

Abstract
Next-generation sequencing is making sequence-based molecular pathology and personalized oncology viable. We selected an individual initially diagnosed with conventional but aggressive prostate adenocarcinoma and sequenced the genome and transcriptome from primary and metastatic tissues collected prior to hormone therapy. The histology-pathology and copy number profiles were remarkably homogeneous, yet it was possible to propose the quadrant of the prostate tumour that likely seeded the metastatic diaspora. Despite a homogeneous cell type, our transcriptome analysis revealed signatures of both luminal and neuroendocrine cell types. Remarkably, the repertoire of expressed but apparently private gene fusions, including C15orf21:MYC, recapitulated this biology. We hypothesize that the amplification and over-expression of the stem cell gene MSI2 may have contributed to the stable hybrid cellular identity. This hybrid luminal-neuroendocrine tumour appears to represent a novel and highly aggressive case of prostate cancer with unique biological features and, conceivably, a propensity for rapid progression to castrate-resistance. Overall, this work highlights the importance of integrated analyses of genome, exome and transcriptome sequences for basic tumour biology, sequence-based molecular pathology and personalized oncology.
Keywords: RNA sequencing, DNA sequencing, prostate cancer, fusion genes, neuroendocrine, personalized medicine, cancer genetics
Introduction
The application of next-generation sequencing (NGS) to cancer promises to usher in the era of personalized oncology, where a patient's tumour is managed based on the unique constellation of ‘omic’ properties of the tumour and its host [1–3]. The comprehensive molecular characterization afforded by NGS will be particularly relevant in prostate cancer (PCa), where clinical and biological heterogeneity make it difficult to accurately predict tumour aggressiveness.
Normal prostate epithelium consists of two histologically distinct layers of basal cells and secretory luminal cells, along with sparsely scattered neuroendocrine cells. Evidence suggests that all three cell types originate from progenitor cells within the prostate, although the nature and location of the stem cell reservoir remains controversial [4–7]. Adenocarcinoma of the prostate is generally believed to arise from the luminal epithelial cell layer, although a basal cell-of-origin model does exist [8]. However, there is increasing interest in neuroendocrine (NE) differentiation: the concurrent presence of NE-like cells in adenocarcinoma foci correlates with progression, poor prognosis and castrate-resistance [9–11].
NE-like cells are distinct from normal NE cells, which are rare, mostly AR-negative, exhibit an anti-apoptotic phenotype and are involved in mediating epithelial cell growth and differentiation [12]. Although their origin is not fully elucidated, a significant body of evidence suggests that under certain conditions (eg androgen depletion) adenocarcinoma cells undergo a trans-differentiation process to acquire NE-like characteristics [9,10,13,14]. Since modern therapeutic strategies select for the lethal castrate-resistant phenotype, understanding the role of NE-like cells is critical for treating advanced stage disease that has adapted to androgen ablation.
We aimed to test the hypothesis that high dimensional data derived from NGS will afford personalized molecular pathology, the antecedent to personalized medicine. To do this, we selected an extreme but nonetheless broadly conventional prostate adenocarcinoma and performed a comprehensive analysis of the genome and transcriptome in primary and metastatic tissue collected prior to hormone therapy. The genome copy number architecture was extremely homogeneous, both within the primary tumour and between primary and metastatic tumours, yet the underlying fusion gene and expression profile revealed a hybrid phenotype of luminal and neuroendocrine cells. This hybrid adenocarcinoma–NE tumour may represent a novel molecular case of PCa, revealed by NGS.
Materials and methods
Clinical details and tissue processing
The proband was diagnosed with high-risk prostate cancer in May 2009 at the age of 45 years. Initial PSA levels were 6.3 μg/l, and all six core biopsies of the prostate revealed adenocarcinoma with a Gleason score of 10 (see Supporting information, Table S1). Staging investigations with a bone scan and CT of the abdomen and pelvis were negative for metastases and alkaline phosphatase was normal. He underwent a radical prostatectomy in June 2009 and pathology demonstrated seminal vesicle involvement, extracapsular spread and lymph node involvement with Gleason score 9 adenocarcinoma, although the vast majority of the specimen was Gleason grade 5 disease. In October 2009, the patient began to experience bone pain and a bone scan confirmed bone metastases, despite low PSA levels of 0.35 μg/l. He began androgen withdrawal therapy with bicalutamide and a GnRH analogue and within 1 month pain symptoms resolved and PSA fell to undetectable levels. Prostate tissue was collected from the patient when he underwent radical prostatectomy, according to the current Vancouver General Hospital pathology protocol, and processed for histological analysis (for details, see Supporting information, Supplementary materials and methods). The study numbers for consent forms and tissue banking were: UBC Ethics Board No. H09–01 628; VCHRI Nos V09-0320 and V07-0058.
aCGH analysis
DNA and RNA were isolated and purified and 0.5 μg of each genomic DNA was processed for aCGH (for details, see Supporting information, Supplementary materials and methods). CGH processed signal was uploaded into Biodiscovery Nexus CGH software v 5.1, where quality was assessed and data was visualized and analysed. To establish relationships between the primary tumour regions and the lymph node metastasis, we compared aCGH data for each pair of samples. For each probe we subtracted the signal of one sample from the other before importing all values to Nexus and generating segment calls. A histogram of segment value, weighted by segment length, allowed definition of clear troughs (for examples, see Supporting information, Figure S3). These values were used as cut-offs in Nexus to define a difference of CN between two samples. The total amount of ‘aberration’ was divided by the number of bases in hg18 to generate the difference percentages in Figure 1. The distance matrix and phylogenetic tree in Figure S1 (see Supporting information) was assembled using T-Rex software [15] and the neighbour-joining method. aCGH data are publicly available at NCBI GEO under Accession No. GSE34649.
Figure 1.
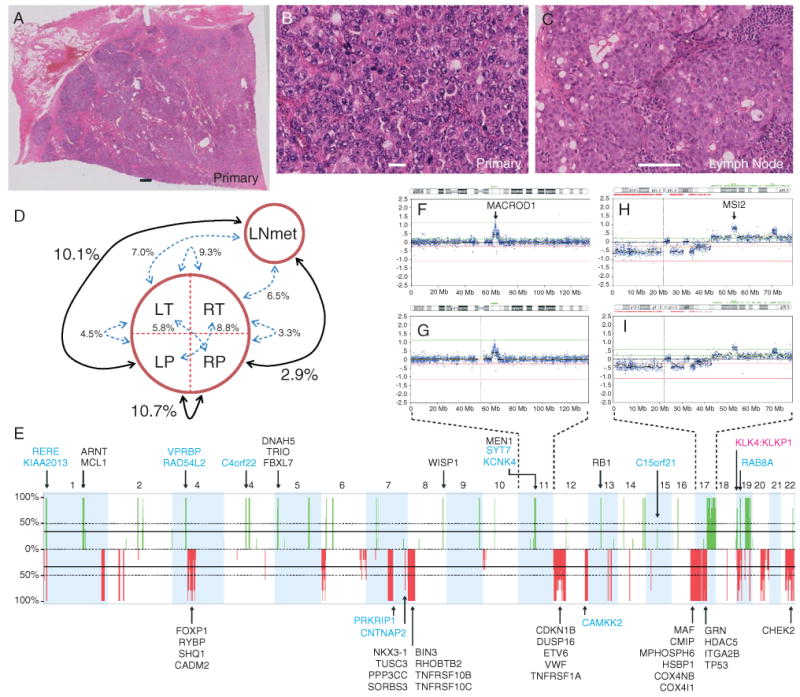
Histopathology and copy number analysis. Haematoxylin and eosin stains showing uniform adenocarcinoma with a Gleason score of 10 in patient 963's radical prostatectomy specimen (A, ×0.5; B, ×40) and a lymph node metastasis (C, ×20). No normal prostate structure can be discerned in (A, B). Lymphocytes can be seen in (C). Scale bars = 1 mm (A); 20 μm (B); 100 μm (C). (D) Differences between the samples, i.e. RP differs in signal intensity from LNmet at 2.9% of aCGH probe loci. The most and least similar pairs are emphasized. (E) Frequency plot showing the copy number (CN) aberrations in the primary tumour quadrants and the LNmet (green, gain; red, loss). A frequency of 100% indicates a CN aberration detected in all five samples; the majority of CN aberrations are detected in all samples. CN aberrant genes previously associated with PCa are annotated. Blue, fusion genes (the genomic breakpoints of 12 genes involved in fusion events coincided with aCGH segment breaks); pink, readthrough events. Note: Chromosomes X and Y demonstrated no aberrations and are not shown. (F, G) Chr11 CN profile in LT (F) and RP (G), indicating the marginal differences. (H, I) Chr17 CN profile in LP (H) and LNmet (I).
Sequencing
Sequencing of the samples was performed at BCCA Genome Sciences Centre (Vancouver, BC, Canada) using the the Illumina Genome Analyser II according to established protocols, as described in [16]. Sequence read details are provided in Table S2 (see Supporting information). Approximately 100 million reads were obtained and mapped to the NCBI 36.1 human genome reference sequence using MAQ 0.7.1 [17] and the following parameters: –n 1 –N –e 100 –a 700. The total number of sequenced bases in a given genomic window and the average sequencing depth across the window (10 or 30 kb) was then calculated. CN was approximated by the ratio of average sequencing depth in a given window to the average sequencing depth across the genome. This value was transformed into log2 space and normalized to corresponding values in buccal epithelial cell DNA from the same patient (DNA-Seq performed in parallel) in order to minimize the sequencing biases. CN profiles were visualized using Nexus. To compare sequencing CN data to aCGH data, segment calls from each technology were exported and the difference between each call plotted. The raw sequence data are available in FASTQ format at: http://www.lagapc.ca/FTP_HybridPCa.html
Identification of read-throughs and fusion genes
Read-throughs were identified from RNA-Seq using deFuse with default parameters, the hg18 reference genome, and ensembl 54 genes [18]. Gene fusions were identified from matched DNA-Seq and RNA-Seq, using Comrad with default parameters, the hg18 reference genome and ensemble 54 genes [19].
Gene expression
Sequence reads for each transcriptome library were aligned to the ALEXA reference sequence database, using MAQ 0.7.1 [17,20] allowing no more than two mismatches. The expression levels using RNA-Seq reads were quantified as previously described [21] and normalized using quantile normalization. Gene expression was estimated using the average of all exons and junctions expression from that gene.
Experimental validation
To validate fusion transcripts, we amplified the site of fusion by RT–PCR from cDNA using standard techniques (for primers, see Supporting information, Table S3). We amplified predicted genomic breakpoints underlying fusion transcripts by PCR from genomic DNA (see Supporting information, Table S2). All amplification products were sequenced using ABI PRISM 310 Genetic Analyser with standard techniques to confirm identity.
Results
Histopathology and copy number analysis
Patient 963 was a 45 year-old man diagnosed with a clinically ‘localized’ prostate cancer and low serum PSA that rapidly progressed to a metastatic disease (see Supporting information, Table S1). Histopathology after a radical prostatectomy and prior to hormone therapy demonstrated adenocarcinoma with a Gleason score of 10 (5 + 5), and lymph node involvement (left and right) with a Gleason score of 9 (5 + 4). The primary tumour was over 5 cm in size and visually homogeneous, with no preservation of the normal prostate tissue structure. In both the primary tumour and lymph node metastases there was a uniform cell type, morphologically resembling luminal cells (Figure 1A–C).
The unusually large primary tumour presented a unique opportunity to sample multiple regions and assess copy number (CN) heterogeneity. We generated CN profiles of four regions of the primary tumour (left and right posterior or transitional: LP, RP, LT, RT, respectively) as well as a lymph node metastasis (LNmet), and identified a remarkably high degree of homogeneity between all five samples (Figure 1E–I; see also Supporting information, Figure S1, and list of all CN changes in Table S4). Nevertheless, we used the few regions which differed in copy number states (see Supporting information, Table S5) to establish relationships between the primary tumour regions and LNmet. LNmet was most similar to RP, differing in probe signal levels at only 2.9% of the genome (Figure 1D; see also Supporting information, Figure S2). To put this in context, LP differed from LNmet at 10.7% and from RP at 10.1% of the genome. Furthermore, ranking the samples by the percentage of their genome mapped as CN aberrant places LNmet (6.58%) and RP (6.96%) as the least aberrant, with LP (8.76%) as the most. This may suggest that the primary tumour initiated in the RP quadrant and the original LNmet clone arose from RP.
CN aberrations were concordant between primary tumour regions and the LNmet and included loss at 8p (lost in ∼30–50% of PCa and associated with advanced stage), 16q23.1, RYBP (3p13), MAF (16q23.1), ETV6 (12p13.2), TP53 (17p13.1) gain of MEN1 (11q13.1) (Figure 1E), all consistent with previous PCa studies [22]. We observed focal CN gain at WISP1, a downstream regulator in the Wnt signalling pathway. Two regions, on chromosome 11 and chromosome 17, demonstrated the highest levels of CN gain and contained the genes MACROD1 and MSI2, respectively (Figure 1F, H). MACROD1 binds to the androgen receptor (AR) and amplifies the transactivation function of AR in response to androgen [23], whilst MSI2 is an RNA-binding protein required to maintain stem cell identity [24].
The primary tumour region with the most CN aberrations (LP) and the LNmet sample were selected for genome and transcriptome sequencing. We used DNA-Seq reads to produce CN profiles, which correlated strongly with the aCGH-derived profiles, demonstrating the reliability of our sequencing experiments (see Supporting information, Figure S3).
Identification of novel fusion genes
Chromosomal rearrangements in cancerous cells can lead to the fusion of two genes, resulting in the production of a fusion protein which may have altered activity, with implications for cancer initiation, progression and treatment [25,26]. We identified 15 novel chimeric transcripts and associated genomic breakpoints (Figure 2; see also Supporting information, Table S3). Remarkably, the fusion gene dataset includes both genes expressed in normal luminal cells (purple genes in Figure 2) and those expressed in normal NE or NE-like cells (orange). RT-PCR revealed that these fusion genes were absent from five PCa cell lines and five patient prostate tumours sequenced in parallel, suggesting that the fusion events may be private to the tumours of patient 963. To confirm, we examined fusion gene predictions from a cohort of 102 prostate tissues and cells lines and found no evidence to suggest that any of the fusion genes detected in patient 963's tumours were recurrent [27]. Overall there was strong concordance between the expression levels of the fusion genes between LP and LNmet (Figure 2), and furthermore PCR and Sanger sequencing revealed that the underlying genome breakpoints for every fusion gene were present in all four primary tumour quadrants and in both the left and right lymph node metastases, highlighting the genomic homogeneity (see Supporting information, Figure S4). The genomic breakpoints of 12 genes involved in fusion events coincided with aCGH segment breaks, indicating a pronounced change in CN (annotated in Figure 1F). The complex nature of genomic breakpoints underlying these fusion events is explored in a separate methods manuscript (in preparation).
Figure 2.
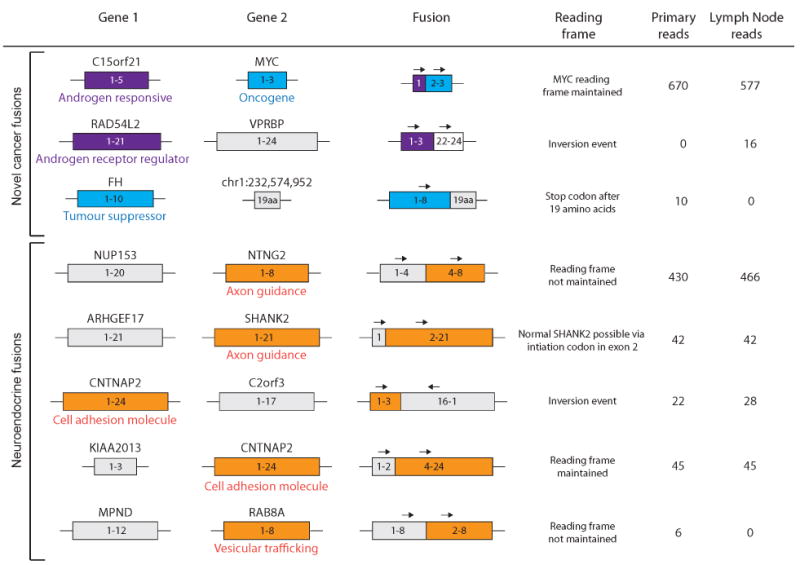
Fusion genes validated in the primary tumour and lymph node metastasis. The top three fusions involve either an androgen-related gene, a tumour suppressor or an oncogene. The bottom five fusions involve genes with a neuroendocrine function. Note that despite absence of RNA-Seq reads (ie expression) for some fusion genes, the underlying genomic breakpoints for each fusion were detected in both primary and metastatic tumours (see Supporting information, Figure S4).
We also identified previously reported fusion transcripts unrelated to chromosomal rearrangements (read-throughs) including three involving 5′ androgen-responsive genes (SLC45A3:ELK4; TPD52:MRPS28 and KLK4:KLKP1) [19,26,28,29].
Gene expression analysis
Our RNA-Seq data revealed high expression of standard histological markers of NE cells, including CHGA, CHGB and SYP. To explore further, we compiled a panel of genes which were differentially expressed in patient 963's tumours compared to adenocarcinoma cell lines C4-2 and LNCaP and benign prostate tissue (Figure 3A). This panel included genes involved in synapse vesicle structure and function, ligand-gated ion channel components, axon guidance and cell adhesion molecules, as well as RNA-binding proteins and neuronal-specific transcription factors. Many of these are expressed in normal NE cells or NE-like cells in PCa [9,30,31]. However, both LP and LNmet also expressed a panel of characteristic markers of secretory luminal prostate cells, including KRT8, KRT18, NKX3-1, AR and KLK3. We used single and dual immunohistochemical staining to confirm uniform co-expression of AR and CHGA in 100% of tumour cells (Figure 3C–G, I, J; see also Supporting information, Figure S5). MACROD1 and MSI2, located in regions of high copy number gain (Figure 1F, H), were expressed at high levels.
Figure 3.
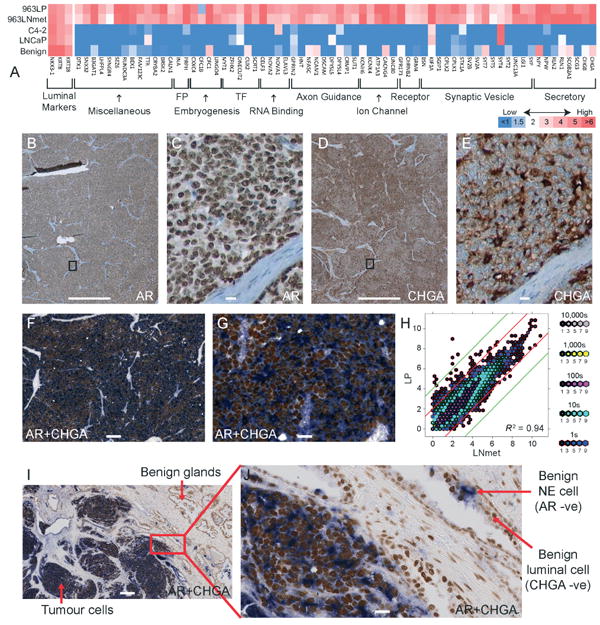
Analysis of gene expression levels. (A) Heat map demonstrating expression of genes with a neural/endocrine function in the primary tumour (LP) and the lymph node metastatic tumour (LNmet) of patient 963 compared to adenocarcinoma cell lines and benign prostate. FP, filament protein; TF, transcription factor. (B–E) Antibody stains showing strong expression of AR (B, ×2; C, ×40) and CHGA (D, ×2; E, ×40) in the primary tumour. Bands of stromal cells are unstained. (F, G, I, J) Dual antibody stains of AR and CHGA (F, ×8; G, ×40; I, ×10; J, ×40) confirming co-expression in 100% of tumour cells. Note that in (I, J) benign prostate glands are visible, demonstrating that normal luminal cells are AR-positive only, while normal NE cells are CHGA-positive only. Scale bars = 1 mm (B, D); 200 μm (I); 100 μm (C, E, F); 20 μm (G, J). (H) Hexbin plot illustrating correlation of gene expression levels between LP and LNmet (log2 scale). Red and green lines indicate a four- and 16-fold expression difference, respectively, between LP and LNmet.
Overall we observed remarkable concordance of gene expression levels between LP and LNmet (R2 = 0.94; Figure 3H), consistent with the genomic homogeneity described above. To our surprise, only 110 genes were differentially expressed in LP compared to LNmet (outside red line in Figure 3H) and many of these are markers of stromal and basal cells [32,33], likely reflecting the small amount of normal prostate tissue in the LP sample. Given the large size of the lymph node metastasis (>2 cm), it is possible that gene expression was no longer significantly influenced by the lymph node microenvironment.
Discussion
Prostate cancer is a complex, clinically and biologically heterogeneous disease that is often multifocal [34]. The aggressive and histologically homogeneous tumour that at initial diagnosis had fully colonized the normal prostate space and that of multiple pelvic lymph nodes therefore represented a unique opportunity for study. Remarkably, the copy number profiles and gene expression levels were highly concordant between all tumour foci sampled. Furthermore, analysis revealed that even the chromosomal rearrangements underlying the novel fusion genes were present in all tumour foci, including the metastases obtained from both left and right pelvic lymph nodes. Therefore, we hypothesize that the spectrum of genomic alterations revealed here occurred in an early clone that expanded to dominate the prostate and seed the metastatic diaspora.
Integrated analyses of RNA-Seq and DNA-Seq data revealed 15 novel fusion genes with underlying genomic rearrangements. The genes involved in fusion events fell into two categories: androgen-responsive/AR-regulatory genes normally expressed in luminal cells (eg C15orf21, RAD54L2) or those normally expressed in NE cells. C15orf21 (also known as HMGN2P46) is a prostate-specific androgen-responsive gene, previously associated with ETS fusions in PCa [35], while RAD54L2 is predicted to modulate AR-dependent transactivation, interacting with AR via its N-terminus (part of which is present in the gene fusion). It is also worth noting that three androgen-responsive readthroughs highly expressed in other androgen-sensitive prostate adenocarcinomas were detected. However, the second category of genes involved in fusion events, those normally expressed in NE cells (eg NTNG2, SHANK2, CNTNAP2 and RAB8A), has not been previously associated with adenocarcinoma. NTNG2 is a membrane-bound axon guidance molecule involved in synapse formation and maintenance [36]. SHANK2, a member of a family of scaffold proteins that localize to postsynaptic sites, is also implicated in the axon-guidance signalling pathway and is normally highly expressed in the brain [37]. RAB8A has been implicated in vesicular trafficking and neurotransmitter release at the synapse [38], while CNTNAP2 is highly expressed in the nervous system and is part of the neurexin family functioning as cell adhesion molecules [39]. Even the seven fusion transcripts involving intronic and intergenic regions (see Supporting information, Table S2) showed enrichment for NE genes, including KCNK4 (potassium channel) and SYT7 (involved in synapse function). Therefore, the fusion gene profile indicates a hybrid phenotype of both luminal androgen-responsive secretory cells and NE cells.
Gene expression analysis provides further support for this hybrid phenotype. A panel of characteristic markers of androgen-responsive secretory cells, including AR and KLK3, were highly expressed and IHC staining demonstrated AR protein expression in the nuclei of all tumour cells. However, a panel of over 65 genes known to be expressed in NE or NE-like cells, including CHGA, CHGB and SYP as well as axon-guidance and cell-adhesion molecules, were also expressed at high levels. Neuroendocrine differentiation (NED), where NE-like cells (expressing CHGA, CHGB and SYP) and adenocarcinoma cells are concurrent, is thought to arise secondary to adenocarcinoma via a transdifferentiation process [9,10,13]. However, the tumours in patient 963 appear distinct from typical NED, since they were comprised of a single cell type at initial diagnosis before hormone therapy. To our knowledge, this molecular phenotype has not been previously reported and, since conventional adenocarcinoma is not routinely stained for NE markers, it is possible that tumours with this hybrid molecular phenotype are currently under-reported. Alternatively, this phenotype may represent a rare or even nonrecurrent phenomenon. Patient 963 responded well to androgen withdrawal—further confirmation that his tumours exhibited androgen dependence. However, one may hypothesize that his metastatic bone tumours will rapidly develop castrate resistance, due to the NE phenotype.
The cell type specificity of some chromosomal translocations is well documented even in PCa, where recent studies have indicated that AR signalling induces gene proximity of TMPRSS2 and ERG, thereby promoting the TMPRSS2:ERG fusion event detected in approximately 50% of PCa [40,41]. Furthermore, a growing body of evidence suggests that binding of transcription machinery predisposes genome regions to breakage and translocations [42,43]. Therefore, the dual nature of the fusion genes and underlying genomic rearrangements detected here suggest that tumourigenesis occurred in a cell co-expressing genes associated with both luminal and NE cells. Studies in mice have indicated that aggressive prostate tumours can arise from bipotent prostatic progenitor cells, and co-express luminal and NE markers at early stages of neoplastic development [44,45]. Similarly, therefore, it is possible that the aggressive clone which emerged to dominate the prostate in patient 963 arose from the epithelial stem cell population proposed to be the progenitors of luminal and NE cells, but remained frozen in a relatively undifferentiated state [4–7]. Interestingly, the MSI2 gene on chromosome 17 is the most highly amplified locus in the patient's tumour and it is highly expressed. The MSI2 gene is required to maintain stem cell identity [24]. In chronic myelogeneous leukaemia, high levels of MSI2 result in loss of the capacity to differentiate, leading to arrested development (blast crisis phase), an aggressive phenotype and poor prognosis [46,47]. MSI2 may therefore contribute to the seemingly frozen state of the tumour cells in patient 963.
The highly expressed C15orf21:MYC fusion gene may also contribute to the aggressive nature of the tumour. Although this is the first report of a MYC fusion gene in PCa, MYC is up-regulated in the majority of PCa, even in the absence of 8q amplification [48]. MYC is also over-expressed in prostatic intraepithelial neoplasia, suggesting a role in cancer initiation [48]. Furthermore, prostate tumours induced by co-expressing MYC with the oncogenic PIM1 kinase show evidence of NED [11].
Other genes involved in fusion events have links to cancer. Fumarate hydratase (FH) is a tumour suppressor gene, germline mutations in which cause hereditary renal cancer [49]. ARHGEF17 is a member of the Rho guanine nucleotide exchange factor (GEF) family, which have a crucial role in activating small GTPases and regulate various cellular functions, including the Rho signalling pathway, which is important in breast cancer [50,51]. Mutations in other GEF family members have been linked to metastatic PCa of the lung (ARHGEF12 [52]) and melanoma (ARHGEF16, -19 and -14 [50]). Both SHANK2 (melanoma) and CNT-NAP2 (glioma) have been previously identified in gene fusion events [39,53]. In fact, CNTNAP2 is a potential tumour suppressor, as over-expression in glioma cell lines resulted in decreased proliferation rates due to increased apoptosis [39].
While the tumours of patient 963 share specific properties with other prostate tumours (eg over-expression of MYC), we could not find evidence that the hybrid phenotype was recurrent in other tumours. However, patient 963's young age and high Gleason score make it difficult to find analogous datasets. For example, in a publicly available dataset of 230 prostate cancer samples [22], only 22 patients were <50 years old, and only 1/22 had a Gleason score >7. Nevertheless, examination of this dataset revealed that MACROD1 was over-expressed or amplified in 17/230 tumours, while MSI2 was over-expressed or amplified in 3/230. There was no correlation with survival or tumour grade in either example. Perhaps of importance is that MSI1 (highly related to MSI2) was over-expressed or amplified in 16/230 tumours and associated with poor clinical outcome (p = 0019).
This study represents a comprehensive characterization of the genome and transcriptome of a patient's prostate tumours and illustrates the potential of sequence-based pathology. Analysis of the transcriptome and genome of the tumour revealed a hybrid tumour expressing luminal and neuroendocrine gene signatures. Remarkably, the repertoire of expressed but private gene fusions recapitulated this biology, while the amplification and over-expression of the MSI2 gene was hypothesized to contribute to the stable hybrid state. Although the primary tumour and lymph node metastasis were highly homogeneous, it was possible to hypothesize on the quadrant of the prostate tumour that likely seeded the metastatic diaspora. In conclusion, our highly integrated ‘omic’ approach appears to have molecularly defined a novel and highly aggressive hybrid case of PCa and highlights the potential NGS technology has to transform oncology with sequence-based molecular pathology and personalized diagnosis.
Supplementary Material
Acknowledgments
This study was supported by the Canadian Institutes of Health Research (to MEG), the Centres of Excellence for Commercialization and Research (to MEG), PNW Prostate Cancer SPORE, Prostate Cancer Canada, Genome BC (to CCC), the Prostate Cancer Foundation (to CCC and AVL) and the Ontario Institute for Cancer Research (through funding provided by the Government of Ontario to RGB, PCB and CCC via the CPC-GENE prostate cancer genome-network).
Footnotes
Illumina sequence data are available from the Vancouver Prostate Centre web-site at: http://www.lagapc.ca/FTP_HybridPCa.html; aCGH data are available at NCBI GEO, Accession No. GSE34649.
No conflicts of interest were declared.
Author contributions: FFCCC funded and directed the project; CCC, CW and AWW conceived experiments and analysed data; AVL, SVV, AM, SA and FM conducted bioinformatics analyses; CW, BJM, RS, AH and SB carried out experiments; LF, AH-C, ECJ and EL identified tumours and performed histopathology and analysis; RHB coordinated statistical analyses; YSB, SJMJ, MH and MAM coordinated the next-generation sequencing and assisted in bioinformatics analyses; FHa, FHo, IM, CAM and AMC assisted in bioinformatics analyses; SCS and MEG co-directed the project and reviewed the manuscript; PCB and RGB aided data analyses and reviewed the manuscript; and AWW, CW and CCC wrote the manuscript.
Supporting information on the internet
The following supporting information may be found in the online version of this article:
Supplementary materials and methods
Figure S1. Homogeneity of the copy number profiles within the primary tumour and between the primary tumour and a lymph node metastasis.
Figure S2. Copy number differences between tumour samples from patient 963.
Figure S3. Correlation of sequencing and aCGH data.
Figure S4. Detection of fusion gene breakpoints demonstrates homogeneity.
Figure S5. Antibody stains showing expression of AR and CHGA in the primary tumour and lymph node metastasis of patient 963.
Table S1. Case 963 clinical characteristics.
Table S2. Summary of sequence data.
Table S3. Primers used for fusion gene validation.
Table S4. Copy number changes in the five tumour foci subjected to aCGH.
Table S5. Copy number differences between the five tumour foci subjected to aCGH.
References
- 1.Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- 2.Link DC, Schuettpelz LG, Shen D, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. J Am Med Assoc. 2011;305:1568–1576. doi: 10.1001/jama.2011.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch JS, Westervelt P, Ding L, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. J Am Med Assoc. 2011;305:1577–1584. doi: 10.1001/jama.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hudson DL. Epithelial stem cells in human prostate growth and disease. Prostate Cancer Prostat Dis. 2004;7:188–194. doi: 10.1038/sj.pcan.4500745. [DOI] [PubMed] [Google Scholar]
- 5.Long RM, Morrissey C, Fitzpatrick JM, et al. Prostate epithelial cell differentiation and its relevance to the understanding of prostate cancer therapies. Clin Sci (Lond) 2005;108:1–11. doi: 10.1042/CS20040241. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZA, Shen MM. Revisiting the concept of cancer stem cells in prostate cancer. Oncogene. 2011;30:1261–1271. doi: 10.1038/onc.2010.530. [DOI] [PubMed] [Google Scholar]
- 7.Leong KG, Wang BE, Johnson L, et al. Generation of a prostate from a single adult stem cell. Nature. 2008;456:804–808. doi: 10.1038/nature07427. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein AS, Huang J, Guo C, et al. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568–571. doi: 10.1126/science.1189992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cindolo L, Cantile M, Vacherot F, et al. Neuroendocrine differentiation in prostate cancer: from lab to bedside. Urol Int. 2007;79:287–296. doi: 10.1159/000109711. [DOI] [PubMed] [Google Scholar]
- 10.Nelson EC, Cambio AJ, Yang JC, et al. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostat Dis. 2007;10:6–14. doi: 10.1038/sj.pcan.4500922. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Kim J, Roh M, et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene. 2010;29:2477–2487. doi: 10.1038/onc.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer. 2007;14:531–547. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 13.Cox ME, Deeble PD, Lakhani S, et al. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59:3821–3830. [PubMed] [Google Scholar]
- 14.Williamson SR, Zhang S, Yao JL, et al. ERG–TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol. 2011;24:1120–1127. doi: 10.1038/modpathol.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makarenkov V. T-REX: reconstructing and visualizing phylogenetic trees and reticulation networks. Bioinformatics. 2001;17:664–668. doi: 10.1093/bioinformatics/17.7.664. [DOI] [PubMed] [Google Scholar]
- 16.Shah SP, Morin RD, Khattra J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McPherson A, Hormozdiari F, Zayed A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McPherson A, Wu C, Hajirasouliha I, et al. Comrad: a novel algorithmic framework for the integrated analysis of RNA-Seq and WGSS data. Bioinformatics. 2011;27:1481–1488. doi: 10.1093/bioinformatics/btr184. [DOI] [PubMed] [Google Scholar]
- 20.Griffith M, Griffith OL, Mwenifumbo J, et al. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 21.Lapuk A, Marr H, Jakkula L, et al. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res. 2010;8:961–974. doi: 10.1158/1541-7786.MCR-09-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Zhao YL, Wu ZQ, et al. The single-macro domain protein LRP16 is an essential cofactor of androgen receptor. Endocr Relat Cancer. 2009;16:139–153. doi: 10.1677/ERC-08-0150. [DOI] [PubMed] [Google Scholar]
- 24.Siddall NA, McLaughlin EA, Marriner NL, et al. The RNA-binding protein Musashi is required intrinsically to maintain stem cell identity. Proc Natl Acad Sci USA. 2006;103:8402–8407. doi: 10.1073/pnas.0600906103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubin CM, Carrino JJ, Dickler MN, et al. Heterogeneity of genomic fusion of BCR and ABL in Philadelphia chromosome-positive acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1988;85:2795–2799. doi: 10.1073/pnas.85.8.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prensner JR, Iyer MK, Balbin OA, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nacu S, Yuan W, Kan Z, et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genomics. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kannan K, Wang L, Wang J, et al. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci USA. 2011;108:9172–9177. doi: 10.1073/pnas.1100489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirano D, Okada Y, Minei S, et al. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45:586–592. doi: 10.1016/j.eururo.2003.11.032. discussion, 592. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Ippolito JE, Garabedian EM, et al. Molecular characterization of a metastatic neuroendocrine cell cancer arising in the prostates of transgenic mice. J Biol Chem. 2002;277:44462–44474. doi: 10.1074/jbc.M205784200. [DOI] [PubMed] [Google Scholar]
- 32.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 33.Gregg JL, Brown KE, Mintz EM, et al. Analysis of gene expression in prostate cancer epithelial and interstitial stromal cells using laser capture microdissection. BMC Cancer. 2010;10:165. doi: 10.1186/1471-2407-10-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andreoiu M, Cheng L. Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum Pathol. 2010;41:781–793. doi: 10.1016/j.humpath.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 36.Berg A, Zelano J, Cullheim S. Netrin G-2 ligand mRNA is down-regulated in spinal motoneurons after sciatic nerve lesion. NeuroReport. 2010;21:782–785. doi: 10.1097/WNR.0b013e32833cadd8. [DOI] [PubMed] [Google Scholar]
- 37.Berkel S, Marshall CR, Weiss B, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 38.Dong C, Yang L, Zhang X, et al. Rab8 interacts with distinct motifs in α2B- and β2-adrenergic receptors and differentially modulates their transport. J Biol Chem. 2010;285:20369–20380. doi: 10.1074/jbc.M109.081521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bralten LB, Gravendeel AM, Kloosterhof NK, et al. The CASPR2 cell adhesion molecule functions as a tumor suppressor gene in glioma. Oncogene. 2010;29:6138–6148. doi: 10.1038/onc.2010.342. [DOI] [PubMed] [Google Scholar]
- 40.Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bastus NC, Boyd LK, Mao X, et al. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70:9544–9548. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nambiar M, Raghavan SC. How does DNA break during chromosomal translocations? Nucleic Acids Res. 2011;39:5813–5825. doi: 10.1093/nar/gkr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 2007;67:5683–5690. doi: 10.1158/0008-5472.CAN-07-0768. [DOI] [PubMed] [Google Scholar]
- 45.Chiaverotti T, Couto SS, Donjacour A, et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. Am J Pathol. 2008;172:236–246. doi: 10.2353/ajpath.2008.070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito T, Kwon HY, Zimdahl B, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466:765–768. doi: 10.1038/nature09171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kharas MG, Lengner CJ, Al-Shahrour F, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16:903–908. doi: 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gurel B, Iwata T, Koh CM, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sudarshan S, Sourbier C, Kong HS, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1α stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bloethner S, Mould A, Stark M, et al. Identification of ARHGEF17, DENND2D, FGFR3, and RB1 mutations in melanoma by inhibition of nonsense-mediated mRNA decay. Genes Chromosomes Cancer. 2008;47:1076–1085. doi: 10.1002/gcc.20598. [DOI] [PubMed] [Google Scholar]
- 51.Bourguignon LY, Singleton PA, Zhu H, et al. Hyaluronan-mediated CD44 interaction with RhoGEF and Rho kinase promotes Grb2-associated binder-1 phosphorylation and phosphatidylinositol 3-kinase signaling leading to cytokine (macrophage-colony stimulating factor) production and breast tumor progression. J Biol Chem. 2003;278:29420–29434. doi: 10.1074/jbc.M301885200. [DOI] [PubMed] [Google Scholar]
- 52.Robbins CM, Tembe WA, Baker A, et al. Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome Res. 2011;21:47–55. doi: 10.1101/gr.107961.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berger MF, Levin JZ, Vijayendran K, et al. Integrative analysis of the melanoma transcriptome. Genome Res. 2010;20:413–427. doi: 10.1101/gr.103697.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.