

Abstract
Tumors are supported by blood vessels and it has long been debated whether their response to irradiation is affected by radiation damage to the vasculature. We have shown in preclinical models that, indeed, radiation is damaging to the tumor vasculature and strongly inhibits tumor angiogenesis. However, the vasculature can recover by colonization from circulating cells, primarily pro-angiogenenic CD11b+ monocytes/macrophages from the bone marrow. This secondary pathway of blood vessel formation, known as vasculogenesis, thus acts to restore the tumor vasculature and allows the tumor to recur following radiation. The stimulus for the influx of these CD11b+ cells into tumors following irradiation is increased levels of hypoxia inducible factor-1 (HIF-1) in the tumor due to induced tumor hypoxia secondary to blood vessel loss. This increases tumor levels of the chemokine stromal cell-derived factor-1 (SDF-1), which has chemokine receptors CXCR4 and CXCR7 on monocytes and endothelial cells thereby capturing these cells in the tumors. The increase in CD11b+ monocytes in tumors following irradiation can be prevented using antibodies or small molecules that inhibit HIF-1 or the interaction of SDF-1 with its receptors. We show that the effect of inhibiting these chemokine/chemokine receptor interactions is a marked increase in the radiation response of transplanted or chemically induced tumors in mice and rats. This strategy of inhibiting vasculogenesis following tumor irradiation is a new paradigm in radiotherapy and suggests that higher levels of local control of tumors in several sites will be achievable with this strategy.
Endothelial Cells in Tumors: Are they a Target for Radiotherapy?
It is now widely appreciated that tumors comprise many cells of host origin in addition to tumor cells and these can influence tumor progression. Among the most important of these are macrophages, endothelial cells, pericytes, dendritic cells, neutrophils, fibroblasts and lymphocytes. Some of these can promote and some can inhibit tumor growth, survival and spread (see recent review (1). Yet, until recently radiobiologists and radiation oncologists have ignored the presence of such cells, calculating the dose needed to control tumors from log cell kill using the radiation survival characteristics of the tumor cells derived either from in vitro or in vivo data and from the number of tumor cells needed to transplant the tumors. In some cases this has been successful (2-4), but in others less so (5). Nonetheless, the dogma in radiation oncology circles has been (and largely remains) that tumor control depends solely on the survival of the tumor cells to radiation, with accommodation being made to the possibility of an immune response, which is considered not to affect the survival of the tumor cells but rather the number of tumor cells needed to regrow the tumor.
Some years ago a major challenge to this dogma was mounted by Juliana Denekamp who pointed out that the vasculature, and in particular the endothelial cells, could be the critical target for tumor control (6). There were good reasons for this: notably each endothelial cell supports some 2000 cancer cells, and the proliferation rates of endothelial cells in tumors is rapid and similar to that of the tumor cells themselves. Thus, unlike the endothelial cells in normal tissues, they are likely to die rapidly from radiation damage by mitotically linked death. Given also that there are considerably fewer endothelial cells than tumor cells in tumors, it makes very good sense that the tumor endothelial cells could be the critical limiting factor in tumor cure by irradiation.
However plausible is the hypothesis that the radiation dose to eliminate tumors is determined by killing of the tumor endothelial cells, data published in 1993 provides strong evidence against it. In this classic study Budach and colleagues determined the TCD50 of 9 different tumors, of both mouse and human origin, in two immunodeficient mouse strains, nude and SCID (7). The data (Figure 1) show no significant differences between the TCD50’s in the two strains. The significance of this is the fact that the SCID is immunodeficient because of an inactivating mutation in the key DNA repair gene DNAPKcs (which is required for VDJ recombination during T and B cell development), and consequently all the tissues of the mouse are highly radiosensitive (8). Therefore, as all the stromal cells of the tumors in the SCID mice, including the endothelial cells, are much more radiosensitive than those of the nude mice, it follows from these data that the endothelial cells in particular, and the stromal cells in general, do not contribute to control of these tumors by irradiation.
Figure 1. Stromal radiosensitivity does not affect tumor control by irradiation.

Tumor control dose (TCD50) for the same tumor cell lines growing either in the nude or C3H mouse or in the SCID mouse, the latter being some 3-fold more sensitive to irradiation. Error bars indicate the 95% confidence intervals for the TCD50. + = no local control observed at the highest dose administered (90 Gy) from (7) with permission.
But there is a second conclusion that can be drawn from the data in Figure 1. Note that the TCD50 values are between 40 and 110 Gy – massive single doses even accounting for the fact that the tumors (and stroma) were made 100% hypoxic by clamping during irradiation. Given the extreme sensitivity of SCID endothelial cells (9), none of the tumor endothelial cells could have survived the radiation dose (50 Gy would kill at least 10 logs of SCID endothelial cells under hypoxia). So how did 50% of the tumors recur following these doses? The conclusion is inescapable that the surviving tumor cells formed their own blood vessels or blood vessel forming cells must have migrated to the tumor from outside the irradiation field.
In 1993 neither of these phenomena had been reported, but 4 years later work from Isner’s laboratory provided definitive evidence that there were circulating endothelial progenitor cells (EPCs) that would home to ischemic areas and contribute to neovascularization(10). Since then there have been numerous preclinical and clinical studies using EPCs to speed the revascularization of damaged tissue such as occurs in myocardial infarction (11). However, the significance of the presence of such cells cannot be overstated. First it explains the data in Figure 1 – the sensitivity of the cells of the tumor vasculature cannot contribute to the radiosensitivity of the tumor if the vasculature of the tumor can be “rescued” by circulating EPCs. Second, it means that the dose to control solid tumors might be significantly less if the EPCs could be prevented from colonizing the tumor following irradiation.
Bone marrow derived myelomonocytic cells colonize irradiated tumors and stimulate tumor regrowth
We performed several studies to identify bone marrow derived endothelial cells that incorporate into the tumor vasculature involving bone marrow transplantation from sex mismatched donors, from lacZ mice (used as a reporter gene) and from GFP (green fluorescent protein) expressing mice. In neither unirradiated tumors, nor irradiated tumors regrowing after irradiation nor tumors growing in an irradiated bed did we find significant number of bone marrow derived EPCs in the tumor vasculature (12, 13). This is in line with work of other investigators who have found few if any bone marrow derived EPCs in the tumor vasculature with or without irradiation (14-16). However, other investigators have shown using either orthotopic aortic allografting (17), or a parabiotic mouse system (two mice joined so as to have a common blood supply) combined with reverse bone marrow transplantation that non-bone marrow derived circulating ECs home to a sites of active angiogenesis (18). Thus it is still an open question, and an area of active investigation, as to the source of the circulating ECs (or EPCs) that may colonize tumors after treatment.
But if the question is still open of whether EPCs colonize tumors after irradiation this is not the case with bone marrow derived monocytes/macrophages. In 2008 we reported that bone marrow derived CD11b monocytes (which differentiate into macrophages) were increased in irradiated mouse tumors as they commenced regrowth and in tumors growing in a pre-irradiated tissue (12) (Figure 2). These cells are highly pro-angiogenic (19-21). We further showed that pharmacological or genetic depletion of these cells or of their ability to express MMP-9 (matrix metallopeptidase-9) reduced the ability of tumors to regrow after irradiation or grow in a preirradiated site (12, 22). We made similar findings in a human glioblastoma (GBM) implanted intracranially in nude mice (13), as have others with mouse and xenograft models after local irradiation (16, 23). Importantly we also found a large increase in CD11b monocytes in GBM recurrences in patients after irradiation (13). An increase in tumor associated macrophages (TAMs) has also been seen in experimental tumors and in cancer patients following chemotherapy (24), and these limit the response to chemotherapy as well as to vascular disrupting agents (25) and to anti-angiogenic therapy (26). At least for irradiation the mechanism for the influx of TAMs appears to be the radiation-induced loss of tumor endothelial cells leading to tumor hypoxia (13, 23). We have shown that inhibition of HIF-1 (hypoxia inducible factor-1) abrogates this influx of macrophages into irradiated GBM xenografts and enhances the efficacy of radiation (13).
Figure 2. Irradiation produces influx of bone marrow derived CD11b+ monocytes into tumors.

(A) Immunostaining for CD11b (red) in tumors with no IR (control), irradiated tumors (IR tumor), or tumors grown in the irradiated bed (pre-IR bed) for MT1A2 (upper panel) and RIF (lower panel). Nucleus staining with DAPI is shown in blue. (B) Quantification of CD11b+ myelomonocytic cells in (B) for MT1A2 (left) and RIF (right) tumors. Symbols and error bars represent the mean ± SEM for n>4 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001, respectively, determined by one-way ANOVA. From (13) with permission.
Targeting the Vasculogenesis Pathway as a Novel Strategy in Radiotherapy
The above studies have led us to formulate a new theory that post-irradiation tumor recurrences can be prevented, or markedly delayed, by blocking the vasculogenesis pathway resulting from the influx of circulating proangiogenic cells-CD11b+ monocytes and endothelial cells - into the tumor. This strategy is based on two concepts as follows:
Tumors have two main ways to grow their vasculature: by angiogenesis, or the sprouting of vessels from pre-existing vessels in the surrounding normal tissues, and by vasculogenesis, or the formation of blood vessels de novo from circulating cells. Under normal conditions the angiogenesis pathway is dominant with the vasculogenesis pathway a minor “backup” pathway.
The high doses of radiation delivered in radiotherapy are more than sufficient to kill the tumor endothelial cells (ECs) in and surrounding the tumor, thereby abrogating local angiogenesis. Though previous investigators have reported that radiation inhibits angiogenesis (27-29), the fact that this is an essentially total inhibition following radiation doses comparable to those given in radiotherapy, this can only be seen if the vasculogenesis pathway is blocked at the same time as the delivery of local irradiation. This we did recently and demonstrated that a single dose of 15 Gy totally abrogated local angiogenesis in the mouse (12). It is important to note that our finding that radiation blocks local angiogenesis is not the same as the proposal of Fuks and Kolesnick that radiation produces a rapid apoptosis of tumor ECs and vascular shutdown (30). We, and others who have measured tumor ECs after irradiation, do not see this rapid apoptosis. Rather ECs disappear slowly after irradiation over several days, consistent with a mitotically linked death (13, 16, 23, 31). This loss of vasculature appears to be the cause of the increased tumor hypoxia days after irradiation and the subsequent increase in the transcription factor HIF-1.
Our theory depends on the supposition that bone marrow derived cells (BMDCs) can contribute to the vascularization of tumors thereby compensating for the abrogation of angiogenesis. The evidence for this is substantial. Several authors have shown that bone marrow-derived endothelial progenitor cells (EPCs) can restore tissue vascularization after ischemic events in limbs, retina, and myocardium (32, 33). Also mice that cannot mobilize bone marrow-derived EPCs due to defective Id genes (Id1+/− Id3−/− mice) show a marked inhibition of the growth of transplanted tumors (34, 35), and normal tumor growth can be restored by transplanting normal bone marrow into these mice. Nonetheless, there is substantial controversy as to the functional significance of EPC given the enormous variation of incorporation into tumor vessels (36). However, there is more established evidence by several laboratories and by us that innate BMDCs contribute to the formation and maintenance of tumor vessels (37, 38). Despite this controversy, there is no doubt that BMDCs contribute to, and/or promote, the tumor vasculature.
How can the influx of these proangiogenic monocytes/macrophages into tumors after irradiation be prevented? The mobilization and trafficking of bone marrow derived cells to sites of ischemic injury has been shown to be regulated by HIF-1(39), and the recruitment of bone BMDCs is markedly reduced in HIF-1 knockout orthotopic GBM (40). Further HIF-1 knockout sensitizes tumors to irradiation without any effect on the intrinsic radiosensitivity of the tumor cells (41) and pharmacological HIF-1 inhibition also sensitizes tumors to irradiation (42). Consistent with these results from the literature we showed that post-irradiation treatment of mice with the HIF-1 inhibitor NSC-134754 markedly sensitizes intracranial U251 GBM tumors to irradiation and blocks the radiation-induced uptake of CD11b+ monocytes into the tumors (Figure 3).
Figure 3. HIF-1 inhibitor prevents the influx of monocytes/macrophages into irradiated U251 intracranial GBM and increases their radiation response.
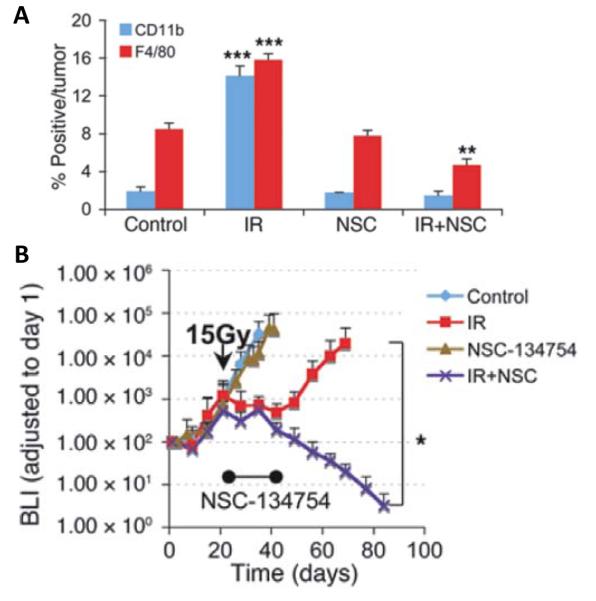
(A) Quantification of CD11b+monocytes and F4/80+macrophage cell influx into GBM tumors. Error bars indicate SEM. **P < 0.01, ***P < 0.001 versus control. (B) Growth curves of i.c. tumors treated with irradiation, NSC-134754, irradiation+NSC-134754, and control (n = 5 per group). Error bars indicate SD. *P < 0.05. From (13) with permission.
However, inhibiting HIF-1 is not an ideal solution as there are no specific inhibitors of this molecule and even if there were the effects would be highly pleotropic because HIF-1 regulates many genes. So the question becomes “What is the downstream pathway regulated by HIF-1 that is responsible for the mobilization and capture of the BMDCs into the tumor after irradiation?” Here again the literature is very helpful. Several investigators have shown that BMDCs are recruited to and retained in hypoxic normal tissues and in tumors by the hypoxia-dependent secretion of SDF-1 (stromal cell-derived factor-1, CXCL12), which binds to its receptor, CXCR4, on BMDCs (39, 43-46). Following these results, we found that SDF-1 secretion was upregulated under hypoxic condition in vitro, and this upregulation was abolished by NSC-134754. In our brain implanted GBM, SDF-1 levels in the tumors rose to a maximum level by 2 weeks after irradiation and then returned to control levels at 4 weeks (13). In addition, we found that local irradiation increased phosphorylation of CXCR4 in the tumor, indicating interaction of SDF-1 with CXCR4.
How could this Strategy be Applied in Radiotherapy?
In order to exploit our findings in experimental systems and ultimately in the clinic highly specific inhibitors of the pathway recruiting vasculature-forming cells are needed, preferably ones either approved, or being developed, for clinical use. One such inhibitor is AMD3100 (“Plerixafor”), a bicyclam that is a highly specific inhibitor of CXCR4 (47). Originally developed as a therapy against HIV, AMD3100 was unexpectedly found to mobilize CD34+ hematopoietic stem cells from the bone marrow into the blood stream. The mechanism of this phenomenon is through its antagonism of the interaction of SDF-1 with CXCR4, an interaction that is responsible for the retention of hematopoietic stem cells in the bone marrow. This has led to the major indication for clinical use of the drug, namely mobilization of hematopoietic stem cells from the bone marrow into the circulation for use in bone marrow transplantation.
We tested AMD3100 given continuously for 21 days following either single dose (15 Gy) or fractionated irradiation (5 × 2 Gy) given to the whole brains of mice with intracranial transplantation of the U251 GBM. Whereas the drug had no affect on the growth of the unirradiated tumor, the effect on the irradiated tumor was quite dramatic, with all the mice achieving local control with no recurrences even after the infusion ceased (Figure 4). Similar results were found using neutralizing antibodies against CXCR4 (13). We also obtained a significant (though less dramatic) increase in tumor regrowth delay in the more radioresistant U87 tumor (13). These results therefore suggest a novel approach for the treatment of GBM: in addition to radiotherapy, the vasculogenesis pathway needs to be blocked, which can be accomplished using administration of AMD3100 after irradiation.
Figure 4. Therapeutic effect of blocking the interaction of SDF-1 with CXCR4 after whole brain irradiation.

(A) Growth curves of i.c. U251 early tumor model after fractionated irradiation (5 daily doses of 2 Gy starting on day 11 after transplantation) with or without infusion of AMD3100 for 21 days *P < 0.05. (B) BLI images after fractionated irradiation treated with or without AMD3100. (C and D) Growth curves of i.c. U251 advanced tumor model after a single dose of irradiation (15 Gy on day 22 after transplantation), treated with AMD3100 (21 day infusion; (C), *P < 0.05) or with neutralizing anti-CXCR4 Ab (D), *P < 0.05), starting immediately after irradiation. From (13) with permission.
However, SDF-1 has a second receptor, CXCR7, which has been implicated in endothelial cell migration(48), is present on tumor vasculature(48), and is potentially also able to activate vasculogenesis. We hypothesized therefore that blocking the interaction of SDF-1 with CXCR7 could also block vasculogenesis and improve tumor control by irradiation. To test this hypothesis we have used two specific inhibitors of CXCR7, CCX771 and CCX2066 and tested these with the intracranial U251 GBM in mice (CCX771) and with chemically induced brain tumors in rats (CCX2066). In the latter case pregnant rats are given a single injection of the carcinogen ethylnitrosourea (ENU) on day 17-18 of gestation. After birth the pups appear healthy for over 100 days at which time they begin to die progressively from brain tumors from day 120 after birth. The key advantages of this model are that the tumors arise spontaneously in immune competent rats and have a genetic diversity and aggressiveness similar to that seen in human brain cancers (49). In both cases we found that the CXCR7 inhibitor did not affect the tumor growth or survival of the rodents that did not receive local brain irradiation but significantly enhanced tumor growth delay and survival of the animals that received local brain irradiation (Walters et al, unpublished).
Finally, the involvement of both CXCR4 and CXCR7 suggests that SDF-1 could be the master regulator of vasculogenesis with CXCR4 being its receptor on CD11b+ monocytes and CXCR7 being its receptor on ECs. To test this we have used the highly specific inhibitor of SDF-1, NOX-A12, an L-enantiomeric RNA oligonucleotide (Spiegelmer), on brain tumor recurrences after irradiation. NOX-A12 inhibits SDF-1 with subnanomolar affinity (50) and should therefore inhibit SDF-1 mediated activation of both receptors, CXCR4 and CXCR7. We used the ENU-induced brain tumors in the rat as described above and delivered a single dose of whole brain irradiation (20 Gy) on day 115 of age and began treatment with NOX-A12 immediately following irradiation and continued every 2 days with either 5 or 20 mg/kg injected subcutaneously for either 4 or 8 weeks These doses and times were chosen as equivalent to human doses and times that, based on existing data, have been found to be safe and well tolerated in human volunteers and which are effective in inhibiting the action of SDF-1. We found that neither 20 Gy nor NOX-A12 alone prolonged the lifespan of the tumor-bearing rats. However, the addition of NOX-A12 to 20 Gy prolonged the lifespan of the rats particularly at the highest dose and for the longer treatment period of 8 weeks (median lifespans of 20 Gy alone and 20 Gy + 5 and 20 mg/kg of NOX-A12 were 196, 291 and 349 days respectively with p values for NOX-A12 treated rats <0.05 vs. radiation or controls alone) (51).
Summary.
Tumors have more than one way to form blood vessels and after radiotherapy, which largely abrogates local angiogenesis. The dominant way in which the surviving cells regrow into a recurrent tumor is through blood vessel formation by vasculogenesis, a process involving the capture by the tumor of circulating blood vessel forming cells. These are CD11b+ monocytes from the bone marrow and probably also endothelial cells or endothelial progenitor cells which come from sites other than the bone marrow. The driving force for vasculogenesis is increased tumor hypoxia after radiation caused by the loss of blood vessels in the tumor. This leads to increased levels of the hypoxic inducible factor-1 (HIF-1) transcription factor which upregulates stromal cell-derived factor -1 (SDF-1, CXCL12), the ligand for the two receptors CXCR4 and CXCR7 on monocytes and endothelial cells. We hypothesize that it is increased levels of SDF-1 in the tumor (and possibly in the plasma) that is responsible for the increased levels of the proangiogenic monocytes (and possibly endothelial cells) in tumors after irradiation and it is these increased levels that promote post-irradiation reconstitution of the tumor vasculature and tumor recurrence. Inhibitors of CXCR4, CXCR7 and SDF-1 all inhibit the post-irradiation formation of the vasculature and delay or abrogate local recurrences. A cartoon of the mechanism is shown in Figure 5. This new strategy of inhibiting vasculogenesis after irradiation could be tested clinically within the next 1-2 years as several of the inhibitors could obtain clinical approval shortly.
Figure 5.

Model showing the vasculogenesis pathway that restores the tumor vasculature after irradiation and the various inhibitors of this pathway that can improve the radiation response of the tumor
Acknowledgments
Supported by USPHS grants R01 CA128873, R01 CA149318 and Stanford University SPARK grant.
Dr Brown has received grant support from Genzyme and Noxxon Pharma AG who own products relevant to the reported studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinhold HS, De Bree C. Tumour cure rate and cell survival of a transplantable rat rhabdomyosarcoma following x-irradiation. Eur J Cancer. 1968;4:367–74. doi: 10.1016/0014-2964(68)90026-1. [DOI] [PubMed] [Google Scholar]
- 3.Baumann M, Dubois W, Suit HD. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiat Res. 1990;123:325–30. [PubMed] [Google Scholar]
- 4.Gerweck LE, Zaidi ST, Zietman A. Multivariate determinants of radiocurability. I: Prediction of single fraction tumor control doses. Int J Radiat Oncol Biol Phys. 1994;29:57–66. doi: 10.1016/0360-3016(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 5.Taghian A, Ramsay J, Allalunis-Turner J, Budach W, Gioioso D, Pardo F, et al. Intrinsic radiation sensitivity may not be the major determinant of the poor clinical outcome of glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1993;25:243–9. doi: 10.1016/0360-3016(93)90345-v. [DOI] [PubMed] [Google Scholar]
- 6.Denekamp J. Vascular endothelium as the vulnerable element in tumours. Acta radiologica Oncology. 1984;23:217–25. doi: 10.3109/02841868409136015. [DOI] [PubMed] [Google Scholar]
- 7.Budach W, Taghian A, Freeman J, Gioioso D, Suit HD. Impact of stromal sensitivity on radiation response of tumors. J Natl Cancer Inst. 1993;85:988–93. doi: 10.1093/jnci/85.12.988. [DOI] [PubMed] [Google Scholar]
- 8.Biedermann KA, Sun J-r, Giaccia AJ, Tosto LM, Brown JM. Scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proc Natl Acad Sci USA. 1991;88:1394–7. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E, et al. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res. 2005;65:4987–92. doi: 10.1158/0008-5472.CAN-04-4250. [DOI] [PubMed] [Google Scholar]
- 10.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 11.Kawamoto A, Losordo DW. Endothelial progenitor cells for cardiovascular regeneration. Trends Cardiovasc Med. 2008;18:33–7. doi: 10.1016/j.tcm.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, et al. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–77. doi: 10.1182/blood-2003-11-3952. [DOI] [PubMed] [Google Scholar]
- 15.Purhonen S, Palm J, Rossi D, Kaskenpaa N, Rajantie I, Yla-Herttuala S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105:6620–5. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679–85. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hillebrands JL, Klatter FA, van Dijk WD, Rozing J. Bone marrow does not contribute substantially to endothelial-cell replacement in transplant arteriosclerosis. Nat Med. 2002;8:194–5. doi: 10.1038/nm0302-194. [DOI] [PubMed] [Google Scholar]
- 18.Aicher A, Rentsch M, Sasaki K, Ellwart JW, Fandrich F, Siebert R, et al. Nonbone marrow-derived circulating progenitor cells contribute to postnatal neovascularization following tissue ischemia. Circ Res. 2007;100:581–9. doi: 10.1161/01.RES.0000259562.63718.35. [DOI] [PubMed] [Google Scholar]
- 19.Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107:1164–9. doi: 10.1161/01.cir.0000058702.69484.a0. [DOI] [PubMed] [Google Scholar]
- 20.Rohde E, Malischnik C, Thaler D, Maierhofer T, Linkesch W, Lanzer G, et al. Blood monocytes mimic endothelial progenitor cells. Stem Cells. 2006;24:357–67. doi: 10.1634/stemcells.2005-0072. [DOI] [PubMed] [Google Scholar]
- 21.Awad O, Dedkov EI, Jiao C, Bloomer S, Tomanek RJ, Schatteman GC. Differential healing activities of CD34+ and CD14+ endothelial cell progenitors. Arterioscler Thromb Vasc Biol. 2006;26:758–64. doi: 10.1161/01.ATV.0000203513.29227.6f. [DOI] [PubMed] [Google Scholar]
- 22.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen FH, Chiang CS, Wang CC, Tsai CS, Jung SM, Lee CC, et al. Radiotherapy decreases vascular density and causes hypoxia with macrophage aggregation in TRAMP-C1 prostate tumors. Clin Cancer Res. 2009;15:1721–9. doi: 10.1158/1078-0432.CCR-08-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011 doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b(+)Gr1(+) myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 27.Prionas SD, Kowalski J, Fajardo LF, Kaplan I, Kwan HH, Allison AC. Effects of X irradiation on angiogenesis. Radiat Res. 1990;124:43–9. [PubMed] [Google Scholar]
- 28.Imaizumi N, Monnier Y, Hegi M, Mirimanoff RO, Ruegg C. Radiotherapy suppresses angiogenesis in mice through TGF-betaRI/ALK5-dependent inhibition of endothelial cell sprouting. PLoS One. 2010;5:e11084. doi: 10.1371/journal.pone.0011084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Udagawa T, Birsner AE, Wood M, D’Amato RJ. Chronic suppression of angiogenesis following radiation exposure is independent of hematopoietic reconstitution. Cancer Res. 2007;67:2040–5. doi: 10.1158/0008-5472.CAN-06-2877. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–9. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 31.Park HJ, Griffin RJ, Hui S, Levitt SH, Song CW. Radiation-induced vascular damage in tumors: implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS) Radiat Res. 2012;177:311–27. doi: 10.1667/rr2773.1. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 33.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–5. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 34.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–7. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 35.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 36.Aghi M, Chiocca EA. Contribution of bone marrow-derived cells to blood vessels in ischemic tissues and tumors. Mol Ther. 2005;12:994–1005. doi: 10.1016/j.ymthe.2005.07.693. [DOI] [PubMed] [Google Scholar]
- 37.De Palma M, Naldini L. Role of haematopoietic cells and endothelial progenitors in tumour angiogenesis. Biochim Biophys Acta. 2006;1766:159–66. doi: 10.1016/j.bbcan.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 39.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 40.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams KJ, Telfer BA, Xenaki D, Sheridan MR, Desbaillets I, Peters HJ, et al. Enhanced response to radiotherapy in tumours deficient in the function of hypoxia-inducible factor-1. Radiother Oncol. 2005;75:89–98. doi: 10.1016/j.radonc.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–41. doi: 10.1016/s1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Haider H, Ahmad N, Zhang D, Ashraf M. Evidence for ischemia induced host-derived bone marrow cell mobilization into cardiac allografts. J Mol Cell Cardiol. 2006;41:478–87. doi: 10.1016/j.yjmcc.2006.06.074. [DOI] [PubMed] [Google Scholar]
- 44.Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–67. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aghi M, Cohen KS, Klein RJ, Scadden DT, Chiocca EA. Tumor stromal-derived factor-1 recruits vascular progenitors to mitotic neovasculature, where microenvironment influences their differentiated phenotypes. Cancer Res. 2006;66:9054–64. doi: 10.1158/0008-5472.CAN-05-3759. [DOI] [PubMed] [Google Scholar]
- 46.Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Yung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 47.De Clercq E. The AMD3100 story: the path to the discovery of a stem cell mobilizer (Mozobil) Biochem Pharmacol. 2009;77:1655–64. doi: 10.1016/j.bcp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 48.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–40. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jang T, Savarese T, Low HP, Kim S, Vogel H, Lapointe D, et al. Osteopontin expression in intratumoral astrocytes marks tumor progression in gliomas induced by prenatal exposure to N-ethyl-N-nitrosourea. Am J Pathol. 2006;168:1676–85. doi: 10.2353/ajpath.2006.050400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sayyed SG, Hagele H, Kukarni OP, Endlich K, Segerer S, Eulberg D, et al. Podocytes produce homeostatic chemokine stromal cell-derived factor-1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia. 2009;52:2445–54. doi: 10.1007/s00125-009-1493-6. [DOI] [PubMed] [Google Scholar]
- 51.Liu SC, Alomran R, Quan F, Merchant M, Zollner S, Kruschinski A, et al. Inhibition of SDF-1 (CXCL12) using the Spiegelmer NOX-A12 markedly delays the recurrence of ENU-induced rats brain tumors following irradiation. AACR 2012 Annual Meeting. 2012 Abstract #4382. [Google Scholar]