats: the role of genetic factors and life events. Eur J Pharmacol. 2005;526:251–258. doi: 10.1016/j.ejphar.2005.09.032. [DOI] [PubMed] [Google Scholar]
- EMCDDA (2011) Cost and financing of drug treatment services in Europe. Lisbon, 28 pages
- Ettenberg A. Opponent process properties of self-administered cocaine. Neurosci Biobehav Rev. 2004;27:721–728. doi: 10.1016/j.neubiorev.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Evenden JL. Varieties of impulsivity. Psychopharmacology (Berl) 1999;146:348–361. doi: 10.1007/pl00005481. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME. Psychomotor stimulant addiction: a neural systems perspective. J Neurosci. 2002;22:3312–3320. doi: 10.1523/JNEUROSCI.22-09-03312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Robinson TE. Amphetamine pretreatment accelerates the subsequent escalation of cocaine self-administration behavior. Eur Neuropsychopharmacol. 2007;17:352–357. doi: 10.1016/j.euroneuro.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Foroud T, Edenberg HJ, Crabbe JC. Genetic research: who is at risk for alcoholism. Alcohol Res Heal J Natl Inst Alcohol Abuse Alcohol. 2010;33:64–75. [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Goeders NE. Stress and cocaine addiction. J Pharmacol Exp Ther. 2002;301:785–789. doi: 10.1124/jpet.301.3.785. [DOI] [PubMed] [Google Scholar]
- Goldberg SR, Woods JH, Schuster CR. Morphine: conditioned increases in self-administration in rhesus monkeys. Science. 1969;166:1306–1307. doi: 10.1126/science.166.3910.1306. [DOI] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Yang CR, Otani S. Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biol Psychiatry. 2010;67:199–207. doi: 10.1016/j.biopsych.2009.08.026. [DOI] [PubMed] [Google Scholar]
- Harrison TR. Principles of internal medicine. New York City: McGraw Hill; 2011. [Google Scholar]
- Helie S, Roeder JL, Ashby FG. Evidence for cortical automaticity in rule-based categorization. J Neurosci. 2010;30:14225–14234. doi: 10.1523/JNEUROSCI.2393-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogarth L, Dickinson A, Duka T. The associative basis of cue-elicited drug-taking in humans. Psychopharmacology (Berl) 2010;208:337–351. doi: 10.1007/s00213-009-1735-9. [DOI] [PubMed] [Google Scholar]
- Hogarth L, Balleine BW, Corbit LH, Killcross S. Associative learning mechanisms underpinning the transition from recreational drug use to addiction. Ann N Y Acad Sci. 2013;1282:12–24. doi: 10.1111/j.1749-6632.2012.06768.x. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Smith AD, Neill DB, Justice JB., Jr Response to novelty predicts the locomotor and nucleus accumbens dopamine response to cocaine. Synapse. 1991;9:121–128. doi: 10.1002/syn.890090206. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Colvin AC, Juncos JL, Justice JB., Jr Individual differences in basal and cocaine-stimulated extracellular dopamine in the nucleus accumbens using quantitative microdialysis. Brain Res. 1992;587:306–312. doi: 10.1016/0006-8993(92)91012-4. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Taylor JR. Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl) 1999;146:373–390. doi: 10.1007/pl00005483. [DOI] [PubMed] [Google Scholar]
- Jhou TC, Good CH, Rowley CS, Xu SP, Wang H, Burnham NW, Hoffman AF, Lupica CR, Ikemoto S. Cocaine drives aversive conditioning via delayed activation of dopamine-responsive habenular and midbrain pathways. J Neurosci. 2013;33:7501–7512. doi: 10.1523/JNEUROSCI.3634-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Balster RL, Bonese K. Self-administration of psychomotor stimulant drugs: the effects of unlimited access. Pharmacol Biochem Behav. 1976;4:45–51. doi: 10.1016/0091-3057(76)90174-x. [DOI] [PubMed] [Google Scholar]
- Jueptner M, Frith CD, Brooks DJ, Frackowiak RS, Passingham RE. Anatomy of motor learning. II. Subcortical structures and learning by trial and error. J Neurophysiol. 1997;77:1325–1337. doi: 10.1152/jn.1997.77.3.1325. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. How do we determine which drug-induced neuroplastic changes are important? Nat Neurosci. 2005;8:1440–1441. doi: 10.1038/nn1105-1440. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Karni A, Meyer G, Jezzard P, Adams MM, Turner R, Ungerleider LG. Functional MRI evidence for adult motor cortex plasticity during motor skill learning. Nature. 1995;377:155–158. doi: 10.1038/377155a0. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest J-M, Berson N, Balado E, Fiancette J-F, Renault P, Piazza PV, Manzoni OJ. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry. 2012;18(6):729–737. doi: 10.1038/mp.2012.59. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelleher RT. Fixed-ratio schedules of conditioned reinforcement with chimpanzees. J Exp Anal Behav. 1958;1:281–289. doi: 10.1901/jeab.1958.1-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl M, Lemaire V, Mayo W, Abrous DN, Maccari S, Piazza PV, Le Moal M, Vallée M. Individual vulnerability to substance abuse and affective disorders: role of early environmental influences. Neurotox Res. 2002;4:281–296. doi: 10.1080/1029842021000010866. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the “dark side” of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci. 2008;363:3113–3123. doi: 10.1098/rstb.2008.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Stinus L, Le Moal M, Bloom FE. Opponent process theory of motivation: neurobiological evidence from studies of opiate dependence. Neurosci Biobehav Rev. 1989;13:135–140. doi: 10.1016/s0149-7634(89)80022-3. [DOI] [PubMed] [Google Scholar]
- Laties VG, Weiss B. Influence of drugs on behavior controlled by internal and external stimuli. J Pharmacol Exp Ther. 1966;152:388–396. [PubMed] [Google Scholar]
- Le Moal M, Stinus L, Simon H. Increased sensitivity to (+)amphetamine self-administered by rats following meso-cortico-limbic dopamine neurone destruction. Nature. 1979;280:156–158. doi: 10.1038/280156a0. [DOI] [PubMed] [Google Scholar]
- Leshner AI. Addiction is a brain disease, and it matters. Science. 1997;278:45–47. doi: 10.1126/science.278.5335.45. [DOI] [PubMed] [Google Scholar]
- Lopez-Quintero C, de los Lopez-Quintero C, de Cobos JP, Hasin DS, Okuda M, Wang S, Grant BF, Blanco C. Probability and predictors of transition from first use to dependence on nicotine, alcohol, cannabis, and cocaine: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Drug Alcohol Depend. 2011;115:120–130. doi: 10.1016/j.drugalcdep.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantsch JR, Yuferov V, Mathieu-Kia A-M, Ho A, Kreek MJ. Effects of extended access to high versus low cocaine doses on self-administration, cocaine-induced reinstatement and brain mRNA levels in rats. Psychopharmacology (Berl) 2004;175:26–36. doi: 10.1007/s00213-004-1778-x. [DOI] [PubMed] [Google Scholar]
- Marinelli M, Piazza PV. Interaction between glucocorticoid hormones, stress and psychostimulant drugs. Eur J Neurosci. 2002;16:387–394. doi: 10.1046/j.1460-9568.2002.02089.x. [DOI] [PubMed] [Google Scholar]
- Marusich JA, Bardo MT. Differences in impulsivity on a delay-discounting task predict self-administration of a low unit dose of methylphenidate in rats. Behav Pharmacol. 2009;20:447–454. doi: 10.1097/FBP.0b013e328330ad6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Rahman S, Charnigo RJ, Dwoskin LP, Crabbe JC, Bardo MT. Genetics of novelty seeking, amphetamine self-administration and reinstatement using inbred rats. Genes Brain Behav. 2010;9:790–798. doi: 10.1111/j.1601-183X.2010.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. Psychiatry. Beyond DSM: seeking a brain-based classification of mental illness. Science. 2010;327:1437. doi: 10.1126/science.327.5972.1437. [DOI] [PubMed] [Google Scholar]
- Morse WH, Skinner BF. Some factors involved in the stimulus control of operant behavior. J Exp Anal Behav. 1958;1:103–107. doi: 10.1901/jeab.1958.1-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman T, Loewenstein Y. Covariance-based synaptic plasticity in an attractor network model accounts for fast adaptation in free operant learning. J Neurosci. 2013;33:1521–1534. doi: 10.1523/JNEUROSCI.2068-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12:2439–2450. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Molecular neurobiology of drug addiction. Neuropsychopharmacology. 1994;11:77–87. doi: 10.1038/npp.1994.37. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Genes and addiction. Nat Genet. 2000;26:277–281. doi: 10.1038/81570. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- Nutt D, King LA, Saulsbury W, Blakemore C. Development of a rational scale to assess the harm of drugs of potential misuse. Lancet. 2007;369:1047–1053. doi: 10.1016/S0140-6736(07)60464-4. [DOI] [PubMed] [Google Scholar]
- O’Brien CP, Ehrman RN, Terns JN. Classical conditioning in human opioid dependence. In: Goldberg SR, Stolerman IP, editors. Behavioral analysis of drug dependence. London: Academic; 1986. p. 329. [Google Scholar]
- O’Neill B, Tilley MR, Gu HH. Cocaine produces conditioned place aversion in mice with a cocaine-insensitive dopamine transporter. Genes Brain Behav. 2013;12:34–38. doi: 10.1111/j.1601-183X.2012.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmstead MC, Lafond MV, Everitt BJ, Dickinson A. Cocaine seeking by rats is a goal-directed action. Behav Neurosci. 2001;115:394–402. [PubMed] [Google Scholar]
- Peer K, Rennert L, Lynch KG, Farrer L, Gelernter J, Kranzler HR. Prevalence of DSM-IV and DSM-5 alcohol, cocaine, opioid, and cannabis use disorders in a largely substance dependent sample. Drug Alcohol Depend. 2013;127:215–219. doi: 10.1016/j.drugalcdep.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelloux Y, Everitt BJ, Dickinson A. Compulsive drug seeking by rats under punishment: effects of drug-taking history. Psychopharmacology (Berl) 2007;194:127–137. doi: 10.1007/s00213-007-0805-0. [DOI] [PubMed] [Google Scholar]
- Perry JL, Larson EB, German JP, Madden GJ, Carroll ME. Impulsivity (delay discounting) as a predictor of acquisition of IV cocaine self-administration in female rats. Psychopharmacology (Berl) 2005;178:193–201. doi: 10.1007/s00213-004-1994-4. [DOI] [PubMed] [Google Scholar]
- Perry JL, Nelson SE, Carroll ME. Impulsive choice as a predictor of acquisition of IV cocaine self- administration and reinstatement of cocaine-seeking behavior in male and female rats. Exp Clin Psychopharmacol. 2008;16:165–177. doi: 10.1037/1064-1297.16.2.165. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Le Moal M. Pathophysiological basis of vulnerability to drug abuse: role of an interaction between stress, glucocorticoids, and dopaminergic neurons. Annu Rev Pharmacol Toxicol. 1996;36:359–378. doi: 10.1146/annurev.pa.36.040196.002043. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Le Moal M. Glucocorticoids as a biological substrate of reward: physiological and pathophysiological implications. Brain Res Brain Res Rev. 1997;25:359–372. doi: 10.1016/s0165-0173(97)00025-8. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Deminière JM, Le Moal M, Simon H. Factors that predict individual vulnerability to amphetamine self-administration. Science. 1989;245:1511–1513. doi: 10.1126/science.2781295. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Maccari S, Deminière JM, Le Moal M, Mormède P, Simon H. Corticosterone levels determine individual vulnerability to amphetamine self-administration. Proc Natl Acad Sci U S A. 1991;88:2088–2092. doi: 10.1073/pnas.88.6.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza PV, Rougé-Pont F, Deminière JM, Kharoubi M, Le Moal M, Simon H. Dopaminergic activity is reduced in the prefrontal cortex and increased in the nucleus accumbens of rats predisposed to develop amphetamine self-administration. Brain Res. 1991;567:169–174. doi: 10.1016/0006-8993(91)91452-7. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Marinelli M, Rougé-Pont F, Deroche V, Maccari S, Simon H, Le Moal M. Stress, glucocorticoids, and mesencephalic dopaminergic neurons: a pathophysiological chain determining vulnerability to psychostimulant abuse. NIDA Res Monogr. 1996;163:277–299. [PubMed] [Google Scholar]
- Piazza PV, Deroche-Gamonent V, Rouge-Pont F, Le Moal M. Vertical shifts in self-administration dose–response functions predict a drug-vulnerable phenotype predisposed to addiction. J Neurosci. 2000;20:4226–4232. doi: 10.1523/JNEUROSCI.20-11-04226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens R, Thompson T. Cocaine-reinforced behavior in rats: effects of reinforcement magnitude and fixed-ratio size. J Pharmacol Exp Ther. 1968;161:122–129. [PubMed] [Google Scholar]
- Pickens CL, Airavaara M, Theberge F, Fanous S, Hope BT, Shaham Y. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–420. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre PJ, Vezina P. Predisposition to self-administer amphetamine: the contribution of response to novelty and prior exposure to the drug. Psychopharmacology (Berl) 1997;129:277–284. doi: 10.1007/s002130050191. [DOI] [PubMed] [Google Scholar]
- Risner ME, Jones BE. Characteristics of unlimited access to self-administered stimulant infusions in dogs. Biol Psychiatry. 1976;11:625–634. [PubMed] [Google Scholar]
- Robbins TW, Ersche KD, Everitt BJ. Drug addiction and the memory systems of the brain. Ann N Y Acad Sci. 2008;1141:1–21. doi: 10.1196/annals.1441.020. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ, Nutt DJ. Introduction. The neurobiology of drug addiction: new vistas. Philos Trans R Soc Lond B Biol Sci. 2008;363:3109–3111. doi: 10.1098/rstb.2008.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DCS, Brebner K, Vincler M, Lynch WJ. Patterns of cocaine self-administration in rats produced by various access conditions under a discrete trials procedure. Drug Alcohol Depend. 2002;67:291–299. doi: 10.1016/s0376-8716(02)00083-2. [DOI] [PubMed] [Google Scholar]
- Roberts DCS, Morgan D, Liu Y. How to make a rat addicted to cocaine. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1614–1624. doi: 10.1016/j.pnpbp.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The psychology and neurobiology of addiction: an incentive-sensitization view. Addict Abingdon Engl. 2000;95(Suppl 2):S91–S117. doi: 10.1080/09652140050111681. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addict Abingdon Engl. 2001;96:103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Addiction. Annu Rev Psychol. 2003;54:25–53. doi: 10.1146/annurev.psych.54.101601.145237. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Review. The incentive sensitization theory of addiction: some current issues. Philos Trans R Soc Lond B Biol Sci. 2008;363:3137–3146. doi: 10.1098/rstb.2008.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Fabbricatore AT, Barker DJ, Ma S, Pawlak AP, West MO. Evidence for habitual and goal-directed behavior following devaluation of cocaine: a multifaceted interpretation of relapse. PLoS One. 2009;4:e7170. doi: 10.1371/journal.pone.0007170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougé-Pont F, Piazza PV, Kharouby M, Le Moal M, Simon H. Higher and longer stress-induced increase in dopamine concentrations in the nucleus accumbens of animals predisposed to amphetamine self-administration. A microdialysis study. Brain Res. 1993;602:169–174. doi: 10.1016/0006-8993(93)90260-t. [DOI] [PubMed] [Google Scholar]
- Rougé-Pont F, Marinelli M, Le Moal M, Simon H, Piazza PV. Stress-induced sensitization and glucocorticoids. II. Sensitization of the increase in extracellular dopamine induced by cocaine depends on stress-induced corticosterone secretion. J Neurosci. 1995;15:7189–7195. doi: 10.1523/JNEUROSCI.15-11-07189.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougé-Pont F, Deroche V, Le Moal M, Piazza PV. Individual differences in stress-induced dopamine release in the nucleus accumbens are influenced by corticosterone. Eur J Neurosci. 1998;10:3903–3907. doi: 10.1046/j.1460-9568.1998.00438.x. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon DP, Butters N. Neurobiology of skill and habit learning. Curr Opin Neurobiol. 1995;5:184–190. doi: 10.1016/0959-4388(95)80025-5. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- Schechter MD. Rats bred for differences in preference to cocaine: other behavioral measurements. Pharmacol Biochem Behav. 1992;43:1015–1021. doi: 10.1016/0091-3057(92)90475-u. [DOI] [PubMed] [Google Scholar]
- Schramm-Sapyta NL, Cauley MC, Stangl DK, Glowacz S, Stepp KA, Levin ED, Kuhn CM. Role of individual and developmental differences in voluntary cocaine intake in rats. Psychopharmacology (Berl) 2011;215:493–504. doi: 10.1007/s00213-011-2216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- Sheeran P, Aarts H, Custers R, Rivis A, Webb TL, Cooke R. The goal-dependent automaticity of drinking habits. Br J Soc Psychol Br Psychol Soc. 2005;44:47–63. doi: 10.1348/014466604X23446. [DOI] [PubMed] [Google Scholar]
- Siegel RK. Intoxication: the universal drive for mind-altering substances. Rochester: Park Street Press; 2005. [Google Scholar]
- Skinner BF. On the conditions of elicitation of certain eating reflexes. Proc Natl Acad Sci U S A. 1930;16:433–438. doi: 10.1073/pnas.16.6.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner BF. Are theories of learning necessary? Psychol Rev. 1950;57:193–216. doi: 10.1037/h0054367. [DOI] [PubMed] [Google Scholar]
- Stewart J, de Wit H, Eikelboom R. Role of unconditioned and conditioned drug effects in the self-administration of opiates and stimulants. Psychol Rev. 1984;91:251–268. [PubMed] [Google Scholar]
- Suto N, Austin JD, Tanabe LM, Kramer MK, Wright DA, Vezina P. Previous exposure to VTA amphetamine enhances cocaine self-administration under a progressive ratio schedule in a D1 dopamine receptor dependent manner. Neuropsychopharmacology. 2002;27:970–979. doi: 10.1016/S0893-133X(02)00379-2. [DOI] [PubMed] [Google Scholar]
- Suto N, Tanabe LM, Austin JD, Creekmore E, Vezina P. Previous exposure to VTA amphetamine enhances cocaine self-administration under a progressive ratio schedule in an NMDA, AMPA/kainate, and metabotropic glutamate receptor-dependent manner. Neuropsychopharmacology. 2003;28:629–639. doi: 10.1038/sj.npp.1300075. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T, Schuster CR. Morphine self-administration, food-reinforced, and avoidance behaviors in rhesus monkeys. Psychopharmacologia. 1964;5:87–94. doi: 10.1007/BF00413045. [DOI] [PubMed] [Google Scholar]
- Tiffany ST. A cognitive model of drug urges and drug-use behavior: role of automatic and nonautomatic processes. Psychol Rev. 1990;97:147–168. doi: 10.1037/0033-295x.97.2.147. [DOI] [PubMed] [Google Scholar]
- Tornatzky W, Miczek KA. Cocaine self-administration “binges”: transition from behavioral and autonomic regulation toward homeostatic dysregulation in rats. Psychopharmacology (Berl) 2000;148:289–298. doi: 10.1007/s002130050053. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault J-A, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Van der Kam EL, Ellenbroek BA, Cools AR. Gene–environment interactions determine the individual variability in cocaine self-administration. Neuropharmacology. 2005;48:685–695. doi: 10.1016/j.neuropharm.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJMJ, Ahmed SH. Animal studies of addictive behavior. Cold Spring Harb Perspect Med. 2013;3(4):a011932. doi: 10.1101/cshperspect.a011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJMJ, Everitt BJ. Drug seeking becomes compulsive after prolonged cocaine self-administration. Science. 2004;305:1017–1019. doi: 10.1126/science.1098975. [DOI] [PubMed] [Google Scholar]
- Veeneman MMJ, Broekhoven MH, Damsteegt R, Vanderschuren LJMJ. Distinct contributions of dopamine in the dorsolateral striatum and nucleus accumbens shell to the reinforcing properties of cocaine. Neuropsychopharmacology. 2012;37:487–498. doi: 10.1038/npp.2011.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P, Pierre PJ, Lorrain DS. The effect of previous exposure to amphetamine on drug-induced locomotion and self-administration of a low dose of the drug. Psychopharmacology (Berl) 1999;147:125–134. doi: 10.1007/s002130051152. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Baler RD, Goldstein RZ. Addiction: pulling at the neural threads of social behaviors. Neuron. 2011;69:599–602. doi: 10.1016/j.neuron.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Willuhn I, Clark JJ, Phillips PEM. Phasic dopamine release in appetitive behaviors and drug addiction. Curr Drug Abuse Rev. 2009;2:195–213. doi: 10.2174/1874473710902020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks JR. Experimental morphine addiction: method for automatic intravenous injections in unrestrained rats. Science. 1962;138:143–144. doi: 10.1126/science.138.3537.143. [DOI] [PubMed] [Google Scholar]
- Weiss B, Laties VG. Effects of amphetamine, chlorpromazine, pentobarbital, and ethanol on operant response duration. J Pharmacol Exp Ther. 1964;144:17–23. [PubMed] [Google Scholar]
- Wenzel JM, Waldroup SA, Haber ZM, Su ZI, Ben-Shahar O, Ettenberg A. Effects of lidocaine-induced inactivation of the bed nucleus of the stria terminalis, the central or the basolateral nucleus of the amygdala on the opponent-process actions of self-administered cocaine in rats. Psychopharmacology (Berl) 2011;217:221–230. doi: 10.1007/s00213-011-2267-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- Wikler A, Pescor FT. Classical conditioning of a morphine abstinence phenomenon, reinforcement of opioid-drinking behavior and “relapse” in morphine-addicted rats. Psychopharmacologia. 1967;10:255–284. doi: 10.1007/BF00401386. [DOI] [PubMed] [Google Scholar]
- Wikler A, Pescor FT, Miller D, Norrell H. Persistent potency of a secondary (conditioned) reinforcer following withdrawal of morphine from physically dependent rats. Psychopharmacologia. 1971;20:103–117. doi: 10.1007/BF00404365. [DOI] [PubMed] [Google Scholar]
- Willuhn I, Burgeno LM, Everitt BJ, Phillips PEM. Hierarchical recruitment of phasic dopamine signaling in the striatum during the progression of cocaine use. Proc Natl Acad Sci U S A. 2012;109:20703–20708. doi: 10.1073/pnas.1213460109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winocur G, Moscovitch M, Bontempi B. Memory formation and long-term retention in humans and animals: convergence towards a transformation account of hippocampal-neocortical interactions. Neuropsychologia. 2010;48:2339–2356. doi: 10.1016/j.neuropsychologia.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Winstanley CA, Olausson P, Taylor JR, Jentsch JD. Insight into the relationship between impulsivity and substance abuse from studies using animal models. Alcohol Clin Exp Res. 2010;34:1306–1318. doi: 10.1111/j.1530-0277.2010.01215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Catecholamine theories of reward: a critical review. Brain Res. 1978;152:215–247. doi: 10.1016/0006-8993(78)90253-6. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neural mechanisms of the reinforcing action of cocaine. NIDA Res Monogr. 1984;50:15–33. [PubMed] [Google Scholar]
- Wise RA. The role of reward pathways in the development of drug dependence. Pharmacol Ther. 1987;35:227–263. doi: 10.1016/0163-7258(87)90108-2. [DOI] [PubMed] [Google Scholar]
- Wise RA. Cocaine reward and cocaine craving: the role of dopamine in perspective. NIDA Res Monogr. 1994;145:191–206. [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. Action of drugs of abuse on brain reward systems: an update with specific attention to opiates. Pharmacol Biochem Behav. 1982;17:239–243. doi: 10.1016/0091-3057(82)90076-4. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Wolf ME. Addiction: making the connection between behavioral changes and neuronal plasticity in specific pathways. Mol Interv. 2002;2:146–157. doi: 10.1124/mi.2.3.146. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–398. doi: 10.1016/j.tins.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. Regulation of AMPA receptor trafficking in the nucleus accumbens by dopamine and cocaine. Neurotox Res. 2010;18:393–409. doi: 10.1007/s12640-010-9176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffgramm J. An ethopharmacological approach to the development of drug addiction. Neurosci Biobehav Rev. 1991;15:515–519. doi: 10.1016/s0149-7634(05)80142-3. [DOI] [PubMed] [Google Scholar]
- Wolffgramm J, Heyne A. Social behavior, dominance, and social deprivation of rats determine drug choice. Pharmacol Biochem Behav. 1991;38:389–399. doi: 10.1016/0091-3057(91)90297-f. [DOI] [PubMed] [Google Scholar]
- Wolffgramm J, Galli G, Thimm F, Heyne A. Animal models of addiction: models for therapeutic strategies? J Neural Transm Vienna Austria. 2000;107:649–668. doi: 10.1007/s007020070067. [DOI] [PubMed] [Google Scholar]
- Wu T, Chan P, Hallett M. Modifications of the interactions in the motor networks when a movement becomes automatic. J Physiol. 2008;586:4295–4304. doi: 10.1113/jphysiol.2008.153445. [DOI] [PMC free article] [PubMed] [Google Scholar]