

Abstract
Context
Depression in bipolar disorder is clinically indistinguishable from that observed in major depressive disorder. As in major depression, selective serotonin reuptake inhibitors targeting brain serotonin transporters are first-line treatments for bipolar depression. Associations of serotonin transporter promoter polymorphisms and bipolarity have been reported; however, research on alterations in serotonergic neurotransmission in bipolar depression remains scant.
Objectives
To assess in vivo brain serotonin transporter binding potential (BP1, proportional to serotonin transporter number) in patients with bipolar depression and controls and to examine the relationship between serotonin transporter binding and genotype.
Design
Case-control study.
Setting
University hospital.
Participants
A sample of 18 medication-free patients with bipolar depression and 41 controls.
Main Outcome Measures
In vivo brain serotonin transporter binding was measured using positron emission tomography and radiolabeled trans-1,2,3,5,6,10-β-hexahydro-6-[4-(methylthio) phenyl]pyrrolo-[2,1-a]-isoquinoline ([11C](+)-McNeil 5652). Participants were genotyped assessing biallelic and triallelic 5-HTTLPR polymorphisms.
Results
Patients with bipolar disorder had 16% to 26% lower serotonin transporter BP1 in the midbrain, amygdala, hippocampus, thalamus, putamen, and anterior cingulate cortex. Triallelic 5-HTTLPR genotypes were unrelated to serotonin transporter BP1.
Conclusions
Lower serotonin transporter BP1 in bipolar depression overlaps with that observed in major depression and suggests that serotonergic dysfunction is common to depressive conditions.
BIPOLAR DISORDER IS A BRAIN disorder characterized by recurrent manic and major depressive episodes (MDEs), the latter being clinically indistinguishable from those observed in unipolar or major depressive disorder. Bipolar disorder ranks sixth in terms of disease burden worldwide,1 yet its neurobiological underpinnings have received scant attention, and most research has focused on functional abnormalities rather than on alterations in neurotransmission.2 However, pharmacotherapy development remains focused on testing molecules that target neuroreceptors, although the pathophysiologic mechanism of bipolar disorder is likely also to involve disturbances in second messenger systems and other subcellular abnormalities.3 Furthermore, treatments for bipolar depression show remission rates only modestly better than placebo.4 As a potential aid to rational drug development and to elucidate its pathophysiologic mechanism, neurotransmission deficits present in bipolar depression require attention.
Dysfunction in serotonin neurotransmission is postulated to have a critical role in mood disorders.5 Serotonin transporters terminate serotonin’s action by reuptake into neurons. In bipolar disorder, platelets have been shown to have lower serotonin reuptake.6-10 Positron emission tomographic (PET) studies of brain serotonin transporter binding have not been published with samples composed exclusively of depressed individuals with bipolar disorder. In major depressive disorder, results are disparate. Three studies11-13 demonstrated lower serotonin transporter binding (serotonin transporter BP) in several brain regions, 1 study14 identified no difference in binding, and 2 studies,15,16 1 of which included 6 individuals with bipolar disorder, found higher serotonin transporter BP. These inconsistencies may be due to methodological issues such as small sample size, insufficient imaging duration, use of different ligands, or use of a reference tissue with measurable specific binding.17 A potential cause of lower serotonin transporter binding is a biallelic functional polymorphism, 5-HTTLPR, which has a short S allele resulting in lower expression of the serotonin transporter in lymphoblastoid cell lines.18 Indeed, 2 meta-analyses19,20 note a modest but statistically significant association of bipolar disorder and the short-allele 5-HTTLPR polymorphism. A recently described triallelic functional polymorphism of the serotonin transporter gene promoter21 has not been studied in bipolar populations yet but may be more closely related to serotonin transporter expression in the brain.
We studied brain serotonin transporter binding using PET and radiolabeled trans-1,2,3,5,6,10-β-hexahydro-6-[4-(methylthio)phenyl]pyrrolo-[2,1-a]-isoquinoline ([11C](+)-McNeil 5652) in 18 medication-free patients with bipolar depression compared with 41 controls. Participants were genotyped assessing biallelic18 and triallelic 5-HTTLPR polymorphisms.21 Given the clinical similarities in MDEs in bipolar and major depressive disorder, we hypothesized that depressed patients with bipolar disorder would show low serotonin transporter BP in the amygdala and midbrain compared with controls, as we have shown in major depressive disorder.13 We also determined serotonin transporter BP in the anterior cingulate cortex, hippocampus, putamen, and thalamus because of their potential role in affective disorders22,23 and the relationship between 5-HTTLPR genotype and BP in the 6 regions of interest (ROIs).
METHODS
PARTICIPANTS
Eighteen patients (10 women and 8 men) who met DSM-IV24 criteria for bipolar disorder with an MDE and 41 controls (19 women and 22 men) participated in this study. Inclusion and exclusion criteria were evaluated by means of clinical history, medical record review, Structured Clinical Interview for DSM-IV Axis I Disorders,25 Hamilton Depression Rating Scale,26 review of systems, physical examination, routine blood tests, pregnancy tests, and urine toxicologic tests. The control group was free of a lifetime psychiatric history and had no first-degree relatives with mood disorders.
Criteria for study entry for bipolar patients included (1) age 18 to 65 years, (2) DSM-IV MDE, bipolar disorder, (3) absence of alcohol or substance abuse or dependence in the previous 6 months, (4) absence of a family history of schizophrenia, (5) absence of exposure to 3,4-methylenedioxymethamphetamine (“ecstasy”), (6) absence of significant medical conditions, (7) absence of pregnancy, (8) capacity to provide informed consent, and (9) ability to tolerate a washout of current psychotropic medications and to be medication free for at least 2 weeks before the positron emission tomographic studies (6 weeks for fluoxetine and 3 weeks for oral neuroleptics), except benzodiazepines, which were discontinued 24 hours before the study. Two patients (11%) had a remission of MDEs during medication washout, but all the patients underwent imaging within a month of full MDEs. Only 2 of the patients with bipolar disorder were medication naive, and 5 were not taking medication before enrollment. Recent medication regimens varied: 4 patients were taking a selective serotonin reuptake inhibitor (SSRI) only, 1 was taking nefazodone hydrochloride only, and 1 was taking methylphenidate hydrochloride only. Several patients were taking an antidepressant (a monoamine oxidase inhibitor or an SSRI) plus other drugs: plus lamotrigine (n=1), plus valproate sodium (n=2), plus atypical antipsychotics (n=2), plus carbamazepine (n=1), and plus gabapentin (n=1). Also, 2 patients received lithium in addition to other medications. The mean±SD number of days without medication use was 72±232 days (range, 14-950 days, excluding 2 medication-naive individuals). Pharmacologic classes administered in the past included lithium, various antiepileptic drugs, atypical antipsychotics, SSRIs, monoamine oxidase inhibitors, other antidepressants, neuroleptics, and benzodiazepines.
Study criteria for controls were similar except for the absence of a psychiatric history, including alcohol and substance abuse, and the absence of mood or psychotic disorders in their first-degree relatives. Controls were recruited concurrently with patients who had bipolar disorder, and their data have previously been reported.27 The institutional review boards of Columbia University Presbyterian Hospital and the New York State Psychiatric Institute approved the protocol. Participants gave written informed consent after detailed explanation of the study.
RADIOCHEMISTRY
The precursor for the production of [11C](+)-McNeil 5652, (+)-McNeil butyryl thioester tartrate, was prepared from (+)-McNeil 5652 using a modification of the published procedure.28
PET PROTOCOL
After an Allen test and subcutaneous administration of 2% lidocaine hydrochloride, a catheter was inserted into the radial artery. A venous catheter was inserted into a forearm vein on the opposite side. Head movement was minimized using a polyurethane head immobilizer system (Soule Medical, Tampa, Fla) molded around the head of the individual. The PET was performed using the ECAT EXACT HR+ (Siemens/CTI, Knoxville, Tenn) (63 slices covering an axial field of view of 15.5 cm, axial sampling of 2.46 mm, 3-dimensional mode, and in-plane and axial resolution of 4.4 and 4.0 mm at full-width, half-maximum at the center of the field of view, respectively). After injection of [11C](+)-McNeil 5652 as an intravenous bolus for 45 seconds using an injection pump, emission data were collected in 3-dimensional mode for 130 minutes as 22 successive frames of increasing duration (3×20 seconds, 3×1 minute, 3×2 minutes, 2×5 minutes, and 11×10 minutes). Images were reconstructed to a 128×128 matrix (pixel size, 2.5×2.5 mm2) with attenuation correction using a 10-minute transmission scan and a Shepp 0.5 filter (cutoff value of 0.5 cycle/projection rays).
MAGNETIC RESONANCE IMAGING ACQUISITION
Magnetic resonance images were acquired using a 1.5-T Signa Advantage system (General Electric Medical Systems, Milwaukee, Wis). A sagittal scout (localizer) was used to identify the anterior commissure–posterior commissure plane (1 minute). A transaxial T1-weighted sequence with 1.5-mm slice thickness was acquired in a coronal plane orthogonal to the anterior commissure–posterior commissure plane over the whole brain using the following parameters: 3-dimensional spoiled gradient recalled acquisition in the steady state; repetition time, 34 milliseconds; echo time, 5 milliseconds; flip angle, 45°; slice thickness, 1.5 mm and zero gap; 124 slices; field of view, 22×16 cm; width, 256×192 matrix reformatted to 256×256, yielding a voxel size of 1.5×0.9×0.9 mm; and time of acquisition, 11 minutes.
IMAGE ANALYSIS
Image analysis was performed using MEDX software (Sensor Systems Inc, Sterling, Va). Each of the 22 frames was coregistered to the eighth frame using the functional magnetic resonance imaging Linear Image Registration Tool, version 5.0.29 Mean PET images were coregistered to the corresponding magnetic resonance image using the same tool. No attempt was made to correct for transmission emission mismatch in reconstruction. Six ROIs and 1 region of reference (cerebellum, see next paragraph) were traced on the magnetic resonance image. The ROIs were traced using brain atlases,30,31 were based on published studies,32,33 and were approved by a neuroanatomist (V.A.). The ROIs were drawn on the left and right sides of the brain except for the midbrain. The other regions included the thalamus, putamen, amygdala, hippocampus, and anterior cingulate cortex. Data analysts were blind to participant diagnosis. Magnetic resonance images underwent brain extraction using the exbrain version 2 utility34 and were then segmented using FMRIB’s Automated Segmentation Tool.29 In the anterior cingulate cortex, only voxels classified as gray matter were used to measure PET activity distribution.
The reference tissue in brain imaging studies is ideally a region devoid of all specific radioligand binding. To avoid as much specific binding as possible, a mean±SD sample of cerebellar cortex of 12.1±1.5 cm3 was used as the reference region.13
INPUT FUNCTION MEASUREMENT
Arterial input indices were obtained as previously described.35 Briefly, after radiotracer injection, for the first 2 minutes 18 arterial samples were collected every 5 seconds using an automated sampling system and manually thereafter at longer intervals. Six samples (collected at 2, 20, 50, 80, 110, and 130 minutes) were further processed by means of high-pressure liquid chromatography to measure the fraction of plasma activity representing unmetabolized parent compound. Increasing volumes of plasma were processed as a function of time to increase the number of radioactive disintegrations counted by the well counter. In addition, the eluents from the high-pressure liquid chromatography column were collected into 5 separate tubes to increase counting statistics. A biexponential function was fitted to the 6 measures of unmetabolized parent compound and was used to interpolate values between the measurements. The smallest exponential of the unmetabolized parent curve was constrained as described elsewhere.36 Input functions (arterial concentration across time, microcuries per milliliter) were calculated as the product of total plasma counts and interpolated unmetabolized parent compound, fitted to a sum of 3 exponentials, and then used as input to the graphical analysis. Determination of [11C](+)-McNeil 5652 free fraction in the plasma using the ultracentrifugation technique37 is prevented by high-filter binding (>80%) of free [11C](+)-McNeil 5652 and thus was not obtained in these studies.
DERIVATION OF OUTCOME MEASURES
We previously showed that [11C](+)-McNeil 5652 binding to serotonin transporter in vivo can be quantified by either a 2-compartment kinetic model or the graphical method of Logan.35,38 The Logan method provides excellent reproducibility and time stability but has a noise-dependent bias.39 Therefore, derivation of [11C](+)-McNeil 5652 regional distribution volumes was performed using the likelihood approach to the graphical method.40,41 This method estimates parameters in the graphical analysis setting using standard likelihood theory. This removes the bias present when using the ordinary least squares method. Brain activity was corrected for the contribution of plasma activity assuming a 5% blood volume in the ROIs.42 The total regional distribution volume (milliliters of plasma per gram of tissue) was defined as the ratio of the tracer concentration in this region to the metabolite-corrected plasma concentration at equilibrium. The likelihood approach to the graphical method was used to calculate the sum of the specific (V3) and nondisplaceable (free plus nonspecific binding=V2) distribution volumes (VT). A mean±SD sample of the cerebellar gray matter of 12.1±1.5 cm3 was the ROI43 used to determine nonspecific binding.
We calculated binding potential with the following equation:
where f1 is the free fraction of [11C](+)-McNeil 5652 in the plasma, Bmax is the total number of available transporters, and 1/KD is the affinity. Ideally, Bmax/KD measurements would be obtained, but f1 cannot be measured with this ligand.
For kinetic modeling, kinetic parameters were derived using procedures implemented in MATLAB (The Math Works Inc, South Natick, Mass). Given the unequal sampling across time (increasing frame acquisition time from the beginning to the end of the study), the fitting procedure was weighted by the square root of the frame acquisition time.
GENOTYPING
The serotonin transporter promoter gene polymorphic region (5-HTTLPR) was genotyped for the biallelic classification (long and short, or L and S)18 and for a triallelic genotype.21 Briefly, DNA was extracted from whole blood or buccal mucosa cheek swabs (BuccalAmp DNA Extraction Kit; Epicenter, Madison, Wis). For triallelic genotyping,21 2 fluorogenic probes specific for the LA and LG alleles were used. Polymerase chain reaction was performed in a 25-μL volume: 25 to 50 ng of DNA in 1 μL, 24 μL of polymerase chain reaction mastermix including probes (120 nmol of adenosine diphosphate and 60 nmol of internal control probe), polymerase chain reaction primers (200 nmol of each), 4% dimethyl sulfoxide by volume, magnesium chloride (5 mmol/L), 1× ABI Corebuffer (Applied Biosystems, Foster City, Calif), deoxyadenosine triphosphate (0.2 mmol/L), deoxyguanosine triphosphate (0.2 mmol/L), deoxycytidine triphosphate (0.2 mmol/L), deoxyuridine triphosphate (0.4 mmol/L), Taq Gold (0.25 U/mL), and AmpErase uracil-N-glycosylase (0.01 U/mL) (Applied Biosystems). Each well of the 96-well optical plates (model 4306737; Applied Biosystems) was sealed with an optical cap (model 4323032; Applied Biosystems). Amplification conditions were 2 minutes at 50°C, 10 minutes at 95°C, then 40 cycles at 96°C for 15 seconds and 62.5°C for 90 seconds. Genotypes were generated using ABI PRISM 7700 Sequence Detection system software (Applied Biosystems). Genotyping standards were LALA, LALG and LGLG genomic DNA, whose genotypes were known by sequencing. In this study, genotypes acquired from these 2 procedures were combined to classify samples as 1 of 6 genotypes: SS, SLA, SLG, LALA, LALG, or LGLG. Because LG and S alleles have comparable expression, which is lower than that of LA, we classified these alleles as L’ (LA) and S’ (LG or S). To evaluate genotyping accuracy, a quarter of the samples selected randomly were genotyped in duplicate. The error rate was less than 0.005.
STATISTICS
Clinical and demographic variables were compared using the 2-tailed t test and the Fisher exact test as appropriate. The Fisher exact test was used to assess genotype and allele frequencies between groups. For imaging data, analysis was performed on the natural log of the data to stabilize the variance and ensure that modeling assumptions were met, after first adding a quantity to all measures to ensure positivity. Considering 6 ROIs at once, data were analyzed using linear mixed-effects models with region and diagnostic group as fixed effects and subject as the random effect. Additional fixed effects considered were age, sex, and genotype. Similarly, for bipolar patients, several exploratory analyses were conducted using linear mixed-effects models with patient as the random effect and region as a fixed effect. Additional fixed effects considered were suicide attempt status (attempter/nonattempter) and bipolar subtype (bipolar I or II). Pearson product-moment correlation was used to evaluate the relationship between BP1 and depression scores on the Hamilton Depression Rating Scale and the Beck Depression Inventory. All P values reported correspond to 2-sided tests.
RESULTS
The control and bipolar disorder groups were similar in terms of age (range, 18-65 years; t=0.35; P=.73) and sex (P = .58 by Fisher exact test). Most of the patients with bipolar disorder (13 of 18) were studied as inpatients. Eight patients met the criteria for bipolar II disorder, and 10 met the criteria for bipolar I disorder. Other Axis I disorders included binge eating (n=2), obsessive-compulsive disorder (n=3), simple phobia (n=1), generalized anxiety disorder (n=1), and posttraumatic stress disorder (n=2). None of the participants had active substance abuse or dependence in the 6 months before imaging, but 5 had a history of substance abuse (3 with marijuana abuse; 1 with alcohol, marijuana, and cocaine abuse; and 1 with marijuana and phencyclidine abuse).
Nine patients with bipolar disorder had attempted suicide (lifetime attempts: mean±SD, 2.33±1.9; range, 1-7). Patients with bipolar disorder had a mean±SD score of 23.7±6.3 on the Hamilton Depression Rating Scale (24 items), whereas controls had a score of 0.61±0.89.
The mean±SD injected dose of [11C](+)-McNeil 5652 was similar in controls (11.89±4.10 mCi) and patients with bipolar disorder (12.8±4.9 mCi; t=−0.73; P=.47). Mean ± SD specific activity (controls: 0.97 ± 0.69 mCi/nmol; patients with bipolar disorder: 0.81±0.40 mCi/nmol; t=0.92; P=.36) and injected mass (controls: 70.5±69.1 μmol; patients with bipolar disorder: 50.4±23.2 μmol; t=1.20; P=.24) also did not differ.
A linear mixed-effects model of regional BP1 demonstrated that patients with bipolar disorder had lower serotonin transporter BP1 (F1,58=5.41; P=.02) across all 6 brain regions examined: midbrain (27%), amygdala (26%), hippocampus (23%), thalamus (23%), putamen (16%), and anterior cingulate cortex (23%) (Figures 1, 2, and 3). There was no evidence that the deficit differed across regions (F5,285=1.34; P=.25), nor was there a correlation between BP1 and depression severity in patients with bipolar disorder as rated by either self-report (Beck Depression Inventory) or clinical assessment (Hamilton Depression Rating Scale) in any of the 6 regions (r=−0.37 to −0.06; P=.13-.80). Diagnostic plots from the linear mixed-effects model identified 2 outliers. This was the case for 1 region in 1 patient with bipolar disorder and 1 region in a control. When the analysis was repeated after removing data from these patients, the conclusions were unchanged, with patients with bipolar disorder having lower binding across the 6 regions considered (F1,56=4.14; P=.047) and no evidence of a region × diagnosis interaction (F5,275=0.54; P=.75). Results also did not change when 3 patients with comorbid obsessive-compulsive disorder were excluded (F1,55=5.27; P=.03). Mean±SD cerebellar serotonin transporter VT was higher in controls than in patients with bipolar disorder (21.49±5.11 vs 17.04±2.58 mL/g; t56=4.43; P<.001). Consequently, analysis with BP2 as the outcome measure did not reveal the difference between patients with bipolar disorder and controls (F1,58=0.68; P=.41).
Figure 1.
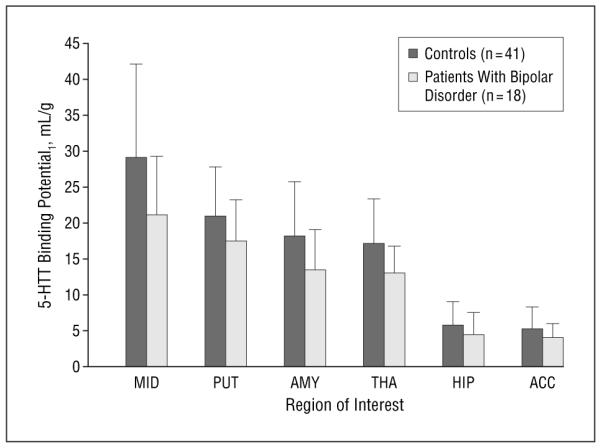
Lower radiolabeled trans-1,2,3,5,6,10-β-hexahydro-6-[4-(methylthio)-phenyl]pyrrolo-[2,1-a]-isoquinoline ([11C](+)-McNeil 5652) binding potential (BP1) to the serotonin transporter (5-HTT) in nonmedicated patients with bipolar disorder in 6 regions of interest compared with controls (F1,58=5.41; P=.02). ACC indicates anterior cingulate cortex; AMY, amygdala; HIP, hippocampus; MID, midbrain; PUT, putamen; and THA, thalamus. Vertical bars represent the mean likelihood approach to the graphical method modeled BP1 (VT[region]–VT[cerebellum]) for each region; error bars, ±1 SD.
Figure 2.
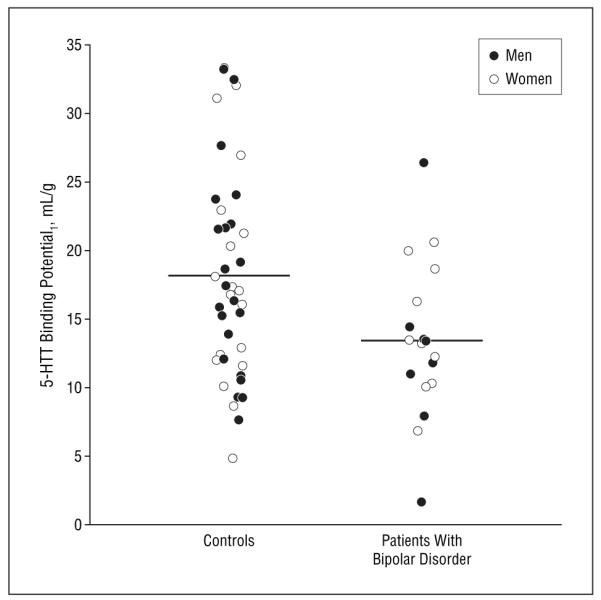
The radiolabeled trans-1,2,3,5,6,10-β-hexahydro-6-[4-(methylthio)-phenyl]pyrrolo-[2,1-a]-isoquinoline ([11C](+)-McNeil 5652) binding to serotonin transporter (5-HTT) in the amygdala. Each circle represents a single measurement of likelihood approach to the graphical method modeled binding potential. Horizontal bars indicate mean.
Figure 3.
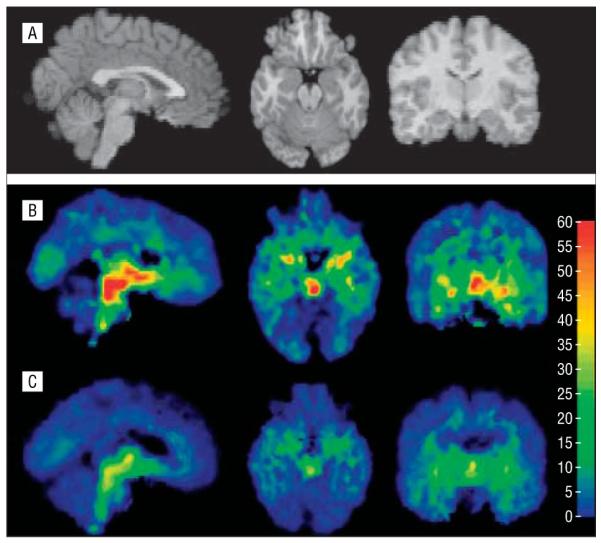
Magnetic resonance images from a control (A) were used to coregister the maps of serotonin transporter binding potential (BP1) from a control (B) and a patient with bipolar disorder (C) whose midbrain BP1 was closest to the respective group mean. Each voxel intensity is a single BP1 measurement. The color bar represents milliliters per gram. Serotonin transporter BP1 was significantly lower in the midbrain (columns 1 and 2), amygdala (column 3), thalamus (columns 1 and 3), anterior cingulate cortex (column 1), hippocampus (column 2), and putamen (data not shown).
As previously noted among unipolar patients and controls,27 there was no effect of sex on BP1. However, there was an indication of an age effect on BP1 after fitting region and diagnosis with all data (F1,56=6.61; P=.01) and after removing the 2 extreme outliers (F1,54=4.05; P=.049), although the age effect was not significant when considering each group separately. Including age in the model did not affect the conclusions drawn on diagnosis (F1,56=6.42; P=.01 with the full data set and F1,54=4.70; P=.04 after removing the outliers). Similarly, no effect of past substance abuse was present (F1,56=2.53; P=.12), as was the case for previous exposure to medication (F1,55=2.29; P=.14 for the full data set after including the effect of age and F1,53=0.07; P=.79 after excluding the outliers), possibly due to the small number of medication-naive patients (n=2). Exploratory analyses revealed no differences in BP1 between suicide attempters and nonattempters among patients with bipolar disorder (F1,17=1.84; P=.19) or between individuals with bipolar I and bipolar II disorder in the full model (F1,56=0.063; P=.80).
Triallelic genotyping showed that 4 patients with bipolar disorder had the low-expressing genotype and that genotype and allele distribution were not different from those observed in controls (Table). Biallelic genotyping yielded a difference between patients with bipolar disorder and controls (Table). Nonetheless, there was no relationship between triallelic genotype based on functional classification (low-, medium-, and high-expressing genotype) and BP1 in the 6 regions considered in patients with bipolar disorder (F2,52=0.24; P=.80) (Figure 4).
Table.
Demographics of the Study Groups
| Controls (n = 41) |
Depressed Patients With Bipolar Disorder (n = 18) |
t Test | P Value | |
|---|---|---|---|---|
| Sex, M/F, No. | 22/19 | 8/10 | NA | .58* |
| Age, mean ± SD, y | 38.1 ± 9.7 | 39.3 ± 16.0 | 0.35 | .73 |
| Bipolar I/bipolar II, No. | NA | 10/8 | NA | NA |
| Hamilton Depression Rating Scale (24 items) score, mean ± SD | 0.61 ± 0.89 | 23.72 ± 6.27 | 15.58 | <.001 |
| Beck Depression Inventory score, mean ± SD | 1.5 ± 2.1 | 28.9 ± 10.1 | 16.71 | <.001 |
| Global Assessment Scale score, mean ± SD | 89.5 ± 4.7 | 48.7 ± 10.1 | 21.26 | <.001 |
| First-degree relative with MDD, No. (%) | NA | 16 (89) | NA | NA |
| First-degree relative with bipolar depression, No. (%) | NA | 3 (17) | NA | NA |
| Age at first MDE, mean ± SD, y | NA | 18.9 ± 7.89 | NA | NA |
| Age at first manic/hypomanic episode, mean ± SD, y | NA | 28.59 ± 11.36 | NA | NA |
| Previous suicide attempts, No. (%) | NA | 9 (50) | NA | NA |
| 5-HTTLPR biallelic genotype, No. (%)† | NA | .03* | ||
| LL | 11 (27.5) | 8 (44.4) | ||
| LS | 18 (45.0) | 10 (55.6) | ||
| SS | 11 (27.5) | 0 | ||
| 5-HTTLPR triallelic genotype, No. (%)† | NA | .40* | ||
| L ’ L ’ | 9 (22.5) | 6 (33.3) | ||
| L ’ S ’ | 15 (37.5) | 8 (44.4) | ||
| S ’ S ’ | 16 (40.0) | 4 (22.2) | NA | |
| 5-HTTLPR triallelic allele frequency, No. (%)† | NA | .52* | ||
| L ’ | 9 (22.5) | 6 (33.3) | ||
| S’ (at least 1 copy of S’) | 31 (77.5) | 12 (66.7) |
Abbreviations: MDD, major depressive disorder; MDE, major depressive episode; NA, not applicable.
Fisher exact test.
Based on 40 controls (1 control refused to undergo genotyping).
Figure 4.
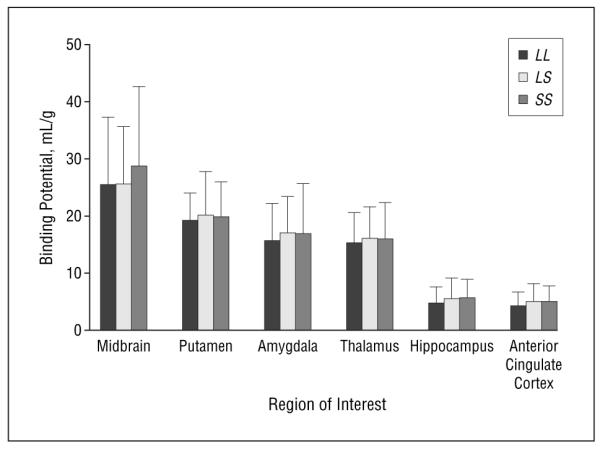
Mean serotonin transporter binding potential according to 5-HTTLPR triallelic polymorphism genotype in patients with bipolar disorder. There are no statistically significant differences among genotypes. Error bars represent SD. LL indicates mean BP1 in patients homozygous for LG; LS, mean BP1 in heterozygous patients (LGS’); and SS, mean BP1 in patients homozygous for S’ (LA or S).
COMMENT
This study shows that patients with bipolar disorder have 16% to 26% lower BP1 in all 6 brain regions examined (amygdala, midbrain, anterior cingulate cortex, hippocampus, putamen, and thalamus), suggesting that lower serotonin transporter binding is pronounced and widespread in bipolar depression. All 6 of these regions receive dense serotonergic projections from the midbrain raphe nuclei. We previously reported13 that BP1 is 16% lower in the amygdala and midbrain in unipolar patients using the same method.
There are several possible explanations for lower serotonin transporter BP1 in patients with bipolar disorder. One is that there are low concentrations of intrasynaptic serotonin in patients with bipolar disorder, thus facilitating serotonin transporter internalization.44 Deficits in synaptic serotonin may be consequent to alterations in binding of other neurotransmitters. Norepinephrine, glutamate, and γ-aminobutyric acid regulate serotonin transporter binding45 and are neurotransmitters implicated in the pathophysiologic mechanism of bipolar disorder.
A second possibility is that lower serotonin transporter binding arises from a primary deficit in serotonin transporter gene expression involving a functional polymorphism of the promoter for the serotonin transporter, such as the triallelic polymorphism of 5-HTTLPR. However, there is no significant association between the triallelic 5-HTTLPR genotype and serotonin transporter binding in the present sample or previous in vivo13 or postmortem46 studies. This, together with the present data, suggests that there are yet to be identified modulators of gene expression or that other effects, such as increased serotonin transporter internalization, are at work in normal and pathological states. There was no difference in genotype in patients with bipolar disorder compared with controls using the triallelic genotype, although the biallelic 5-HTTLPR genotype misclassified several S’S’ individuals as LS. Given the small sample size, findings regarding genotype or allele frequency cannot be extrapolated to bipolar populations in general.
Finally, because serotonin transporter binding has been suggested to be a marker for serotonin innervation,47 reduced serotonin transporter binding could reflect fewer serotonin neuronal processes or neurons in individuals with bipolar disorder. The presence of fewer cells among a subpopulation of neurons in the ventrolateral nucleus of the dorsal raphe nucleus is reported in a small sample of patients with mood disorders,48 although we report more serotonin neurons in the raphe nucleus of depressed individuals who committed suicide49 and more tryptophan hydroxylase50 and messenger RNA51 in this area, which could relate to either suicidal behavior or depression. Similar to the findings reported herein, our group13 previously showed that unipolar patients have lower serotonin transporter binding in the midbrain and amygdala and suggested that this deficit may be restricted to a subpopulation of serotonin transporter–expressing serotonin neurons in the dorsal raphe nuclei. Perhaps bipolar disorder is accompanied by larger deficits in raphe neurons that affect a wider distribution of neurons compared with unipolar depression. That there are similarities in the clinical and neurobiological aspects of unipolar and bipolar disorder, with bipolar disorder having additional, more extensive clinical and biological abnormalities, is consistent with the earliest clinical descriptions of bipolar illness. Bleuler52 considered bipolar and unipolar disorder to be along a spectrum, with bipolar disorder being a more severe expression of the condition than unipolar disorder. This hypothesis is partially supported by the observation that unipolar and bipolar disorder often both manifest in affected families.53
Three explanatory models for the neurobiological abnormalities in bipolar disorder have been proposed recently.2,54,55 However, there is no agreement regarding the “primary lesion” in bipolar disorder.2 Nonetheless, these 3 neurocircuitry-focused models implicate prefrontal cortical, subcortical, and limbic structures, suggesting that, at least in part, the inability to assess the emotional salience of external stimuli54,55 or inhibit responses to the stimuli2 is an important component of this disorder. In agreement with the brain areas implicated in these models, we found reduced serotonin transporter BP1 in the anterior cingulate, putamen, thalamus, midbrain, amygdala, and hippocampus.
These results suggest that serotonergic dysfunction has a role in bipolar depression. For example, a single-photon emission computed tomography case study56 showed that while in a manic episode, an individual with bipolar disorder had higher midbrain serotonin transporter binding than any of the controls. Moreover, SSRIs and other antidepressants that block reuptake of serotonin are reported to induce manic episodes in bipolar disorder.4 Thus, changes in serotonin transporter binding in different phases of the illness may provide at least a partial basis for a model for bipolar disorder.
Almost all the patients with bipolar disorder in this sample had been previously medicated (16 of 18). In unipolar depression, previous medication exposure is associated with serotonin transporter levels that are closer to those observed in controls.13 This finding suggests that if a medication-naive sample were available, larger differences between controls and patients with bipolar disorder would be noted. Although we found no evidence that medication status affected BP1, this may be due to the fact that only 2 patients in this sample were medication naive. Additional confounds are that some patients had comorbid conditions, such as obsessive-compulsive disorders, which respond to treatment with SSRIs and may have serotonin transporter abnormalities associated with them, although a small study57 did not demonstrate this. Analyses excluding these patients did not change the results. We also did not find an effect of past substance abuse. Thus, although these findings require replication, they seem to be robust in this sample.
Cerebellar serotonin transporter VT is higher in controls than in patients with bipolar disorder. Such an effect leads to underestimates of serotonin transporter BP1 in the ROIs, with the effect being more pronounced in controls. Thus, observed differences in BP1 between controls and depressed patients with bipolar disorder are likely an underestimation of the actual physiologic difference.
We repeated the analysis using BP2 as the outcome measure, as follows:
where Bmax indicates density of available binding sites; 1/KD, affinity of the radiotracer for available binding sites; f2, radiotracer intracerebral free fraction; VREF, volume distribution of the reference region; V2, distribution volume of nondisplaceable (free and nonspecifically bound) compartment; k3 and k4, rate constants for transit of the radiotracer between nondisplaceable and specific binding compartments; and VT, total tissue distribution volume relative to total radiotracer concentration. There was no evidence of a difference between patients with bipolar disorder and controls. This is to be expected given that cerebellar VT is higher in the control group, and therefore, using the cerebellum to estimate V2 results in lower BP2 in controls, obviating the possibility of detecting a difference between depressed patients with bipolar disorder and controls. This may explain why other researchers58,59 have preliminarily reported no differences in serotonin transporter BP2 in depressed patients with bipolar disorder compared with controls. It may be that lower cerebellar VT in patients with bipolar disorder is due to group differences in f1 or f2. We cannot rule out this possibility. However, the cerebellum is increasingly implicated in mood disorders. Specifically, decreased cerebellar vermis volume has been observed to be related to recurrence of episodes in bipolar disorder (for a review, see Strakowski et al3). Using autoradiography, our group27 demonstrated that the serotonin transporter is present in the postmortem human cerebellum. Moreover, using [11C]N,N-dimethyl-2-(2-amino-4-cyanophenylthio)benzylamine to measure displacement by sertraline in vivo, we showed that every cerebellar subregion has quantifiable serotonin transporters.60 Thus, differences in cerebellar VT in patients with bipolar disorder and controls may be due to differences in specific binding, f1 or f2, in the cerebellum. Nonetheless, the fact remains that this difference likely underestimates BP1 differences in the ROIs between patients with bipolar disorder and controls.
Whether the lower serotonin transporter BP1 observed in depressed patients with bipolar disorder is due to primary deficits in the serotonin system or is secondary to other neurochemical abnormalities and whether this abnormality is a trait or is only present during the depressed state require further inquiry. Studies examining serotonin transporter binding during periods of mania or euthymia or measuring serotonin transporter binding concomitantly with γ-aminobutyric acid or glutamate levels in the brain may elucidate these issues. Notwithstanding, these findings, the first in vivo evidence of a brain abnormality of this magnitude in bipolar disorder, suggest that bipolar depression is associated with extensive serotonergic dysfunction and may be a more pronounced manifestation of illness than unipolar depression along a spectrum of mood disorders.
Acknowledgment
We thank the R.W. Johnson Pharmaceutical Research Institute for providing the (+)-McNeil 5652.
Funding/Support: This work was supported by grants MH62185 and MH59710 from the Public Health Service, the Stanley Medical Research Foundation, the American Foundation for Suicide Prevention, and The Moody’s Foundation.
Footnotes
Financial Disclosure: None reported.
REFERENCES
- 1.Murray CJL. Rethinking DALYs. In: Murray CJL, Lopez AD, editors. The Global Burden of Disease: A Comprehensive Assessment of Mortality and Disability From Diseases, Injuries, and Risk Factors in 1990 and Projected to 2020. Harvard School of Public Health; Boston, Mass: 1996. pp. 1–98. [Google Scholar]
- 2.Haldane M, Frangou S. New insights help define the pathophysiology of bipolar affective disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:943–960. doi: 10.1016/j.pnpbp.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 3.Strakowski SM, DelBello MP, Adler CM. The functional neuroanatomy of bipolar disorder: a review of neuroimaging findings. Mol Psychiatry. 2005;10:105–116. doi: 10.1038/sj.mp.4001585. [DOI] [PubMed] [Google Scholar]
- 4.Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression. Am J Psychiatry. 2004;161:1537–1547. doi: 10.1176/appi.ajp.161.9.1537. [DOI] [PubMed] [Google Scholar]
- 5.Oquendo MA, Mann JJ. Serotonergic dysfunction in mood disorders. In: Soares J, Gershon S, editors. Basic Mechanisms and Therapeutic Implications of Bipolar Disorder. Marcel Dekker Inc; New York, NY: 2000. pp. 121–142. [Google Scholar]
- 6.Meltzer HY, Arora RC, Baber R, Tricou B-J. Serotonin uptake in blood platelets of psychiatric patients. Arch Gen Psychiatry. 1981;38:1322–1326. doi: 10.1001/archpsyc.1981.01780370024002. [DOI] [PubMed] [Google Scholar]
- 7.Born GVR, Grignani G, Martin K. Long-term effect of lithium on the uptake of 5-hydroxytryptamine by human platelets. Br J Clin Pharmacol. 1980;9:321–325. doi: 10.1111/j.1365-2125.1980.tb01057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott M, Reading HW, Loudon JB. Studies on human blood platelets in affective disorders. Psychopharmacol (Berl) 1979;60:131–135. doi: 10.1007/BF00432283. [DOI] [PubMed] [Google Scholar]
- 9.Marazziti D, Lenzi A, Galli L, San Martino S, Cassano GB. Decreased platelet serotonin uptake in bipolar I patients. Int Clin Psychopharmacol. 1991;6:25–30. doi: 10.1097/00004850-199100610-00004. [DOI] [PubMed] [Google Scholar]
- 10.Stahl SM, Woo DJ, Mefford WN, Berger PA, Ciaranello RD. Hyperserotonemia and platelet serotonin uptake and release in schizophrenia and affective disorders. Am J Psychiatry. 1983;140:26–30. doi: 10.1176/ajp.140.1.26. [DOI] [PubMed] [Google Scholar]
- 11.Malison RT, Price LH, Berman R, Van Dyck CH, Pelton GH, Carpenter L, Sanacora G, Owens MJ, Nemeroff CB, Rajeevan N, Baldwin RM, Seibyl JP, Innis RB, Charney DS. Reduced brain serotonin transporter availability in major depression as measured by [123I]-2β-carbomethoxy-3β-(4-iodophenyl)tropane and single photon emission computer tomography. Biol Psychiatry. 1998;44:1090–1098. doi: 10.1016/s0006-3223(98)00272-8. [DOI] [PubMed] [Google Scholar]
- 12.Newberg AB, Plossl K, Mozley PD, Stubbs JB, Wintering N, Udeshi M, Alavi A, Kauppinen T, Kung HF. Biodistribution and imaging with (123)I-ADAM: a serotonin transporter imaging agent. J Nucl Med. 2004;45:834–841. [PubMed] [Google Scholar]
- 13.Parsey RV, Hastings RS, Oquendo MA, Huang YY, Simpson N, Arcement J, Huang Y, Ogden RT, Van Heertum RL, Arango V, Mann JJ. Lower serotonin transporter binding potential in the human brain during major depressive episodes. Am J Psychiatry. 2006;163:48–57. doi: 10.1176/appi.ajp.163.1.52. [DOI] [PubMed] [Google Scholar]
- 14.Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ, Ginovart N, Spencer EP, Cheok A, Houle S. Serotonin transporter occupancy of five selective serotonin reuptake inhibitors at different doses: an [11C]DASB positron emission tomography study. Am J Psychiatry. 2004;161:826–835. doi: 10.1176/appi.ajp.161.5.826. [DOI] [PubMed] [Google Scholar]
- 15.Ichimiya T, Suhara T, Sudo Y, Okubo Y, Nakayama K, Nankai M, Inoue M, Yasuno F, Takano A, Maeda J, Shibuya H. Serotonin transporter binding in patients with mood disorders: a PET study with [11C](+)McN5652. Biol Psychiatry. 2002;51:715–722. doi: 10.1016/s0006-3223(01)01351-8. [DOI] [PubMed] [Google Scholar]
- 16.Reivich M, Amsterdam JD, Brunswick DJ, Shiue C-Y. PET brain imaging with [11C](+)McN5652 shows increased serotonin transporter availability in major depression. J Affect Disord. 2004;82:321–327. doi: 10.1016/j.jad.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Parsey RV, Mann JJ. Applications of positron emission tomography in psychiatry. Semin Nucl Med. 2003;33:129–135. doi: 10.1053/snuc.2003.127302. [DOI] [PubMed] [Google Scholar]
- 18.Lesch K-P, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J, Müller CR, Hamer DH, Murphy DL. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 19.Cho HJ, Meira-Lima I, Cordeiro O, Michelon L, Sham P, Vallada H, Collier DA. Population-based and family-based studies on the serotonin transporter gene polymorphisms and bipolar disorder. Mol Psychiatry. 2005;10:771–781. doi: 10.1038/sj.mp.4001663. [DOI] [PubMed] [Google Scholar]
- 20.Lasky-Su JA, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of the association between two polymorphisms in the serotonin transporter gene and affective disorders. Am J Med Genet B Neuropsychiatr Genet. 2005;133:110–115. doi: 10.1002/ajmg.b.30104. [DOI] [PubMed] [Google Scholar]
- 21.Hu X, Oroszi G, Chun J, Smith TL, Goldman D, Schuckit MA. An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alcohol Clin Exp Res. 2005;29:8–16. doi: 10.1097/01.alc.0000150008.68473.62. [DOI] [PubMed] [Google Scholar]
- 22.Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 23.Drevets WC, Ongür D, Price JL. Neuroimaging abnormalities in the subgenual prefrontal cortex. Mol Psychiatry. 1998;3:220–226. doi: 10.1038/sj.mp.4000370. [DOI] [PubMed] [Google Scholar]
- 24.American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- 25.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. American Psychiatric Press; Washington, DC: 1997. [Google Scholar]
- 26.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parsey RV, Oquendo MA, Ogden RT, Olvet DM, Simpson N, Huang YY, Van Heertum RL, Arango V, Mann JJ. Altered serotonin 1A binding in major depression: a [carbonyl-C-11]WAY100635 positron emission tomography study. Biol Psychiatry. 2006;59:106–113. doi: 10.1016/j.biopsych.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 28.Frankle WG, Huang Y, Hwang DR, Talbot PS, Slifstein M, Van Heertum R, Abi-Dargham A, Laruelle M. Comparative evaluation of serotonin transporter radioligands 11C-DASB and 11C-McN 5652 in healthy humans. J Nucl Med. 2004;45:682–694. [PubMed] [Google Scholar]
- 29.Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Med Image Anal. 2001;5:143–156. doi: 10.1016/s1361-8415(01)00036-6. [DOI] [PubMed] [Google Scholar]
- 30.Duvernoy HM. The Human Brain: Surface, Three-dimensional Sectional Anatomy and MRI. Springer-Verlag Wien; Vienna, Austria: 1991. [Google Scholar]
- 31.Talairach J, Tournoux P. Co-planar Stereotaxic Atlas of the Human Brain: 3-Dimensional Proportional System: An Approach to Cerebral Imaging. Thieme Medical Publishers Inc; New York, NY: 1988. [Google Scholar]
- 32.Kates WR, Abrams MT, Kaufmann WE, Breiter SN, Reiss AL. Reliability and validity of MRI measurement of the amygdala and hippocampus in children with fragile X syndrome. Psychiatry Res. 1997;75:31–48. doi: 10.1016/s0925-4927(97)00019-x. [DOI] [PubMed] [Google Scholar]
- 33.Killiany RJ, Moss MB, Nicholson T, Jolesz F, Sandor T. An interactive procedure for extracting features of the brain from magnetic resonance images: the lobes. Hum Brain Mapp. 1997;5:355–363. doi: 10.1002/(SICI)1097-0193(1997)5:5<355::AID-HBM4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Lemieux L, Hammers A, Mackinnon T, Liu RS. Automatic segmentation of the brain and intracranial cerebrospinal fluid in T1-weighted volume MRI scans of the head, and its application to serial cerebral and intracranial volumetry. Magn Reson Med. 2003;49:872–884. doi: 10.1002/mrm.10436. [DOI] [PubMed] [Google Scholar]
- 35.Parsey RV, Kegeles LS, Hwang D-R, Simpson N, Abi-Dargham A, Mawlawi O, Slifstein M, Van Heertum RL, Mann JJ, Laruelle M. In vivo quantification of brain serotonin transporters in human using [11C]McN 5652. J Nucl Med. 2000;41:1465–1477. [PubMed] [Google Scholar]
- 36.Abi-Dargham A, Simpson N, Kegeles L, Parsey RV, Hwang D-R, Anjilvel S, Zea-Ponce Y, Lombardo I, Van Heertum R, Mann JJ, Foged C, Halldin C, Laruelle M. PET studies of binding competition between endogenous dopamine and the D1 radiotracer [11C]NNC 756. Synapse. 1999;32:93–109. doi: 10.1002/(SICI)1098-2396(199905)32:2<93::AID-SYN3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 37.Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, Innis RB. Evaluation of ultrafiltration for the free-fraction determination of single photon emission computed tomography (SPECT) radiotracers: β-CIT, IBF, and iomazenil. J Pharm Sci. 1994;83:1014–1019. doi: 10.1002/jps.2600830718. [DOI] [PubMed] [Google Scholar]
- 38.Logan J, Wolf AP, Shiue C-Y, Fowler JS. Kinetic modeling of receptor-ligand binding applied to positron emission tomographic studies with neuroleptic tracers. J Neurochem. 1987;48:73–83. doi: 10.1111/j.1471-4159.1987.tb13129.x. [DOI] [PubMed] [Google Scholar]
- 39.Slifstein M, Laruelle M. Effects of statistical noise on graphic analysis of PET neuroreceptor studies. J Nucl Med. 2000;41:2083–2088. [PubMed] [Google Scholar]
- 40.Ogden RT. Estimation of kinetic parameters in graphical analysis of PET imaging data. Stat Med. 2003;22:3557–3568. doi: 10.1002/sim.1562. [DOI] [PubMed] [Google Scholar]
- 41.Parsey RV, Ogden RT, Mann JJ. Determination of volume of distribution using likelihood estimation in graphical analysis: elimination of estimation bias. J Cereb Blood Flow Metab. 2003;23:1471–1478. doi: 10.1097/01.WCB.0000099460.85708.E1. [DOI] [PubMed] [Google Scholar]
- 42.Mintun MA, Raichle ME, Kilbourn MR, Wooten GF, Welch MJ. A quantitative model for the in vivo assessment of drug binding sites with positron emission tomography. Ann Neurol. 1984;15:217–227. doi: 10.1002/ana.410150302. [DOI] [PubMed] [Google Scholar]
- 43.Parsey RV, Arango V, Olvet DM, Oquendo MA, Van Heertum RL, Mann JJ. Regional heterogeneity of 5-HT1A receptors in human cerebellum as assessed by positron emission tomography. J Cereb Blood Flow Metab. 2005;25:785–793. doi: 10.1038/sj.jcbfm.9600072. [DOI] [PubMed] [Google Scholar]
- 44.Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;258:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- 45.Adell A, Celada P, Abellan MT, Artigas F. Origin and functional role of the extracellular serotonin in the midbrain raphe nuclei. Brain Res Brain Res Rev. 2002;39:154–180. doi: 10.1016/s0165-0173(02)00182-0. [DOI] [PubMed] [Google Scholar]
- 46.Mann JJ, Huang Y, Underwood MD, Kassir SA, Oppenheim S, Kelly TM, Dwork AJ, Arango V. A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry. 2000;57:729–738. doi: 10.1001/archpsyc.57.8.729. [DOI] [PubMed] [Google Scholar]
- 47.Soucy J-P, Lafaille F, Lemoine P, Mrini A, Descarries L. Validation of the transporter ligand cyanoimipramine as a marker of serotonin innervation density in brain. J Nucl Med. 1994;35:1822–1830. [PubMed] [Google Scholar]
- 48.Baumann B, Bielau H, Krell D, Agelink MW, Diekmann S, Wurthmann C, Trubner K, Bernstein HG, Danos P, Bogerts B. Circumscribed numerical deficit of dorsal raphe neurons in mood disorders. Psychol Med. 2002;32:93–103. doi: 10.1017/s0033291701004822. [DOI] [PubMed] [Google Scholar]
- 49.Underwood MD, Khaibulina AA, Ellis SP, Moran A, Rice PM, Mann JJ, Arango V. Morphometry of the dorsal raphe nucleus serotonergic neurons in suicide victims. Biol Psychiatry. 1999;46:473–483. doi: 10.1016/s0006-3223(99)00043-8. [DOI] [PubMed] [Google Scholar]
- 50.Boldrini M, Underwood MD, Mann JJ, Arango V. More tryptophan hydroxylase in the brainstem dorsal raphe nucleus in depressed suicides. Brain Res. 2005;1041:19–28. doi: 10.1016/j.brainres.2005.01.083. [DOI] [PubMed] [Google Scholar]
- 51.Bach-Mizrachi H, Underwood MD, Kassir SA, Bakalian MJ, Sibille E, Tamir H, Mann JJ, Arango V. Neuronal tryptophan hydroxylase mRNA expression in the human dorsal and median raphe nuclei: major depression and suicide. Neuropsychopharmacology. 2006;31:814–824. doi: 10.1038/sj.npp.1300897. [DOI] [PubMed] [Google Scholar]
- 52.Bleuler E. Dementia Praecox or the Group of Schizophrenias. International Universities Press; New York, NY: 1950. [Google Scholar]
- 53.Simpson SG, Folstein SE, Meyers DA, DePaulo JR. Assessment of lineality in bipolar I linkage studies. Am J Psychiatry. 1992;149:1660–1665. doi: 10.1176/ajp.149.12.1660. [DOI] [PubMed] [Google Scholar]
- 54.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception, II: implications for major psychiatric disorders. Biol Psychiatry. 2003;54:515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 55.Leibenluft E, Charney DS, Pine DS. Researching the pathophysiology of pediatric bipolar disorder. Biol Psychiatry. 2003;53:1009–1020. doi: 10.1016/s0006-3223(03)00069-6. [DOI] [PubMed] [Google Scholar]
- 56.Tolmunen T, Joensuu M, Saarinen PI, Mussalo H, Ahola P, Vanninen R, Kuikka J, Tiihonen J, Lehtonen J. Elevated midbrain serotonin transporter availability in mixed mania: a case report. BMC Psychiatry. 2004;4:27. doi: 10.1186/1471-244X-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simpson HB, Lombardo I, Slifstein M, Huang HY, Hwang DR, Abi-Dargham A, Liebowitz MR, Laruelle M. Serotonin transporters in obsessive-compulsive disorder: a positron emission tomography study with [(11)C]McN 5652. Biol Psychiatry. 2003;54:1414–1421. doi: 10.1016/s0006-3223(03)00544-4. [DOI] [PubMed] [Google Scholar]
- 58.Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N, Goulding V, Kennedy J, Wilson AA. Brain serotonin transporter binding potential measured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry. 2004;61:1271–1279. doi: 10.1001/archpsyc.61.12.1271. [DOI] [PubMed] [Google Scholar]
- 59.Cannon D, Ichise M, Fromm S, Nugetn AL, Rollis D, Gandhi SK, Drevets WC. Serotonin transporter binding in unmedicated bipolar disorder subjects using [carbon 11] DASB and positron emission tomography. Paper presented at: American College of Neuropsychopharmacology Annual Meeting; Kona, Hi. December 6, 2005. [Google Scholar]
- 60.Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB, Arango V, Mann JJ. Acute occupancy of brain serotonin transporter by sertraline as measured by [(11)C] DASB and positron emission tomography. Biol Psychiatry. 2006;59:821–828. doi: 10.1016/j.biopsych.2005.08.010. [DOI] [PubMed] [Google Scholar]