

Abstract
Electrochemically controlled, reversible assembly of biopolymers into hydrogel structures is a promising technique for on-demand cell or drug encapsulation and release systems. An electrochemically sol-gel transition has been demonstrated in regenerated Bombyx mori silk fibroin, offering a controllable way to generate biocompatible and reversible adhesives and other biomedical materials. Despite the involvement of an electrochemically triggered electrophoretic migration of the silk molecules, the mechanism of the reversible electrogelation remains unclear. It is, however, known that the freshly prepared silk electrogels (e-gels) adopt a predominantly random coil conformation, indicating a lack of crosslinking as well as thermal, mechanical and morphological stabilities. In the present work, the tuning of covalent and physical β-sheet crosslinks in silk hydrogels was studied for programming the structural properties. Scanning electron microscopy (SEM) revealed delicate morphology, including locally aligned fibrillar structures, in silk e-gels, preserved by combining glutaraldehyde-crosslinking and ethanol dehydration. Fourier transform infrared (FTIR) spectroscopic analysis of either electrogelled, vortex-induced or spontaneously formed silk hydrogels showed that the secondary structure of silk e-gels was tunable between non β-sheet dominated and β-sheet dominated states. Dynamic oscillatory rheology confirmed the mechanical reinforcement of silk e-gels provided by controlled chemical and physical crosslinks. The selective incorporation of either chemical or physical or both crosslinks into the electrochemically-responsive, originally unstructured silk e-gel should help in the design for electrochemically-responsive protein polymers.
Keywords: silk, electrogelation, crosslinking, morphological analysis, mechanical reinforcement
1. Introduction
Regenerated Bombyx mori silk fibroin formed into materials possess excellent biological and mechanical properties, including biocompatibility, programmable biodegradability, and remarkable strength and toughness.1–3 Diverse and tunable properties of silk are possible through structural variations under different processing conditions, e.g. hydrogelation into 3-D scaffolds,4,5 casting into 2-D films,6,7 electrospinning for 1-D fibers,8 and post-annealing treatments.3
Among the hydrogelation mechanisms with freshly regenerated silk fibroin, pH-controlled sol-gel transitions are reversible. As the pH approaches the isoelectric point (pI = 4.2), silk proteins aggregate without significant change in conformation.9,10 These acid-induced gels can be reversed by shifting the pH back toward alkaline conditions. However, the overall acidic environment of silk pH-gels is unfavorable for cell-based and other eco- or bio-friendly applications.
Reversible changes in solution pH can also be obtained electrochemically. Controllable deposition and growth of silk hydrogels on electrode surfaces has been demonstrated by passing an electric current through the silk solution.11,12 At sufficient voltage, water ionizes to form hydrogen ions, H+, and hydroxide ions, OH−. The ions migrate under the influence of the electric field and act on the local pH. Finite-element modeling revealed ion electrodiffusion as a key mechanism governing silk electrogelation.13 In this process, the acid exposure of cells can be limited to a few seconds, which suffices for the growth of a bulk gel. The method also facilitates better diffusion of the nutrients for cells to survive, and allows cell recovery from the pH damage at least in local areas where the pH returns to normal upon the removal of the electric current.
Interestingly, the gel phase in the electrogelation, known as silk e-gel, was dominated by random coil and α-helix conformations, and possessed strong adhesion to a variety of surfaces.11,12 The secondary structures of silk after regeneration spontaneously relax toward more ordered states dominated by β-sheets. The formation of thermodynamically stable β-sheet structures serves as physical crosslinks, stabilizing silk materials.14 Sufficient energy to overcome the energy barrier for β-sheet crystallization by heating,15 vortex shearing16 and ultrasonication,17 accelerates the hydrogelation kinetics. β-sheet-enriched silk hydrogels show improved mechanical strength over the unstructured silk hydrogels.14 Therefore, in contrast to its β-sheet dominated counterparts, the unstructured silk e-gel provides a unique opportunity for selective incorporation of crosslinks into the silk network, and thereby programming the degradation for drug/cell release.
The 3-D micro-architecture of aqueous silk hydrogels is difficult to preserve for detailed morphological characterization, such as by electron microscopy, using conventional freeze drying techniques. The appearance of freezing artifacts, caused by ice crystal formation, makes morphological interpretation difficult. This is a complication with any highly hydrated biomaterial structure, prompting the need for alternative techniques to gain insight into processing-induced structural and morphological features. The self-assembly of silk fibroin in dilute and semi-dilute solutions has been studied using noncontact mode atomic force microscopy (AFM).18 The nanomechanical stimulus applied at the tip during the lateral scanning of AFM imaging accelerated β-sheet formation in silk-like proteins.19 Environmental SEM allows sample imaging with spatial resolution in the nanometer range in a water vapor environment, but is limited to surface investigation.20 Cross-sectioning of a cryogenically prepared sample using focused ion beam (FIB) milling is costly and time-intensive compared with conventional SEM.21
In the present work, crosslinking strategies were developed and implemented to facilitate tunable structure and properties of silk e-gels, which, essentially in random coil state, are lacking thermal, mechanical and morphological stability for bio-applications. Glutaraldehyde was used as the chemical crosslinker, and physical β-sheet crosslinks were induced by ethanol dehydration. Fourier self-deconvolution analysis was performed on the Amide I band of Fourier transform infrared (FTIR) spectra with the results of secondary structure formation displaying tunability in the silk e-gel. SEM studies revealed improved morphological preservation in all of the three types of silk hydrogels, produced by electrogelation, vortex shearing, or spontaneously, after crosslinking. Dynamic oscillatory rheology was employed for in-situ induction of β-sheet crosslinks. The reinforcing effects of these chemical and physical crosslinking methods on the mechanical properties of electrogelled silk are also reported. The demonstration presented in this work of the tunability control over the structural properties of regenerated silk should provide insight into the design of novel electrochemically-responsive protein polymers.
2. Materials and Methods
2.1 Materials
Cocoons of Bombyx mori silkworm silk were supplied by Tajima Shoji Co., LTD (Sumiyoshicho, Naka-ku, Yokohama, Japan). Sodium carbonate (Na2CO3, 99 %), lithium bromide (LiBr, 99 %), α-chymotrypsin (Type II, three times crystallized from bovine pancreas) were purchased from Sigma-Aldrich (St. Louis, MO). Glutaraldehyde (GA, 20%) aqueous solution was obtained from Electron Microscopy Sciences (Fort Washington, PA), and Tris-Hydrochloride buffer (Tri-HCl buffer, 1M, pH 8.0) from Boston BioProducts (Ashland, MA). All chemicals were used without further purification. Slide-A-Lyzer® dialysis cassettes (3.5K molecular weight cutoff) were purchased from Pierce Biotechnology (Rockford, IL) and pre-soaked in distilled water for 10 minutes before use.
2.2 Preparation of regenerated silk fibroin solutions
Cocoons of B. mori were cut into small pieces and treated in a boiling aqueous solution of sodium carbonate to remove the sericin. Every 5 grams of the raw silk material were degummed in one liter of 0.02 M Na2CO3 solution for 10 minutes.22 The degummed silk fibers were washed in many changes of distilled water, and air-dried at room temperature overnight. The degummed dry silk was dissolved in a 9.3 M aqueous solution of LiBr, at a ratio of 1 g of silk fibers per 4 mL of LiBr solution. The dissolution process was carried out at 60°C for 4 h. Every 12 mL of the silk fibroin solution was placed in a cellulose membrane based Slide-A-Lyzer® dialysis cassette, and dialyzed against 1 L of distilled water to remove the salt 18. Water used for dialysis was changed after 1, 3, 6, 12, 24, 36, 48, 60 and 72 hr(s). The regenerated silk fibroin stock solution (5 wt %) was stored at 4°C for further use.
2.3 Preparation of silk hydrogels
Three types of silk hydrogels were prepared as described below, and then washed with distilled water to remove any non-gelled silk molecules.
2.3.1 Electrogelation of silk fibroin
A 1 mL aliquot of the freshly regenerated silk solution (5 wt %) was held between two pieces of aluminum foil, the top serving as the negative electrode and the bottom as the positive electrode, respectively. The two parallel electrodes were spaced 5 mm apart by a sheet of silicone rubber (polydimethylsiloxane, PDMS, Sylgard 184, Dow Corning Corp., Midland, MI). A hole with a 15 mm diameter was punched through the PDMS spacer, and served as the solution reservoir. The electrochemical growth of the silk hydrogel was induced at the solution/positive electrode interface by applying a constant voltage of 25 V (electric field of 5 V/mm) across the parallel electrodes using a DC power supply (Agilent E3612A DC power supply, Agilent Technologies, Inc., Englewood, CO). During the 5 min electrogelation process, the interface between the optically clear solution and the yellowish e-gel advanced upwards (from the positive to the negative electrode), as indicated by the arrow shown in Figure 1.
Figure 1.
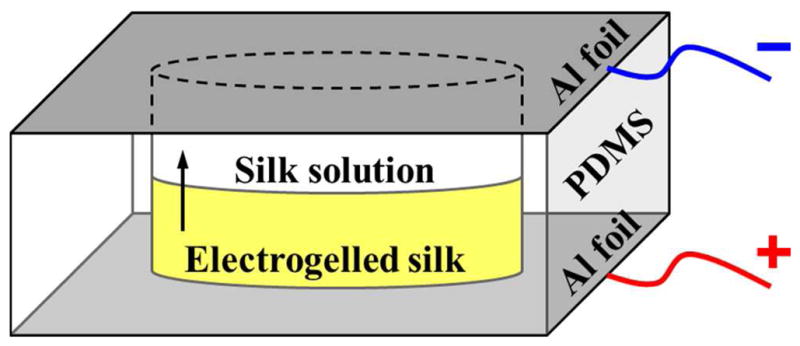
Schematic diagram of the experimental setup for the electrogelation of the regenerated silk. The silk solution was filled in a 15 mm hole in a 5 mm thick PDMS spacer, and sandwiched between two aluminum foil sheets serving as the electrodes. The application of a 25 V DC electric potential across the parallel electrodes induced the formation of yellowish electrogelled silk from the optically transparent solution at the positive electrode surface. During the 5 min of electrogelation process, the gelation front proceeded upward.
2.3.2 Vortex-induced hydro-gelation of silk fibroin
A 1.8 mL aliquot of the regenerated silk solution (5 wt %) was pipetted into a 2 ml Eppendorf tube. The tube was then vortexed for 5 min at 3,200 rpm using a vortexer (Fisher Scientific, Hampton, NH). The silk molecules experiencing large vortex shearing were physically crosslinked with the mechanically-induced β-sheet structures, and underwent a sol-to-gel transition.16
2.3.3 Spontaneous hydro-gelation of silk fibroin
A 5 mL aliquot of the regenerated silk solution (5 wt %) was placed in a 10 mL centrifuge tube, and kept at 4°C. Silk molecules exhibiting random coil and α-helix conformations after regeneration start to self-assemble into β-sheet dominated conformations, and therefore transform into a thermodynamically more stable gel state.15 The hydrogelation kinetics were typically completed after 3 ~ 4 weeks.
2.3.4 Adding chemical crosslinks to silk hydrogels using glutaraldehyde (GA)
Glutaraldehyde was found to react with several side chains in proteins, including the ε-amino groups of lysine23 and the phenol groups of tyrosine24. Covalent modification and crosslinking of silk fibroin with glutaraldehyde have been demonstrated previously.25,26 A 20 % w/w glutaraldehyde solution was added to each of the three silk hydrogels, giving a final crosslinker to silk weight ratio in the range of about 0.2:1.27 The crosslinking reaction was allowed to proceed at room temperature for 20 minutes. All of the crosslinked silk samples were subsequently rinsed with distilled water prior to drying or mechanical testing.
2.4 Morphological analysis of silk hydrogels by scanning electron microscopy
2.4.1 Freeze drying of silk hydrogels
The freshly prepared un-treated and glutaraldehyde-crosslinked silk hydrogels were frozen overnight at −20°C. The frozen hydrogels were then completely dried in a freezer-dryer (Labconco FreeZone® Plus Freeze Dry Systems, 7960042, Labconco Corp., Kansas City, MO) for 24 ~ 48 hrs prior to analysis.
2.4.2 Critical point drying of silk hydrogels
Part of the freshly prepared silk hydrogels were crosslinked with glutaraldehyde (Section 2.3.4), and progressively dehydrated in a graded series of ethanols (30 %, 50 %, 75 %, 95 % and twice in 100 %, 30 min at each concentration). The samples were subsequently dried by critical point drying with a liquid CO2 dryer (AutoSamdri-815, Tousimis Research Corp., Rockville, MD).
2.4.3 Enzymatic degradation of the less crystalline regions in silk
The three types of the glutaraldehyde-crosslinked, progressively dehydrated, and critical point dried silk hydrogels were cut into small pieces. For a silk sample weighing 40 mg, 10 mL of Tris-HCl buffer (pH 8.0, 80 mM) containing 40 mg of α-chymotrypsin (40 units/mg silk) was used for the selective digestion of the less crystalline regions.28 Enzymatic digestion was carried out at 25°C for 3 days. The samples in the crystalline β form after enzymatic degradation were then rinsed three times with distilled water, and dried in air overnight.
2.4.4 Scanning electron microscopy (SEM) of dried silk hydrogels
Morphological analysis of the electrogelled, vortex-induced, and spontaneously formed silk hydrogels was conducted using an SEM(Zeiss UltraPlus SEM or Zeiss Supra 55 VP SEM, Carl Zeiss SMT Inc., Peabody, MA) at a voltage of 2 ~ 3 kV. After drying as described above, all the samples were coated with a thin layer (10 nm thick) of Pt/Pd using a sputter coater (208HR, Cressington Scientific Instruments Inc., Cranberry Twp, PA) prior to imaging.
2.5 Tuning physical β-sheet crosslinks in electrogelled silk by mechanical shearing
2.5.1 Rheological characterization of mechanical reinforcement of electrogelled silk under dynamic shear loading
The electrogelled silk samples, untreated and chemically crosslinked using glutaraldehyde, were analyzed by rheological techniques on an ARES strain-controlled rheometer (TA Instruments, New Castle, DE) with a 25 mm diameter parallel plate geometry. The outer edge of the hydrogel was coated in mineral oil to minimize water evaporation during measurements. Non-equilibrium dynamics were observed on the untreated silk e-gel in rheological studies.12 The linear viscoelastic region of the glutaraldehyde-crosslinked silk e-gel was determined by a strain sweep from 0.1 to 100 % strain amplitude at 1 rad/sec frequency. Mechanical reinforcement on the dynamic shear moduli (G′ and G″) of silk e-gels during large-amplitude oscillatory shear (LAOS) was probed using 1 % strain amplitude at 1 rad/sec frequency.
2.5.2 Measurement of water content in eletro-gelated silk during the oscillatory shear tests
The silk e-gels (either un-treated or crosslinked with glutaraldehyde) were weighed to obtain the hydrated weight (Wh) at different times as the rheological characterization. The gels were then lyophilized and weighed again to obtain the dry weight (Wd). The degree of hydration of the hydrogels was calculated based on the following equation: (Wh − Wd)/Ws × 100 %. The measurements were performed on at least three samples at each time point, and mean and standard deviation are presented.
2.6 Secondary structure analysis of silk hydrogels by Fourier Transform Infrared spectroscopy
Fourier Transform Infrared (FTIR) secondary structure analysis was performed on the silk gel samples prepared as previously described,3,29 either dried by the different drying methods or hydrated after shear tests. For each mid-infrared spectrum (400 ~ 4,000 cm−1), 128 scans were collected in reflection mode at 4 cm−1 resolution using a JASCO FTIR 6200 spectrometer (JASCO, Tokyo, Japan) equipped with a Smart MIRacle™ horizontal attenuated total reflection (ATR) Ge crystal accessory (Pike Technologies, Inc., Madison, WI). The Amide I bands (1595 ~ 1705 cm−1) were resolved by Fourier self-deconvolution (FSD) in the Opus 5.0 software package (Bruker Optics, Billerica, MA) using a Lorentzian line shape and parameters equivalent to 20 cm−1 bandwidth at half-height and a noise suppression factor of 0.3. Gaussian curve-fitting was then conducted on the amide I spectra after resolution-enhancement FSD. The secondary structure content of silk were calculated from the areas of the individual assigned bands and their fraction of the total area in the amide I region. For each type of silk gel, the conformational characterization was carried out on at least three samples prepared independently. Further comparison among the three types of silk gels was made based on the average percentage of secondary structure.
3. Results and Discussion
3.1 Tuning the chemical and physical crosslinks in silk hydrogels for morphological analysis
The interconnected porous network structures in the silk hydrogels: electrogelled, vortex-induced and spontaneously formed, as shown in Figure 2a,c,e, respectively, were damaged during freeze drying. The SEMs of these freeze-dried silk samples revealed the presence of freezing artifacts, including distortions in pore size and shape, and cracks on the surface. The addition of chemical crosslinks into the silk hydrogel networks, using glutaraldehyde as crosslinker, preserved fibril micro-morphology after freeze-drying (Figure 2b,d,f) to different extents. Web-like structures were observed in the electrogelled silk (circled in Figure 2b) and the vortex-induced silk gel (Figure 2d) after glutaraldehyde treatment. The three dimensional pore structures were retained after freeze drying of the crosslinked spontaneously formed silk gel (Figure 2f).
Figure 2.

SEMs of the freeze-dried silk hydrogels showing an improved morphological preservation provided by chemical crosslinking. The network structures, damaged during freeze drying in (a) the electrogelled silk, (c) the vortex-induced silk gel and (e) the spontaneously formed silk gel, were preserved in (b), (d) and (f), respectively, by adding glutaraldehyde crosslinks to the silk structure.
Figure 3 shows the FTIR spectra of the freeze-dried electrogelled silk, untreated (top) and glutaraldehyde-crosslinked (bottom), in the high frequency region between 3500 and 2800 cm−1, characteristic of the N-H and C-H stretching vibrational modes.30,31 The bands at 2972 and 2930 cm−1 were assigned to the CH3 and CH2 asymmetric stretching modes, respectively, as listed in the inset of Figure 3.30,31 Crosslinking of silk was confirmed by a decrease in intensity of the CH3 asymmetric stretching mode relative to that of the CH2 mode after glutaraldehyde treatment. This indicates the introduction of additional CH2 groups into the silk structure by glutaraldehyde, CH2(CH2CHO)2.32 The crosslinking-induced relative change in the νas(CH3)/ νas(CH2) intensity ratio was also seen in the FTIR spectra of the vortex-induced and spontaneously formed silk gels (Figure S1).
Figure 3.

FTIR spectra of the freeze-dried electrogelled silk: untreated (top dashed) and glutaraldehyde-crosslinked (bottom solid) in the 3500 ~ 2800 cm−1 range. The principle IR bands and their assignments are summarized in the inset.26,27 The chemically crosslinked silk e-gel exhibits a decrease in the intensity of the 2972 cm−1 CH3 asymmetric stretching vibration band suggesting the involvement of the side chain methyl groups in the crosslinking. Similar results were obtained on the vortex-induced and spontaneously formed silk gels (Figure S1).
The heavy chain of B. mori silk fibroin is alanine rich,3 with a repeat motif, GAGAGS, that forms stable β-sheet structures. Secondary structure analysis by FTIR spectroscopy provides a measure of the relative percentage of β-sheet content for each of the three types of the freeze-dried silk gels. As shown in Figure 5, self-deconvolution data reveal an increase in β-sheet content of 15 ~ 16 % for the electrogelled silk, 24 ~ 35 % for the vortex-induced silk hydrogel, and 44 ~ 45 % for the spontaneously formed silk hydrogel.
Figure 5.

FTIR analysis of silk secondary structure in dried hydrogels.24 From top to bottom: (a, b) electrogelled silk, (c, d) vortex-induced silk hydrogel, and (e, f) spontaneously formed silk hydrogel. (Left) Amide I region (1585 ~ 1785 cm−1) of the FTIR spectra for silk hydrogels: freeze-dried (dotted line), glutaraldehyde-crosslinked and freeze-dried (dash-dot line), and glutaraldehyde, dehydrated with ethanol and critical point dried (solid line). (Right) The percentage of β-sheet content determined by Fourier self-deconvolution of the amide I band: freeze-dried (⊞), glutaraldehyde-crosslinked and freeze-dried (▧), and glutaraldehyde, dehydrated with ethanol and critical point dried (■).
The SEM images (Figure 2) and FTIR data (Figure 3 and 5) of the freeze dried silk hydrogels show that the mechanical strength, originated from either physical (β-sheet) or chemical (glutaraldehyde) crosslinks, is critical for preserving the morphological structures during the drying process. Ethanol is commonly used in critical point drying to replace water in hydrogels due to its miscibility with both water and liquid carbon dioxide. For silk, the ethanol treatment is known to induce a conformational transition from random coil to β-sheet structures.33 The combination of chemical and physical crosslinking makes the critical point drying a useful method to preserve the ultra-structure and surface structure of silk hydrogels for morphological study. The SEMs in Figure 4 display the distinctive features of silk hydrogels prepared by the three different methods. In the spontaneously formed silk hydrogel (Figure 4e,f), silk molecules self-assembled under thermal fluctuation into a network of randomly oriented fibers. In contrast, in the vortex-induced silk hydrogel (Figure 4c,d), silk molecules were highly oriented along the shearing direction.
Figure 4.

SEMs of the critical point dried silk hydrogels (a, c, e) before enzymatic degradation, and (b, d, f) after 3 days of the enzymatic degradation by α-chymotrypsin. The fibril structures were locally aligned in the electrogelled silk (a, b), and highly oriented in the vortex induced silk gels (c, d). No significant fiber alignment was observed in the spontaneously formed silk hydrogel (e, f).
The electrogelation of silk is accompanied by electrolysis of water. Changes in pH are induced locally near the electrode surface by electrochemical oxidation or reduction. The formation of silk e-gel is governed by the electro-diffusion of the H+ and OH− ions generated in hydrolysis.13 A locally aligned fibrillar structure was observed in the electrogelled silk (Figure 4a,b). As determined by Fourier self-deconvolution of the Amide I FTIR spectra (Figure 5), all of the critical point dried silk gels were dominated by the β-sheet structures (44 ~ 45 %). This is consistent with the SEM observation (Figure 4b,d,f) that the critical point dried samples retained their structural integrity after three days of degradation by α-chymotrypsin, which is known to preferentially attack the amorphous regions in silk fibroin.3
3.2 Tunable physical crosslink (β-sheet) content in electrogelled silk
We have shown that the morphological features of silk hydrogels were preserved during critical point drying and the secondary structures were preserved during freeze drying. The secondary structures of silk hydrogels were characterized using FTIR spectroscopy incorporating resolution-enhancement techniques. There are eleven component bands contributing to the Amide I band contour in silk, as revealed by Fourier self-deconvolution and derivation.29 The relative percentage of β-sheets was calculated from the areas of the β-sheet bands and their fraction of the total area in the amide I region (1585 ~ 1785 cm−1). The electrogelled silk was tuned from less-β-sheet form (15 ~ 16 % of β-sheets) to β-sheet dominated forms (45 % of β-sheets) via the ethanol treatment as a part of the critical point drying process (Figure 5a,b). Chemical crosslinking using glutaraldehyde did not cause significant changes in the secondary structure content.
Less tunability of β-sheet content was found in the vortex-induced silk hydrogels (Figure 5c,d). Vortex sheared silk contains 24 ~ 35 % of mechanically induced β-sheets, varying with thermal and mechanical history. The percentage of β-sheets increased to 44 % after ethanol treatment. The spontaneously formed silk hydrogels consisted predominantly of the β-sheet conformation (44 ~ 45 % of β-sheets), which is thermodynamically more stable (Figure 5e,f).
3.3 Mechanical reinforcement of electrogelled silk through shear-induced physical β-sheet crosslinks
The reinforcement effect of chemical crosslinking on the mechanical properties of electrogelled silk was confirmed by oscillatory shear studies (Figure 6a,b). For silk e-gels without shear history (at time t = 0), both the shear storage (G′) and loss (G″) moduli were enhanced about 10-fold by the glutaraldehyde treatment at a protein/crosslinker ratio of 100:4 (w/w). The shear storage modulus G′, for instance, was ~ 20 Pa for the untreated e-gel and ~ 200 Pa for the crosslinked e-gel. The mechanical strength can be varied by changing the crosslinker-to-protein ratio. Shear induced mechanical reinforcement of the electrogelled silk was monitored in-situ. As shown in Figure 6a, the shear moduli of the untreated silk e-gel increased by two orders of magnitude during the first hour of the dynamic shear loading. The shear storage modulus G′ increased from ~ 20 to ~ 2,000 Pa. The rate of the increase in moduli slowed after the first hour.
Figure 6.

Shear-induced β-sheet formation gave rise to physical crosslinks and mechanical reinforcement of electrogelled silk. Representative time-evolved shear storage (G′) and loss (G″) moduli at the frequency of 1 rad/sec for (a) the untreated e-gel and (b) the glutaraldehyde-crosslinked e-gel. (c) Water loss in silk e-gels forming intra- and inter-molecular hydrogen bonds under shearing. (d) FTIR analysis shows the β-sheet-dominant structures in the electrogelled silk after 10 hours of dynamic shear loading.
FTIR secondary structure analysis (Figure 6d) suggests that the formation of intermolecular hydrogen-bonded β-sheets during mechanical shearing was responsible for the enhancement of mechanical properties in silk e-gel. The untreated sample was dominated by β-sheets (45 %) after 10 hours of mechanical shearing. Similar mechanical behavior of shear reinforcement was observed on the chemically crosslinked silk e-gel (Figure 6b). The shear storage modulus G′ of the crosslinked silk e-gel increased by two orders of magnitude, from ~ 200 to ~ 20,000 Pa, after five hours of mechanical shear loading, and after that reached a plateau. The crosslinked silk e-gel after 10 hours of shearing contained 48 % β-sheets, determined by FTIR secondary structure analysis (Figure 6d).
The formation of β-sheets involves breakage of hydrogen bonds between water and silk and formation of intra-/inter-chain hydrogen bonds. The rate of water loss from silk hydrogels under shear is therefore correlated with the growth rate of β-sheet crosslinks. Measurements of the degree of hydration (Figure 6c) showed that the crosslinked silk e-gel lost water at a lower rate than the untreated silk e-gel. This is consistent with the steeper initial rise of the shear modulus in the untreated sample than in the crosslinked sample.
The results demonstrate that chemical and physical crosslinks can be introduced into silk hydrogels with control or tunablity. The value for this control is in both the analysis of the gels and their potential utility. In terms of analysis, morphological features formed during the gelation process can provide insight into structure-morphology relationship. In terms of utility, control of these features allows matches with mechanical properties and other potential biomaterial applications. Control of structure impacts degradation lifetime due to the impact on kinetics of proteases involved in silk digestion in vitro or in vivo. Options to tune such features, with a foundation based on the control of morphology and structure formed via the various gelation methods, provides materials design parameter space to guide biomedical uses. Since the methods described are amenable to the addition of bioactive compounds due to the ambient, aqueous conditions utilized, refinements in the morphologies and structures formed upon such additions may provide further insights for the utility of these gels in biomedically-relevant applications involving delivery of therapeutics (e.g., drugs, cells) or other compounds.
4. Conclusions
Various methods of post-crosslinking following the electrogelation of silk have been explored for tuning the structural properties of the silk e-gels, dominated by random coil structures as they form. A technique combining chemical and physical crosslinking was demonstrated to enhance the structural stability of silk hydrogels, so that delicate morphological features were preserved after critical point drying for conventional SEM imaging. Silk was randomly oriented in the spontaneously formed hydrogels, but highly aligned along the shear direction in the vortex-induced hydrogel. The electrogelled silk displayed local alignment of fibrillar structures, consistent with the gelation mechanism comprising local pH changes caused by ion migration. The secondary structures of silk hydrogels were also preserved in the dry states using a combination of covalent crosslinking and freeze drying. The relative percentages of β-sheet content in silk hydrogels were determined from the amide I bands of FTIR spectra with assistance of resolution enhancement techniques. The spontaneously formed silk hydrogel consisted predominantly of β-sheets (45 %), and the percent of β-sheets in the vortex-induced silk hydrogel was tunable between 24 % and 45 %. The highest structural tunability, nearly 30 %, was found with the electrogelled silk. Dynamic oscillatory shear testing confirmed significant mechanical reinforcement of the electrogelled silk as a result of crosslinking, induced either covalently by glutaraldehyde, or mechanically in the form of β-sheets. The tunability control over the stability/degradability, demonstrated in the characterization, is essential for applications of electrogelled silk, including on-demand cell encapsulation and programmable drug release. The embodied idea of selective incorporation of chemical/physical crosslinks into the electrogelled silk may inspire new design strategies for stimuli-responsive protein polymers.
Supplementary Material
Acknowledgments
The authors thank Carolyn Marks for help during the initial exploration of the optimal dehydration conditions for silk hydrogels. This work was financially supported by the NIH (EB002520, AR005593), the AFOSR (FA9550-10-1-0172) and DARPA (N68001-12-C-4194). This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Infrastructure Network (NNIN). CNS is part of the Faculty of Arts and Sciences at Harvard University.
Footnotes
Supporting information available
FTIR spectra of the freeze-dried vortex-induced silk gels and spontaneously formed silk gels, and linear rheological measurements of glutaraldehyde-crosslinked silk e-gel. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Omenetto FG, Kaplan DL. Science. 2010;329:528–531. doi: 10.1126/science.1188936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim DH, Viventi J, Amsden JJ, Xiao JL, Vigeland L, Kim YS, Blanco JA, Panilaitis B, Frechette ES, Contreras D, Kaplan DL, Omenetto FG, Huang YG, Hwang KC, Zakin MR, Litt B, Rogers JA. Nat Mater. 2010;9:511–517. doi: 10.1038/nmat2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu X, Shmelev K, Sun L, Gil ES, Park SH, Cebe P, Kaplan DL. Biomacromolecules. 2011;12:1686–1696. doi: 10.1021/bm200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandal BB, Grinberg A, Gil ES, Panilaitis B, Kaplan DL. Proc Natl Acad Sci U S A. 2012;109:7699–7704. doi: 10.1073/pnas.1119474109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rockwood DN, Preda RC, Yucel T, Wang XQ, Lovett ML, Kaplan DL. Nat Protoc. 2011;6:1612–1631. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang CY, Wang XY, Gunawidjaja R, Lin YH, Gupta MK, Kaplan DL, Naik RR, Tsukruk VV. Adv Funct Mater. 2007;17:2229–2237. [Google Scholar]
- 7.Tsioris K, Raja WK, Pritchard EM, Panilaitis B, Kaplan DL, Omenetto FG. Adv Funct Mater. 2012;22:330–335. [Google Scholar]
- 8.Jin HJ, Fridrikh SV, Rutledge GC, Kaplan DL. Biomacromolecules. 2002;3:1233–1239. doi: 10.1021/bm025581u. [DOI] [PubMed] [Google Scholar]
- 9.Fini M, Motta A, Torricelli P, Glavaresi G, Aldini NN, Tschon M, Giardino R, Migliaresi C. Biomaterials. 2005;26:3527–3536. doi: 10.1016/j.biomaterials.2004.09.040. [DOI] [PubMed] [Google Scholar]
- 10.Zhou CZ, Confalonieri F, Medina N, Zivanovic Y, Esnault C, Yang T, Jacquet M, Janin J, Duguet M, Perasso R, Li ZG. Nucleic Acids Res. 2000;28:2413–2419. doi: 10.1093/nar/28.12.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leisk GG, Lo TJ, Yucel T, Lu Q, Kaplan DL. Adv Mater. 2010;22:711–715. doi: 10.1002/adma.200902643. [DOI] [PubMed] [Google Scholar]
- 12.Yucel T, Kojic N, Leisk GG, Lo TJ, Kaplan DL. J Struct Biol. 2010;170:406–412. doi: 10.1016/j.jsb.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kojic N, Panzer MJ, Leisk GG, Raja WK, Kojic M, Kaplan DL. Soft Matter. 2012 doi: 10.1039/C2SM25783A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keten S, Xu ZP, Ihle B, Buehler MJ. Nat Mater. 2010;9:359–367. doi: 10.1038/nmat2704. [DOI] [PubMed] [Google Scholar]
- 15.Kim UJ, Park JY, Li CM, Jin HJ, Valluzzi R, Kaplan DL. Biomacromolecules. 2004;5:786–792. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 16.Yucel T, Cebe P, Kaplan DL. Biophys J. 2009;97:2044–2050. doi: 10.1016/j.bpj.2009.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XQ, Kluge JA, Leisk GG, Kaplan DL. Biomaterials. 2008;29:1054–1064. doi: 10.1016/j.biomaterials.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greving I, Cai MZ, Vollrath F, Schniepp HC. Biomacromolecules. 2012;13:676–682. doi: 10.1021/bm201509b. [DOI] [PubMed] [Google Scholar]
- 19.Chang J, Peng XF, Hijji K, Cappello J, Ghandehari H, Solares SD, Seog J. J Am Chem Soc. 2011;133:1745–1747. doi: 10.1021/ja110191f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Jonge N, Ross FM. Nat Nanotechnol. 2011;6:695–704. doi: 10.1038/nnano.2011.161. [DOI] [PubMed] [Google Scholar]
- 21.Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Nat Methods. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]
- 22.Wray LS, Hu X, Gallego J, Georgakoudi I, Omenetto FG, Schmidt D, Kaplan DL. J Biomed Mater Res, Part B. 2011;99B:89–101. doi: 10.1002/jbm.b.31875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Migneault I, Dartiguenave C, Bertrand MJ, Waldron KC. Biotechniques. 2004;37:790–802. doi: 10.2144/04375RV01. [DOI] [PubMed] [Google Scholar]
- 24.Hayat MA. Principles and techniques of electron microscopy: biological applications. 4. Cambridge University Press; Cambridge, UK: 2000. [Google Scholar]
- 25.Chen X, Li WJ, Shao ZZ, Zhong W, Yu TY. J Appl Polym Sci. 1999;73:975–980. [Google Scholar]
- 26.Zhu L, Hu RP, Wang HY, Wang YJ, Zhang YQ. J Agric Food Chem. 2011;59:10298–10302. doi: 10.1021/jf202036v. [DOI] [PubMed] [Google Scholar]
- 27.Ekici S, Saraydin D. Drug Delivery. 2004;11:381–388. doi: 10.1080/10717540490884804. [DOI] [PubMed] [Google Scholar]
- 28.Numata K, Cebe P, Kaplan DL. Biomaterials. 2010;31:2926–2933. doi: 10.1016/j.biomaterials.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu X, Kaplan D, Cebe P. Macromolecules. 2006;39:6161–6170. [Google Scholar]
- 30.Carter EA, Fredericks PM, Church JS, Denning RJ. Spectrochim Acta, Part A. 1994;50:1927–1936. [Google Scholar]
- 31.Shao JZ, Zheng JH, Liu JQ, Carr CM. J Appl Polym Sci. 2005;96:1999–2004. [Google Scholar]
- 32.Chang MC, Tanaka J. Biomaterials. 2002;23:4811–4818. doi: 10.1016/s0142-9612(02)00232-6. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Shao ZZ, Marinkovic NS, Miller LM, Zhou P, Chance MR. Biophys Chem. 2001;89:25–34. doi: 10.1016/s0301-4622(00)00213-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.