

Abstract
Invasion is a hallmark of malignant gliomas and is the main reason for therapeutic failure and recurrence of the tumor. CXCR4 is a key chemokine receptor implicated in glioma cell migration and whose expression is regulated by hypoxia. Here, we report that, hepatocyte growth factor (HGF) upregulated CXCR4 protein expression in glioma cells. HGF pre-treatment increased migration of U87MG and LN229 glioma cells towards the CXCR4 ligand, stromal cell-derived factor-1α (SDF-1α). AMD3100, a CXCR4 inhibitor, inhibited the increased migration of HGF pre-treated LN229 glioma cells towards SDF-1α. Following exposure to HGF and hypoxia both cell lines showed nuclear translocation of NF-κB (p65). The HGF- and hypoxia-induced nuclear translocation of NF-κB (p65) involved phosphorylation and degradation of IκB-α. Knock-down of NF-κB expression inhibited the induction of CXCR4 expression in response to HGF, but not to hypoxia. However, knock-down of NF-κB expression inhibited the induction of CXCR4 expression in response to hypoxia in the presence of HGF. NF-κB mediated migration towards SDF-1α in response to HGF. Knock-down of NF-κB expression resulted in decreased migration of HGF pre-treated glioma cells towards SDF-1α. Therefore, HGF upregulates CXCR4 expression via NF-κB and leads to enhanced migration. To our knowledge, this is the first report to show that a crosstalk mediated by NF-κB exists between the SDF-1α/CXCR4 and HGF/c-Met axes relevant to glioma cell migration. These findings imply that effective inhibition of glioma invasion should be directed against several ligand/receptor signaling pathways.
Keywords: CXCR4, glioma, hepatocyte growth factor, hypoxia, migration, NF-κB
Introduction
Gliomas are the most common type of primary brain tumor. Histologically, they consist of a tumor mass with an ill-defined border [1]. Instead of metastasizing to distal organs, gliomas characteristically spread through the brain parenchyma as individual cells along different anatomic structures [2]. As a result, after surgical resection of the tumor mass, a recurrent tumor typically occurs within a 2 cm margin of the primary tumor. Evidence suggests that invasive glioma cells display decreased proliferation rate and increased resistance to apoptosis, rendering chemotherapy and radiation therapy ineffective [3]. Thus, there is a need to understand the molecular mechanisms governing glioma invasion in order to develop effective anti-invasive therapies.
Several ligand/receptor systems confer an invasive phenotype to glioma cells. For these studies we will focus on two of them. First, stromal cell-derived factor-1α (SDF-1α)/CXCR4 has emerged as a key axis in glioma invasion. CXCR4 is a G-protein coupled chemokine receptor, also known as “fusin”. CXCR4’s ligand is the small chemokine SDF-1α. CXCR4 expression has been correlated with increased tumor grade and malignancy [4]. Overexpression results in increased migration of tumor cells in various cancers including breast [5], ovarian [6], non-small cell lung [7], prostate [8], renal cell [9] and gliomas [10]. CXCR4 is expressed in glioma cell lines and surgical specimens [11]. SDF-1α has been shown to increase chemotaxis and invasion of CXCR4-positive U251 glioma cells through the activation of downstream intracellular pathways of Erk1/2 and Akt [12]. Studies have also noted that SDF-1α upregulates MT2-matrix metalloproteinase (MMP) expression and enhances the invasiveness of CXCR4-positive glioma cells [13].
A second ligand/receptor system mediating glioma invasion is the hepatocyte growth factor (HGF)/c-Met axis. Tyrosine kinase receptor c-Met is a high affinity receptor for the cytokine HGF, also known as scatter factor. The HGF/c-Met axis is a potential therapeutic target in cancers such as non-small cell lung [14], prostate [15], salivary gland [16], breast [17], hepatocellular carcinoma [18] and glioma [19]. Both HGF and c-Met are overexpressed in gliomas and their expression correlates with increased tumor grade and malignancy [19]. Among 14 different growth factors examined in vitro, HGF emerged as the most potent stimulator of glioma cell migration [20]. Moreover, blocking signaling through the HGF/c-Met axis using antibodies against HGF led to tumor regression in vivo [21].
In the current study, we defined the crosstalk between the SDF-1α/CXCR4 and HGF/c-Met axes relevant to glioma cell migration. Previously, both CXCR4 and c-Met were shown to be upregulated by hypoxia in glioma cell lines, leading to enhanced migration [10, 22]. A study on breast cancer cells reported that HGF was able to induce the expression of CXCR4 and contribute to tumor cell invasiveness [23]. Similarly, we hypothesized that HGF upregulates CXCR4 in glioma cells and this results in increased migration. We therefore tested (1) whether HGF upregulates CXCR4 expression in glioma cells, (2) whether HGF increases glioma cell migration towards SDF-1α, (3) whether NF-κB contributes to HGF and hypoxic induction of CXCR4, and (4) whether NF-κB mediates glioma cell migration towards SDF-1α in response to HGF.
Materials and Methods
Cell Culture and Reagents
Human glioma cell lines U87MG and LN229 were obtained from ATCC (Manassas, VA, USA). Cell lines were cultured in 5% CO2 at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) (Cellgro, Herndon, VA, USA). The medium was supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, GA, USA), 1% penicillin and streptomycin, and 2 mM glutamine (Gibco BRL, Grand Island, NY, USA). For hypoxic exposure, cells were placed in a sealed Modular Incubator Chamber (Billups-Rothenberg Inc., Del Mar, CA, USA) flushed with 1% O2, 5% CO2, and 94% N2. Recombinant human HGF (R&D Systems Inc., Minneapolis, MN, USA) was dissolved in 0.1% bovine serum albumin (BSA) in PBS and stock solution (10 μg/ml) was stored at - 20°C. Recombinant human SDF-1α (R&D Systems Inc.) was prepared in 0.1% BSA in PBS and stock solution (100 μg/ml) was stored at -20°C. AMD3100, a CXCR4 inhibitor [2] (kindly provided by Dr. J.B. Rubin, Washington University, St. Louis, MO, USA), was prepared in PBS (5 mg/ml) and kept at 4°C until used.
Western Blot Analysis
Cells were lysed in RIPA buffer supplemented with protease inhibitors [10]. Protein quantitation and electrophoresis were performed as previously described [10]. Western blot analysis was performed with the following antibodies: rabbit anti-CXCR4 polyclonal antibody 1:1000 (Imgenex, San Diego, CA, USA), rabbit anti-NF-κB (p65) polyclonal antibody 1:1000 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit anti-p-IκB-α polyclonal antibody 1:1000 (Santa Cruz Biotechnology, Inc.), rabbit anti-IκB-α polyclonal antibody 1:1000 (Santa Cruz Biotechnology, Inc.) and mouse anti-actin monoclonal antibody 1:20,000 (clone C4, Chemicon International, Inc., Temecula, CA, USA). Donkey anti-rabbit and anti-mouse IgG horseradish peroxidase-conjugated secondary antibodies (Amersham Life Pharmacia Biotech, Piscataway, NJ, USA) were used at 1:2500 dilution. Immunodetection was carried out with the Supersignal West Pico Chemiluminescent Reagent (Thermo Fisher Scientific, Rockford, IL, USA). Visualization and densitometry of protein bands were performed with the National Institutes of Health (NIH) Image software (version 1.62).
Migration Assay
BD Biocoat chambers (BD Bioscience Discovery Labware, Bedford, MA, USA) with 8-μm pore size polycarbonate filter inserts for 24-well plates were used according to the manufacturer’s instructions and as described [10]. Briefly, cells (5 × 104) either untreated or pre-treated for 16 h with HGF (20 ng/ml) were seeded onto the upper chambers in 400 μl of DMEM medium with 10% FBS in the presence or absence of 100 nM of AMD3100 and placed into wells containing 600 μl of complete medium with or without SDF-1α (100 ng/ml) to induce cell migration. The migration chambers were incubated for 24 h in normoxic or hypoxic conditions at 37°C. After incubation, the inserts were fixed and stained and the number of migrating cells was counted as described [10]. Each assay was performed in duplicate and repeated two times with similar results. The data from independent experiments were pooled for statistical analysis.
Immunofluorescence Microscopy
Glioma cells (3 × 104) were seeded onto poly-D-lysine-coated glass coverslips and incubated overnight. Cells were grown under normoxia or hypoxia and in the presence or absence of HGF for 30 min and processed for immunofluorescence. Cells were first fixed in 100% methanol for 2 min and later permeabilized in 0.5% Triton X-100/PBS for 5 min at room temperature. Nonspecific binding was blocked by incubation in blocking buffer containing 3% BSA for 30 minutes at room temperature. Cells were incubated with primary rabbit anti-NF-κB (p65) polyclonal antibody 1:100 (Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. Cells were then washed in blocking buffer three times for 10 min each before incubation with secondary goat anti-rabbit fluorescein isothiocyanate (FITC)-conjugated antibody (Santa Cruz Biotechnology, Inc.) 1:500 for 1 h at room temperature in the dark. Cells were washed in PBS, mounted onto glass slides, and examined using a Zeiss Axiovert 200 M microscope (63x objective lens, N.A. 1.4, 1.6x Optovar) equipped with a cooled Retiga 2000R CCD (QImaging) and Metamorph Software (Molecular Devices). Two independent experiments were performed.
Transfection of glioma cells with small interfering RNA (siRNA) targeting NF-κB expression
Glioma cells were plated (3 × 105) in 60-mm dishes and after 24 h were transfected. Before transfection the medium was aspirated and 4.6 ml of serum-free medium was added to each plate. Knock-down of NF-κB expression was performed using validated target sequences (SI02654932; Qiagen, Valencia, CA, USA). For transfection, 5 nmol annealed siRNA targeting NF-κB or the AllStars negative control siRNA [a scrambled (Scr) sequence with no significant homology to any known gene sequences from mouse, rat, or human cell lines] was used. The siRNA sequences were diluted in serum-free medium, mixed with 4 μl Hiperfect (Qiagen, Valencia, CA, USA), and incubated at room temperature for 10 min. The mixtures were added dropwise to each dish, mixed by gently swirling the dish, and incubated for 4 h at 37°C when 0.5 ml of FBS was added for a final concentration of 10%. After incubation at 37°C for 48 h, cells were cultured in normoxic or hypoxic conditions in the presence or absence of HGF for 16 h. Cells were harvested for Western blot analysis or migration assay. Two independent experiments were performed.
Results
HGF and hypoxia upregulate CXCR4 protein expression
We first evaluated the effect of HGF on CXCR4 protein expression. U87MG and LN229 glioma cell lines were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 16 h. Total cell lysates were collected and subjected to Western blot analysis (Fig. 1). Although we observed that U87MG and LN229 glioma cells had different endogenous levels of CXCR4, exposure to HGF and hypoxia increased CXCR4 protein expression in both cell lines. HGF upregulated CXCR4 protein expression by three-fold in U87MG and five-fold in LN229 glioma cells. Hypoxia increased CXCR4 expression by four-fold in U87MG and six-fold in LN229 glioma cells. Combining HGF and hypoxia produced a more potent effect in LN229 glioma cells as the CXCR4 protein levels were upregulated by ten-fold.
Fig. 1.
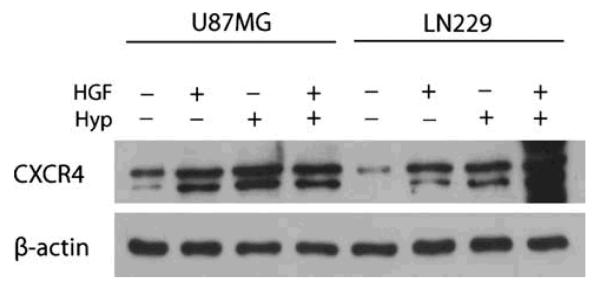
HGF and hypoxia upregulate CXCR4 protein expression
U87MG and LN229 glioma cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 16 h. Lysates were collected and analyzed by Western blot for CXCR4 protein expression. β-actin was used as loading control. Data are representative of three independent experiments with similar results. HGF, hepatocyte growth factor; Hyp, hypoxia.
HGF pre-treatment increases migration towards SDF-1α
To evaluate whether the HGF- and hypoxia-induced CXCR4 expression was of biological significance, we tested the migration of U87MG and LN229 glioma cells untreated or pre-treated with HGF towards SDF-1α in normoxic or hypoxic conditions. Results from two independent experiments are shown (Fig. 2). Cells untreated or pre-treated with 20 ng/ml of HGF for 16 h were seeded in migration chambers and placed into wells in the presence or absence of 100 ng/ml of SDF-1α. They were allowed to migrate for 24 h in normoxic or hypoxic conditions, after which they were fixed, stained and quantitated. In normoxic conditions, HGF pre-treatment or the presence of SDF-1α in the lower well alone had no effect on the number of migrated cells in either cell line. In contrast, when U87MG (Fig. 2a) and LN229 (Fig. 2b) glioma cells were first pre-treated with HGF, they then showed a significant increase in migration towards SDF-1α as compared to cells exposed to SDF-1α alone (P<0.001).
Fig. 2.

HGF pre-treatment increases migration towards SDF-1α
U87MG (a) and LN229 (b and c) glioma cells untreated or pre-treated with 20 ng/ml of HGF for 16 h were seeded in migration chambers in the presence or absence of 100 nM of AMD3100 and placed into wells in the presence or absence of 100 ng/ml of SDF-1α. They were allowed to migrate for 24 h in normoxic or hypoxic conditions. Bar graphs indicate the average number of migrated cells per field. Error bars denote mean ± standard deviation. *P<0.001 versus normoxic control; **P<0.001 versus non-SDF-1α exposed cells; ***P<0.001 versus SDF-1α exposed cells; ΔP<0.001 versus HGF pre-treated, SDF-1α exposed cells. Bar graphs represent pooled data from two independent experiments. N & white bars, normoxic conditions; H & grey bars, hypoxic conditions; hatched bars, AMD3100-treated cells.
In hypoxic conditions, both cell lines increased their migration significantly compared to similar cultures in normoxic conditions (P<0.001). In the absence of HGF pre-treatment, but in the presence of SDF-1α in the lower well, U87MG glioma cells showed a significant increase in migration towards SDF-1α compared to hypoxic control cultures (P<0.001). Migration of HGF pre-treated cell lines towards SDF-1α was significantly increased as compared to cells exposed to SDF-1α alone (P<0.001). When repeated in the presence of 10 mM hydroxyurea, which blocks proliferation [2], HGF pre-treated cells still showed a marked increase in migration towards SDF-1α as compared to cells exposed to SDF-1α alone (data not shown).
To confirm that the HGF-induced CXCR4 expression led to increased migration towards SDF-1α, we blocked CXCR4 expression in LN229 glioma cells with AMD3100, a CXCR4 inhibitor (Fig. 2c). AMD3100 inhibited the increased migration of HGF pre-treated LN229 glioma cells towards SDF-1α compared to control cultures (P<0.001).
NF-κB contributes to HGF-mediated CXCR4 upregulation
In response to an appropriate signal, the cytoplasmic inhibitor IκB-α is phosphorylated on serine and degraded, thus dissociating from the NF-κB (p65-p50) heterodimer. As a result NF-κB heterodimer translocates from the cytosol to the nucleus and induces the expression of target genes containing NF-κB response elements [24]. To determine if HGF and hypoxia activated NF-κB signaling, U87MG and LN229 glioma cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 30 min and then subjected to immunostaining for NF-κB (p65) (Fig. 3a). In the absence of HGF or hypoxia, U87MG and LN229 glioma cells showed cytoplasmic localization of NF-κB (p65). Following exposure to HGF and hypoxia both cell lines showed nuclear translocation of NF-κB (p65). To test whether the HGF- and hypoxia-induced nuclear translocation of NF-κB (p65) involved phosphorylation and degradation of IκB-α, we assessed the phosphorylation state of IκB-α protein. U87MG and LN229 glioma cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 30 min. Total cell lysates were collected and subjected to Western blot analysis (Fig. 3b). We observed that exposure to HGF and hypoxia increased IκB-α phosphorylation levels in both cell lines, which was accompanied by a marked decrease in IκB-α protein expression.
Fig. 3.

NF-κB contributes to HGF-mediated CXCR4 upregulation
U87MG and LN229 glioma cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 30 min and then subjected to (a) immunostaining for NF-κB (p65) and (b) Western blot analysis for p-IκB-α and IκB-α protein expression. (c) Cells were transfected with Scr siRNA or siRNA directed against NF-κB. After 48 h, cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 16 h. Lysates were collected and analyzed by Western blot for NF-κB and CXCR4 protein expression. β-actin was used as loading control. The experiments were repeated twice. HGF, hepatocyte growth factor; Hyp, hypoxia. si NF-κB (−) indicates cells transfected with Scr siRNA and si NF-κB (+) indicates cells transfected with siRNA directed against NF-κB. Original magnifications: x63.
The CXCR4 promoter region has been shown to contain NF-κB response elements [25]. NF-κB has also been demonstrated to stimulate glioma cell invasion [26]. We thus wanted to determine whether NF-κB played a role in the induction of CXCR4 in response to HGF and hypoxia. The results of representative Western blots from two independent experiments are shown (Fig. 3c). U87MG and LN229 glioma cells were transfected with Scr siRNA or siRNA directed against NF-κB. After 48 h, cells were cultured in normoxic or hypoxic conditions in the presence or absence of HGF for 16 h. Total cell lysates were then collected and subjected to Western blot analysis. In both U87MG and LN229 glioma cells transfected with Scr siRNA, NF-κB expression was upregulated following exposure to HGF (lane 2) and hypoxia (lane 5). In contrast, knock-down of NF-κB expression inhibited the induction of NF-κB in response to HGF (lanes 2 vs. 4) and hypoxia (lanes 2 vs. 7). U87MG and LN229 glioma cells transfected with Scr siRNA upregulated CXCR4 expression following exposure to HGF (lane 2) and hypoxia (lane 5). Knock-down of NF-κB expression inhibited the induction of CXCR4 in response to HGF (lanes 2 vs. 4). Knock-down of NF-κB expression, however, did not inhibit the induction of CXCR4 in response to hypoxia (lanes 5 vs. 7). On the other hand, in the presence of HGF, knock-down of NF-κB expression inhibited the induction of CXCR4 in response to hypoxia (lane 8).
NF-κB mediates migration towards SDF-1α in response to HGF
We then determined whether knock-down of NF-κB expression resulted in decreased migration of HGF pre-treated glioma cells towards SDF-1α in normoxic and hypoxic conditions. Having observed similar responses in U87MG and LN229 glioma cells (Figs. 1, 2, 3), we chose LN229 glioma cells as a representative cell line for this investigation. LN229 glioma cells were transfected with Scr siRNA or siRNA directed against NF-κB. After 48 h, cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 16 h. Cells untreated or pre-treated with HGF were seeded in migration chambers and placed into wells in the presence or absence of 100 ng/ml of SDF-1α. They were allowed to migrate for 24 h in normoxic or hypoxic conditions, after which they were fixed, stained and quantitated. Results from two independent experiments are shown (Fig. 4). As we observed previously (Fig. 2b), in normoxic conditions, HGF pre-treated cells showed a significant increase in migration towards SDF-1α as compared to cells exposed to SDF-1α alone (P<0.001). After knock-down of NF-κB expression, migration of the HGF pre-treated cells towards SDF-1α was significantly decreased compared to Scr siRNA transfected cultures (P<0.001). In hypoxic conditions, knock-down of NF-κB expression also led to a decrease in migration compared to Scr siRNA transfected hypoxic control cultures (P<0.001).
Fig. 4.

NF-κB mediates migration towards SDF-1α in response to HGF
LN229 glioma cells were transfected with Scr siRNA or siRNA directed against NF-κB. After 48 h, cells were cultured in normoxic or hypoxic conditions in the presence or absence of 20 ng/ml of HGF for 16 h. Cells untreated or pre-treated with HGF were seeded in migration chambers and placed into wells in the presence or absence of 100 ng/ml of SDF-1α. They were allowed to migrate for 24 h in normoxic or hypoxic conditions. Bar graphs indicate the average number of migrated cells per field. Error bars denote mean ± standard deviation. *P<0.001 versus normoxic control; ***P<0.001 versus SDF-1α exposed cells; □P<0.001 versus hypoxic control; □□P<0.001 versus HGF pre-treated cells exposed to SDF-1α. Bar graphs represent pooled data from two independent experiments. N & white bars, normoxic conditions; H & grey bars, hypoxic conditions. si NF-κB (−) indicates cells transfected with Scr siRNA and si NF-κB (+) indicates cells transfected with siRNA directed against NF-κB.
Discussion
Our findings demonstrate that (1) HGF upregulates CXCR4 expression in glioma cells, (2) HGF increases glioma cell migration towards SDF-1α, (3) NF-κB contributes to HGF-mediated induction of CXCR4, and (4) NF-κB mediates glioma cell migration towards SDF-1α in response to HGF. To our knowledge, this is the first report to show a crosstalk mediated by NF-κB exists between the SDF-1α/CXCR4 and HGF/c-Met axes relevant to glioma cell migration (Fig. 5).
Fig. 5.

Molecular pathways leading to CXCR4 upregulation in glioma
This schema diagrams two potential factors, HGF and hypoxia, that may play a role in the upregulation of CXCR4 in glioma. HGF-mediated CXCR4 induction appears to be via NF-κB. Our results indicate a crosstalk between the SDF-1α/CXCR4 and HGF/c-Met axes relevant to glioma cell migration.
Previous studies have demonstrated that hypoxia is a pivotal factor in the glioma microenvironment that mediates invasion [27] and induces both CXCR4 [2, 10] and c-Met [22, 28]. In this study we observed that HGF, another critical factor of the glioma microenvironment, increased CXCR4 protein levels in U87MG and LN229 glioma cells. Similarly, another study has reported increased SDF-1 and CXCR4 mRNA levels in glioma cells upon treatment with HGF [29]. As documented here, the crosstalk between the SDF-1α/CXCR4 and HGF/c-Met axes appears not to rely entirely on hypoxia. Therefore, our data suggest that although targeting hypoxia might be a promising therapeutic strategy, it is not sufficient to abolish glioma invasion. This further underlies the need to fully understand the molecular mechanisms governing glioma cell migration before effective anti-invasive therapies can be developed.
HGF was previously demonstrated to promote the migration of glioma cell lines [22]. However, whether HGF pre-treated glioma cells migrate more readily towards SDF-1α has never been explored. We observed a marked increase in migration towards SDF-1α in HGF pre-treated glioma cells. This increase was even more pronounced in hypoxic conditions. Our results suggest that HGF pre-treatment “primes” the glioma cells to migrate towards SDF-1α by upregulating CXCR4 expression. Similar findings were previously reported for breast cancer cells [23]. It should be noted that although hypoxia upregulated CXCR4 protein levels in both U87MG and LN299 glioma cells, the level of hypoxia-induced CXCR4 protein expression was low in LN229 compared to U87MG glioma cells. Therefore, whereas neither cell line migrated towards SDF-1α under normoxic conditions, only U87MG glioma cells increased their migration towards SDF-1α under hypoxic conditions.
A variety of signals activate the transcription factor NF-κB, including growth factors, inflammatory cytokines, UV light and oxidative stress [24]. Our data show that HGF and hypoxia activate NF-κB by inducing the nuclear translocation of the p65 subunit via phosphorylation and degradation of the cytoplasmic inhibitor IκB-α. Previous studies suggested that aberrant constitutive activation of NF-κB drives invasion in glioma [26]. In line with this observation, blocking NF-κB activity resulted in a marked decrease in glioma invasion in vitro [30]. NF-κB was also shown to induce the expression of CXCR4 [25] and mediate HGF-induced CXCR4 upregulation and invasion in breast cancer cells [31]. Our results suggest that although CXCR4 is upregulated by both HGF and hypoxia in glioma cells, NF-κB induces CXCR4 only after HGF stimulation. Hypoxic upregulation of CXCR4 is therefore more likely due to the activity of transcriptional activators such as HIF-1 [10]. We observed a marked decrease in migration of cells pre-treated with HGF towards SDF-1α after knock-down of NF-κB expression compared to Scr siRNA transfected cultures. These findings indicate that HGF upregulates CXCR4 expression via NF-κB and promotes enhanced glioma cell migration. In hypoxic conditions, knock-down of NF-κB expression also led to a marked decrease in migration compared to control cultures. NF-κB has been previously shown to be regulated by hypoxia and associated with cell motility [32]. Thus, it is likely that NF-κB contributes to the hypoxia-mediated increase in cell migration by inducing other genes in addition to CXCR4. For instance, NF-κB was shown to be essential in enhancing the expression of the metalloproteinases MMP-2 and MMP-9 and contribute to glioma invasion [33]. However, the possibility of other transcription factors besides NF-κB contributing to HGF-mediated upregulation of CXCR4 should not be ignored. For instance Ets-1, another HGF and hypoxia-inducible transcription factor, is able to activate the transcription of CXCR4 [31] and mediate glioma invasion [34].
In vivo, both HGF and c-Met [35], as well as CXCR4 [10] are expressed by both tumor and endothelial cells of gliomas. This suggests both autocrine and paracrine signaling mechanisms between tumor cells and tumor endothelium affect glioma progression by promoting invasion as well as angiogenesis. HGF released either by tumor or endothelial cells of gliomas may enhance not only HGF/c-Met, but also SDF-1α/CXCR4 signaling on tumor cells, thereby promoting invasion. On the other hand, HGF may similarly enhance angiogenesis due to the crosstalk between these two signaling pathways.
Collectively, our data indicate a crosstalk between two ligand/receptor systems essential for glioma cell migration, i.e. SDF-1α/CXCR4 and HGF/c-Met. This implies that effective inhibition of glioma invasion should be directed against more than one signaling pathway.
Acknowledgments
This work was supported by the National Institutes of Health grant R01 CA100426.
Abbreviations
- HGF
hepatocyte growth factor
- SDF-1α
stromal cell-derived factor-1α
References
- 1.Mueller MM, Werbowetski T, Del Maestro RF. Soluble factors involved in glioma invasion. Acta Neurochir (Wien) 2003;145:999–1008. doi: 10.1007/s00701-003-0132-0. [DOI] [PubMed] [Google Scholar]
- 2.Zagzag D, Esencay M, Mendez O, et al. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1alpha/CXCR4 expression in glioblastomas: one plausible explanation of Scherer’s structures. Am J Pathol. 2008;173:545–560. doi: 10.2353/ajpath.2008.071197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giese A, Bjerkvig R, Berens ME, et al. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21:1624–1636. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 4.Rempel SA, Dudas S, Ge S, et al. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res. 2000;6:102–111. [PubMed] [Google Scholar]
- 5.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 6.Scotton CJ, Wilson JL, Scott K, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62:5930–5938. [PubMed] [Google Scholar]
- 7.Su L, Zhang J, Xu H, et al. Differential expression of CXCR4 is associated with the metastatic potential of human non-small cell lung cancer cells. Clin Cancer Res. 2005;11:8273–8280. doi: 10.1158/1078-0432.CCR-05-0537. [DOI] [PubMed] [Google Scholar]
- 8.Darash-Yahana M, Pikarsky E, Abramovitch R, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. Faseb J. 2004;18:1240–1242. doi: 10.1096/fj.03-0935fje. [DOI] [PubMed] [Google Scholar]
- 9.Pan J, Mestas J, Burdick MD, et al. Stromal derived factor-1 (SDF-1/CXCL12) and CXCR4 in renal cell carcinoma metastasis. Mol Cancer. 2006;5:56. doi: 10.1186/1476-4598-5-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zagzag D, Lukyanov Y, Lan L, et al. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: implications for angiogenesis and glioma cell invasion. Lab Invest. 2006;86:1221–1232. doi: 10.1038/labinvest.3700482. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Larsen PH, Hao C, et al. CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem. 2002;277:49481–49487. doi: 10.1074/jbc.M206222200. [DOI] [PubMed] [Google Scholar]
- 12.Wu M, Chen Q, Li D, et al. LRRC4 inhibits human glioblastoma cells proliferation, invasion, and proMMP-2 activation by reducing SDF-1 alpha/CXCR4-mediated ERK1/2 and Akt signaling pathways. J Cell Biochem. 2008;103:245–255. doi: 10.1002/jcb.21400. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Sarkar S, Yong VW. The chemokine stromal cell derived factor-1 (CXCL12) promotes glioma invasiveness through MT2-matrix metalloproteinase. Carcinogenesis. 2005;26:2069–2077. doi: 10.1093/carcin/bgi183. [DOI] [PubMed] [Google Scholar]
- 14.Masuya D, Huang C, Liu D, et al. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br J Cancer. 2004;90:1555–1562. doi: 10.1038/sj.bjc.6601718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujiuchi Y, Nagakawa O, Murakami K, et al. Effect of hepatocyte growth factor on invasion of prostate cancer cell lines. Oncol Rep. 2003;10:1001–1006. [PubMed] [Google Scholar]
- 16.Hara S, Nakashiro K, Klosek SK, et al. Hypoxia enhances c-Met/HGF receptor expression and signaling by activating HIF-1alpha in human salivary gland cancer cells. Oral Oncol. 2006;42:593–598. doi: 10.1016/j.oraloncology.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Martin TA, Parr C, Davies G, et al. Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis. 2003;24:1317–1323. doi: 10.1093/carcin/bgg072. [DOI] [PubMed] [Google Scholar]
- 18.Miura Y, Kozuki Y, Yagasaki K. Potentiation of invasive activity of hepatoma cells by reactive oxygen species is mediated by autocrine/paracrine loop of hepatocyte growth factor. Biochem Biophys Res Commun. 2003;305:160–165. doi: 10.1016/s0006-291x(03)00725-3. [DOI] [PubMed] [Google Scholar]
- 19.Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-oncol. 2005;7:436–451. doi: 10.1215/S1152851705000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brockmann MA, Ulbricht U, Gruner K, et al. Glioblastoma and cerebral microvascular endothelial cell migration in response to tumor-associated growth factors. Neurosurgery. 2003;52:1391–1399. doi: 10.1227/01.neu.0000064806.87785.ab. discussion 9. [DOI] [PubMed] [Google Scholar]
- 21.Burgess T, Coxon A, Meyer S, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006;66:1721–1729. doi: 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 22.Eckerich C, Zapf S, Fillbrandt R, et al. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer. 2007;121:276–283. doi: 10.1002/ijc.22679. [DOI] [PubMed] [Google Scholar]
- 23.Matteucci E, Locati M, Desiderio MA. Hepatocyte growth factor enhances CXCR4 expression favoring breast cancer cell invasiveness. Exp Cell Res. 2005;310:176–185. doi: 10.1016/j.yexcr.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Vermeulen L, Wilde G De, Notebaert S. Regulation of the transcriptional activity of the nuclear factor-κB p65 subunit. Biochem Pharmacol. 2002;64:963–970. doi: 10.1016/s0006-2952(02)01161-9. [DOI] [PubMed] [Google Scholar]
- 25.Helbig G, Christopherson KW, 2nd, Bhat-Nakshatri P, et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631–21638. doi: 10.1074/jbc.M300609200. [DOI] [PubMed] [Google Scholar]
- 26.Raychaudhuri B, Han Y, Lu T, et al. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007;85:39–47. doi: 10.1007/s11060-007-9390-7. [DOI] [PubMed] [Google Scholar]
- 27.Fujiwara S, Nakagawa K, Harada H, et al. Silencing hypoxia-inducible factor-1alpha inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int J Oncol. 2007;30:793–802. [PubMed] [Google Scholar]
- 28.Pennacchietti S, Michieli P, Galluzzo M, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 29.Tu H, Zhou Z, Liang Q, et al. SDF-1 and CXCR4 production are stimulated by hepatocyte growth factor and promote glioma cell invasion. Onkologie. 2009;32:331–336. doi: 10.1159/000216352. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Gondi CS, Dinh DH, et al. Transfection with anti-p65 intrabody suppresses invasion and angiogenesis in glioma cells by blocking nuclear factor-kappaB transcriptional activity. Clin Cancer Res. 2007;13:2178–2190. doi: 10.1158/1078-0432.CCR-06-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maroni P, Bendinelli P, Matteucci E, et al. HGF induces CXCR4 and CXCL12-mediated tumor invasion through Ets1 and NF-kappaB. Carcinogenesis. 2007;28:267–279. doi: 10.1093/carcin/bgl129. [DOI] [PubMed] [Google Scholar]
- 32.Cummins EP, Comerford KM, Scholz C, et al. Hypoxic regulation of NF-kappaB signaling. Methods Enzymol. 2007;435:479–492. doi: 10.1016/S0076-6879(07)35025-8. [DOI] [PubMed] [Google Scholar]
- 33.Park MH, Ahn BH, Hong YK, et al. Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-kappaB/Sp1-mediated signaling pathways. Carcinogenesis. 2009;30:356–365. doi: 10.1093/carcin/bgn287. [DOI] [PubMed] [Google Scholar]
- 34.Sahin A, Vercamer C, Kaminski A, et al. Dominant-negative inhibition of Ets 1 suppresses tumor growth, invasion and migration in rat C6 glioma cells and reveals differentially expressed Ets 1 target genes. Int J Oncol. 2009;34:377–389. [PubMed] [Google Scholar]
- 35.Kunkel P, Muller S, Schirmacher P, et al. Expression and localization of scatter factor/hepatocyte growth factor in human astrocytomas. Neuro Oncol. 2001;3:82–88. doi: 10.1093/neuonc/3.2.82. [DOI] [PMC free article] [PubMed] [Google Scholar]