

Abstract
Objectives
To address concerns regarding increased risk of prostate cancer (PrCA) among Angiotensin Receptor Blocker users, we used national retrospective data from the Department of Veterans Affairs (VA) through the Veterans Affairs Informatics and Computing Infrastructure (VINCI).
Methods
We identified a total of 543,824 unique Veterans who were classified into either ARB treated or not-treated in 1:15 ratio. The two groups were balanced using inverse probability of treatment weights. A double-robust cox-proportional hazards model was used to estimate the hazard ratio for PrCA incidence. To evaluate for a potential Gleason score stage migration we conducted weighted Cochrane-Armitage test.
Results
Post weighting, the rates of PrCA in treated and not-treated groups were 506 (1.5%) and 8,269 (1.6%), respectively; representing a hazard ratio of (0.91, p-value 0.049). There was no significant difference in Gleason scores between the two groups.
Conclusions
We found a small, but statistically significant, reduction in the incidence of clinically detected PrCA among patients assigned to receive ARB with no countervailing effect on degree of differentiation (as indicated by Gleason score). Findings from this study support FDA’s recent conclusion that ARB use does not increase risk of incident PrCA.
Keywords: Angiotensin Receptor Blockers, Prostate Cancer, Department of Veterans Affairs, Inverse Probability of Treatment Weight, Propensity Score, Survival analysis, Cancer Registry, Drug Safety
Introduction
Angiotensin receptor blocker (ARB) use was reported to increase the risk for solid-cancers.1 For prostate cancer (PrCA) the ARB-treated cohort had a meta-analytic risk ratio of 1.15 (C.I., 0.99,1.34, p = 0.076), that barely missed statistical significance.1 The increased risk was not confirmed in subsequent studies or by the Food and Drug Administration (FDA)2–6. In fact Uemura et.al., have found evidence that ARBs may have anti-proliferative effect of PrCA cells.7–11 Thus, the residual concerns for increased risk of PrCA among ARB users is minimal, but since the majority of the follow-up studies were not done on individual level data, i.e. were meta-analysis, and when done with individual level data were based mainly on claims data from outside the United States, we decided to evaluate the safety signal in the Department of Veterans Affairs (VA).
The VA has provided medical care to >8 million male individuals. The VA researchers have access to claims data linked with individual patient level data from electronic medical records that includes pharmacy dispensation, laboratory results, cancer registry. This data when used with appropriate epidemiological research methods is able to provide valuable insight into the real-world relationship between ARB use and risk of prostate cancer.
Methods
We conducted an intention-to-treat (ITT) inverse-probability-of-treatment-weighted (IPTW)12 retrospective cohort study to evaluate the impact of ARB prescribed for clinically indicated reasons on the incidence and histopathological grade of clinically detectable PrCA. The IPTW method is a propensity-score based method12 that is able to correct mathematically for baseline differences (measured covariates) between comparison groups: in this retrospective study, IPTW is expected to balance differences in measured baseline covariates between the group that was assigned to receive ARB and the group that was assigned to not receive ARB. Regulatory approvals were obtained from the Institutional Review Board (IRB) of the William J.B. Dorn VAMC, the VA National Data Systems, the VA Patient Care Services and the Veterans Affairs Informatics and Computing Infrastructure (VINCI). We obtained linked individual-level data on all eligible Veteran patients from 1999 to 2011 from VA’s Central Cancer Registry (CCR), MedSAS, Decision Support System (DSS),13 Vital status file, health factors (for tobacco exposure),14 and the Corporate Data Warehouse (CDW).15
We based our cohort selection method on the methods developed by Hernan et.al.16 –where the recruitment process of a clinical trial was simulated using observational data. We simulated a randomly allocated intention to treat experiment that randomly assigns recruited patients to either ARB (treated) or non-ARB (not-treated) groups in 1:15 ratio continuously across calendar years 2003–2009 with a-priori end-point of December 31st 2010. The cohort selection was performed in blocks of calendar years and then all individuals were pooled to form the final definitive cohort. As a first step, we identified new users of ARB between 2003 and 2009, who did not have an ARB dispensing in the previous years starting from 1999.
We first created the 2003 cohort. For patients receiving their first ARB dispensation in 2003, their start date of follow-up was defined as the closest date of outpatient VA clinician encounter ≤2 weeks before start date of ARB dispensing, ‘assigned to receive treatment’ (treated). If a patient was found to have started ARB without a corresponding VA clinical encounter, that patient was excluded. The rationale for this exclusion was that it is unlikely for a patient to start a new ARB treatment without a corresponding encounter with a clinician, except in such instances as a Veteran filling a non-VA prescription at a VA pharmacy. Including such individuals may introduce bias from missing information on comorbidity as the predominant care may be at a non-VA setting and thus not captured by VA electronic medical record system. The comparator groups, i.e. ‘assigned to receive no-treatment’ (not-treated) were 15-randomly selected individuals who had a VA clinician encounter in the year 2003, but did not receive ARB in the year of 2003 (from Jan 2003– Dec 2003). The start date of follow-up for not-treated group was a randomly selected date of their many respective actual clinical encounters in the year 2003. We achieved this by selecting the date corresponding to the lowest seed per person for that year, generated using SAS 9.2 Cary, NC function Proc RANUNI).
We then repeated this cohort selection method for the years 2004–2009, except that all patients who were already selected into prior year cohorts were not eligible to be selected into subsequent year cohorts. Finally, we pooled all 7-cohorts to form a single definitive cohort that was analyzed. Thus, a single patient was eligible to be represented in the definitive cohort only once. Baseline covariates were identified based on the date of start of follow-up.
We excluded from the staged selection process patients who were documented to have cancer in VA Central Cancer Registry (excluding non-melanoma skin cancer); had not established VA clinical, pharmacy and laboratory care at least 6-months prior to start date; those without information in VA health factor file for tobacco use; those with age < 55 or > 74 years (to ensure homogeneity between the two group, as both PrCA and ARBs are related to age); and those in the not-treated group who had propensity scores of either less than the 5th percentile or greater than 95th percentile of the treatment group (to reduce instability of IPTW).17,18 All selected patients were then followed till the first of either the last date of VA healthcare benefit, death, date of diagnosis of prostate cancer, or December 31st 2010 (a-priori determined end-point), whichever came first.
We computed propensity scores using all variables listed in Table 1 and weighted the cohort using stabilized IPTW. The weighted cohort may now be expected to be similar to a cohort obtained from a random allocation experiment.19 Incidence curves were drawn for both types of exposures and for the absolute difference between exposures (Figure 1). Double-robust regression with IPTW after checking for Cox-Proportionality assumption was used to derive weighted hazard ratios (HR) with 95% confidence intervals. To evaluate if there was a difference in Gleason scores for the PrCAs diagnosed in the two groups, we conducted weighted CochranArmitage test for trend, as Gleason score is ordinal. All reported p-values are two-sided and all analyses on categorical data used exact methods when possible.
Table 1.
Distribution of baseline covariates between treated and untreated before and after weighting with inverse probability of treatment weights
| TABLE | Treated vs. Untreated (Un-weighted) |
Treated vs. Untreated (Weighted) |
|---|---|---|
| Number of patients | 33,989 vs. 509,835 | 34,275 vs. 509,922 |
| Age | 63.6 ± (5.5) vs. 63.6 ± (5.6) | 63.6 ± (5.5) vs. 63.6 ± (5.6) |
| Male | 33,989 (100%) vs. 509,835 (100%) | 34,275 (100%) vs. 509,922 (100%) |
| Race | ||
| White (European American) | 27,656 (81.4%) vs. 421,829 (82.7%) | 28,444 (83%) vs. 421,484 (82.7%) |
| African American | 4,887 (14.4%) vs. 67,033 (13.1%) | 4,404 (12.8%) vs. 67,414 (13.2%) |
| Hawaiian or Pacific Islander | 176 (0.5%) vs. 2,455 (0.5%) | 163 (0.5%) vs. 2,467 (0.5%) |
| Mixed European- and African- American race | 437 (1.3%) vs. 6,943 (1.4%) | 486 (1.4%) vs. 6,921 (1.4%) |
| Mixed other races | 566 (1.7%) vs. 8,183 (1.6%) | 543 (1.6%) vs. 8,200 (1.6%) |
| Other races | 267 (0.8%) vs. 3,392 (0.7%) | 235 (0.7%) vs. 3,437 (0.7%) |
| Hispanic Ethnicity | 1,825 (5.4%) vs. 26,208 (5.1%) | 1,657 (4.8%) vs. 26,270 (5.2%) |
| Body Mass Index | 31.5 ± (5.7) vs. 30.4 ± (5.4) | 30.4 ± (5.3) vs. 30.5 ± (5.5) |
| Dual benefit patient (VA and Medicare) | 18,324 (53.9%) vs. 270,814 (53.1%) | 18,107 (52.8%) vs. 271,133 (53.2%) |
| Religion | ||
| Catholic | 8,773 (25.8%) vs. 130,919 (25.7%) | 8,563 (25%) vs. 130,970 (25.7%) |
| Protestant | 20,857 (61.4%) vs. 314,581 (61.7%) | 21,245 (62%) vs. 314,517 (61.7%) |
| Jewish | 448 (1.3%) vs. 5,965 (1.2%) | 397 (1.2%) vs. 6,017 (1.2%) |
| Other | 3,911 (11.5%) vs. 58,370 (11.4%) | 4,069 (11.9%) vs. 58,419 (11.5%) |
| Tobacco use | ||
| Current user | 17,811 (52.4%) vs. 277,553 (54.4%) | 18,749 (54.7%) vs. 276,935 (54.3%) |
| Former user | 15,227 (44.8%) vs. 218,653 (42.9%) | 14,553 (42.5%) vs. 219,269 (43%) |
| Never user | 951 (2.8%) vs. 13,629 (2.7%) | 973 (2.8%) vs. 13,719 (2.7%) |
| Alcohol Abuse | 3,762 (11.1%) vs. 59,006 (11.6%) | 4,297 (12.5%) vs. 58,870 (11.5%) |
| Substance Abuse | 2,327 (6.8%) vs. 34,173 (6.7%) | 2,594 (7.6%) vs. 34,252 (6.7%) |
| Baseline Comorbidity | ||
| Diabetes Mellitus | 12,590 (37%) vs. 130,146 (25.5%) | 8,991 (26.2%) vs. 133,898 (26.3%) |
| Essential Hypertension | 33,484 (98.5%) vs. 508,953 (99.8%) | 34,151 (99.6%) vs. 508,431 (99.7%) |
| Myocardial infarction | 720 (2.1%) vs. 7,688 (1.5%) | 520 (1.5%) vs. 7,885 (1.5%) |
| Cardiac dysrhythmia | 5,863 (17.2%) vs. 78,794 (15.5%) | 5,252 (15.3%) vs. 79,377 (15.6%) |
| Congestive Heart Failure | 3,300 (9.7%) vs. 26,376 (5.2%) | 1,845 (5.4%) vs. 27,847 (5.5%) |
| Acute Cerebrovascular disease | 1,674 (4.9%) vs. 23,130 (4.5%) | 1,639 (4.8%) vs. 23,249 (4.6%) |
| Chronic Obstructive Pulmonary Disease | 7,373 (21.7%) vs. 108,041 (21.2%) | 7,375 (21.5%) vs. 108,249 (21.2%) |
| Asthma | 2,042 (6%) vs. 26,781 (5.3%) | 1,753 (5.1%) vs. 27,031 (5.3%) |
| Chronic Renal Failure | 2,169 (6.4%) vs. 13,934 (2.7%) | 960 (2.8%) vs. 15,096 (3%) |
| Ulcerative Colitis | 280 (0.8%) vs. 4,423 (0.9%) | 307 (0.9%) vs. 4,413 (0.9%) |
| Rheumatoid Arthritis | 701 (2.1%) vs. 11,358 (2.2%) | 743 (2.2%) vs. 11,304 (2.2%) |
| Benign Prostatic Hyperplasia | 7,011 (20.6%) vs. 110,520 (21.7%) | 7,345 (21.4%) vs. 110,190 (21.6%) |
| Human Immunodeficiency Virus | 85 (0.3%) vs. 1,116 (0.2%) | 99 (0.3%) vs. 1,130 (0.2%) |
| Hepatitis B | 619 (1.8%) vs. 8,709 (1.7%) | 605 (1.8%) vs. 8,756 (1.7%) |
| Hepatitis C | 1,670 (4.9%) vs. 23,363 (4.6%) | 1603 (4.7%) vs. 23,476 (4.6%) |
| Mood disorder | 9,232 (27.2%) vs. 132,832 (26.1%) | 9,162 (26.7%) vs. 133,239 (26.1%) |
| Schizophrenia | 928 (2.7%) vs. 14,968 (2.9%) | 1,179 (3.4%) vs. 14,911 (2.9%) |
| Personality Disorder | 547 (1.6%) vs. 8,191 (1.6%) | 656 (1.9%) vs. 8,198 (1.6%) |
| Epilepsy | 679 (2%) vs. 10,674 (2.1%) | 789 (2.3%) vs. 10,647 (2.1%) |
| History of Coma | 144 (0.4%) vs. 1,782 (0.3%) | 136 (0.4%) vs. 1,808 (0.4%) |
| History of suicidality | 165 (0.5%) vs. 2,274 (0.4%) | 170 (0.5%) vs. 2,287 (0.4%) |
| Baseline Medication | ||
| Angiotensin Converting Enzyme inhibitor | 14,420 (42.4%) vs. 165,276 (32.4%) | 11356 (33.1%) vs. 168519 (33%) |
| Antidepressants | 5,693 (16.7%) vs. 78,708 (15.4%) | 5,236 (15.3%) vs. 79,159 (15.5%) |
| Beta blockers | 11,051 (32.5%) vs. 148,579 (29.1%) | 9,577 (27.9%) vs. 149,555 (29.3%) |
| Calcium channel blocker | 4,636 (13.6%) vs. 58,201 (11.4%) | 3,637 (10.6%) vs. 58,839 (11.5%) |
| Glucocorticoids | 1,286 (3.8%) vs. 15,558 (3.1%) | 989 (2.9%) vs. 15,795 (3.1%) |
| Insulin | 4,052 (11.9%) vs. 27,145 (5.3%) | 1,925 (5.6%) vs. 29,310 (5.7%) |
| Statins | 2,772 (8.2%) vs. 34,565 (6.8%) | 2,268 (6.6%) vs. 35,002 (6.9%) |
| 5-alpha-reductase inhibitor | 733 (2.2%) vs. 10,686 (2.1%) | 706 (2.1%) vs. 10,704 (2.1%) |
| Thiazide diuretics | 12,368 (36.4%) vs. 150,127 (29.4%) | 9,838 (28.7%) vs. 152,233 (29.9%) |
| Baseline laboratory results | ||
| Prostate specific antigen | 1.8 ± (2) vs. 1.8 ± (2.9) | 1.8 ± (2.7) vs. 1.8 ± (2.9) |
| Alanine amino transferase | 33.3 ± (18.9) vs. 32.8 ± (17.6) | 32.9 ± (17.6) vs. 32.8 ± (18.1) |
| Asparatate aminotransferase | 28.2 ± (16.5) vs. 28.2 ± (13.8) | 28.4 ± (17.4) vs. 28.2 ± (14) |
| International Normalized Ratio | 1.4 ± (0.5) vs. 1.4 ± (0.5) | 1.4 ± (0.5) vs. 1.4 ± (0.5) |
| Platelet count | 158.3 ± (32.9) vs. 157.2 ± (30.8) | 157 ± (29.8) vs. 157.2 ± (31) |
| Albumin | 4.1 ± (0.4) vs. 4.1 ± (0.3) | 4.1 ± (0.4) vs. 4.1 ± (0.3) |
| High Density Lipoprotein | 42.7 ± (7.5) vs. 43.3 ± (7.4) | 43.3 ± (7.6) vs. 43.2 ± (7.4) |
| Hemoglobin | 14.5 ± (1.4) vs. 14.6 ± (1.2) | 14.6 ± (1.3) vs. 14.6 ± (1.2) |
| Low Density Lipoprotein | 103.4 ± (30.3) vs. 106.7 ± (28.6) | 106.5 ± (30.5) vs. 106.5 ± (28.6) |
| Potassium | 4.3 ± (0.5) vs. 4.3 ± (0.4) | 4.3 ± (0.4) vs. 4.3 ± (0.4) |
| Creatinine | 1.2 ± (0.5) vs. 1.1 ± (0.3) | 1.1 ± (0.3) vs. 1.1 ± (0.3) |
| Total Cholesterol | 175.5 ± (38.7) vs. 177.1 ± (36.1) | 177 ± (37.8) vs. 177 ± (36.3) |
| Triglycerides | 167.2 ± (91.7) vs. 160.2 ± (85) | 160.4 ± (86.5) vs. 160.6 ± (85.4) |
Figure 1.
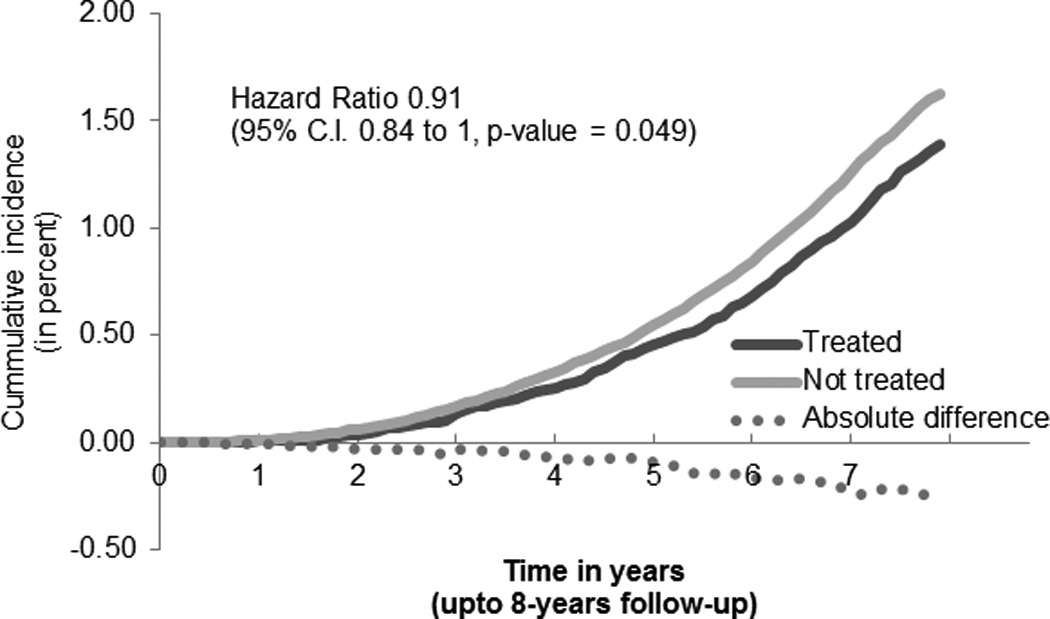
Cumulative incidence of prostate cancer
Results
For the years 2003 to 2009 the un-weighted cohorts had respectively: 95,568; 99,664; 81,600; 70,944; 67,616; 66,080 and 62,352 individual patients. This formed a pooled unweighted cohort of 543,824 individuals. Weighting with IPTW resulted in excellent balance for all 54 variables that was used to compute propensity to receive treatment (Table 1). Propensity to receive treatment was most impacted by diabetes mellitus, serum creatinine, current use of insulin, use of Angiotensin Converting Enzyme inhibitors (ACEi), body mass index [BMI=weight(kg)/height(m)2], chronic renal failure, congestive heart failure, hypertension and low density lipoprotein; and least impacted by race, ethnicity, income, insurance status and religion. As expected, baseline PSA levels, prior utilization of PSA-based testing, prior prostate biopsy or use of 5-α-reductase inhibitor were not found to significantly impact the decision to assign ARB.
The weighted definitive cohort had, in treated and not-treated arms respectively 34,275 and 509,922; with PrCA rates of 506 (1.5%) and 8,269 (1.6%). The weighted hazard ratio (HR) for ARB was 0.91 (95% C.I. 0.84 to 0.99, p-value = 0.049). All independent HRs are reported in Table 2. Current smokers had a HR of 1.69 (95% C.I., 1.38, 2.06, p < 0.001) compared to never smokers, while patients with extremes of BMI where less likely to be diagnosed with PrCA compared to patients with normal BMI. We classified Gleason score based on aggressiveness into either <7, =7 or >7. The distribution of Gleason scores in the treated group was 225 (46.9%), 178 (37.1%) and 77 (16%); this was similar to the not-treated group 3,580 (46.3%), 3,006 (38.9%) and 1,142 (14.8%), with no statistical difference.
Table 2.
Adjusted hazard ratios for Prostate Cancer occurrence, double-robust Inverse Probability treatment weighted survival analysis
| Variables | Hazard Ratio |
|---|---|
| Angiotensin Receptor Blocker | 0.91 (0.84 to 1, p-value 0.049) |
| Age group (Reference '>=70') | |
| 65 to 70 years | 1.03 (0.97 to 1.1, p-value 0.3974) |
| 60 to 65 | 1.51 (1.42 to 1.61, p-value <.0001) |
| < 60 | 1.03 (0.97 to 1.1, p-value 0.3974) |
| Race (reference 'White' or European American) | |
| African American | 2.44 (2.32 to 2.56, p-value <.0001) |
| Hawaiian or Pacific Islander | 0.86 (0.59 to 1.26, p-value 0.4397) |
| Mixed European- and African- American race | 1.79 (1.58 to 2.03, p-value <.0001) |
| Mixed other races | 1.09 (0.81 to 1.46, p-value 0.5655) |
| Other races | 1.09 (0.81 to 1.46, p-value 0.5655) |
| Hispanic ethnicity | 1.19 (1.09 to 1.31, p-value 0.0001) |
| Smoker (reference 'Never') | |
| Current | 1.69 (1.38 to 2.06, p-value <.0001) |
| Former | 1.58 (1.29 to 1.93, p-value <.0001) |
| Body Mass Index | 0.99 (0.98 to 0.99, p-value <.0001) |
| Diabetes Mellitus | 0.99 (0.94 to 1.05, p-value 0.8303) |
Discussion
In this national cohort of veteran patients we observed a slight statistically significant reduction in the rate of clinically detected PrCA among patients assigned to receive ARB. An interesting incidental finding is that, this cancer reduction effect was not associated with PrCA grade migration as is expected in the 5-α reductase inhibitor clinical trials20. The independent pathways by which ARBs are proposed to exert anti-cancer effect may be the reason for the lack of grade progression.7,8,10
In our review of literature, we found that the biological evidence supporting the increased risk of PrCA from use of ARB is limited. Some explanations are related to imbalance in the local tissue level effect of Angiotensin II on inflammation and carcinogenesis. Angiotensin II influences the regulation of cell proliferation, angiogenesis, tissue repair, healing and development, and an imbalance may alter the risk of proliferation of cancer cells.21–23 Unlike angiotensin converting enzyme inhibitors, ARBs do not suppress the production of Angiotensin II: thus in the presence ARB induced Angiotensin type-I receptor blockade, it maybe speculated that circulating Angiotensin II may have enhanced Angiogenesis or pro-inflammatory activity; either directly or through Angiotensin type-II receptors.24,25 However, many researchers suggest a cancer protective effect.
Cancer protective effects of ARBs reported by other researchers include: reduction of basal and squamous cell carcinomas,26 lung cancer metastatic burden,27 and to improve overall lung cancer survival.28 Specifically for PrCA, Candesartan has been shown to decrease PSA levels, improve performance status and decrease the need for analgesics among castration-resistant prostate cancer (CRPC) patients.10 Activation of AT1R has been shown to stimulate the proliferation of prostatic adenocarcinoma,9 while blockade of locally expressed AT1 receptors by ARBs are reported to modulate local growth factors and cytokine expression in tumor tissues (local RAS).29 In addition to the local RAS for cancer prevention, a prostate tissue-specific pathway may exist that would down-regulate the expression of androgen receptors.11
The study limitations are many, but have been addressed to a large extent by careful study design and analysis. While computing propensity scores we have include many measured confounders and many instrumental variables. By balancing instrumental variables, we hope that we will be able to balance many unmeasured variables as well. To avoid errors due to missing information on Veterans who only receive part of their care in the VA, we required all individuals to have already established care in the VA at least 6-months prior to start of follow-up; still there may be some individuals who might receive their ARB dispensation from a non-VA pharmacy. A research study using Veteran only data may not be easily generalizable to non-Veteran population, and this concern is common to all research involving data from the VA. We conducted analysis by strictly adhering to the ITT paradigm, i.e. although we had information on treatment patterns during interval follow-up these data were not analyzed and switches in treatment/compliance was not taken into account. Also not analyzed were cumulative exposures, as these may be affected by the violation of the ITT assumption. Because our purpose was to evaluate the class effect of ARB on PrCA, we did not conduct sub-analyses stratified by ARB-subtype.
The findings of our study are insufficient to recommend the use of ARB as a PrCA chemoprevention modality. However, our finding of an ARB-related weak PrCA protective effect helps assuage any residual concerns of increased PrCA risk1 and supports the conclusions of the FDA.30
Acknowledgements
We wish to thank Dr. Eric Brenner from the University of South Carolina for his support and encouragement to pursue this line of enquiry. Special thanks to Dr. Victoria Barrett, Dr. Jeffrey Scehnet, Mr. Mark Ezzo and Ms. Yiwen Yao of the Veterans Affairs Informatics and Computing Infrastructure (VINCI) program for their assistance through the VINCI program. Special thanks to Ms. Lynnette Nilan, Ms. RayeAnne Dorn and Ms. Audrey Revere from Department of Veterans Affairs (VA) Patient Care Services for their guidance on obtaining and using VA Central Cancer Registry data. Special thanks to entire research department at the William JB Dorn VA Medical Center for allowing use of VA resources for this project.
Dr. Hébert was supported by an Established Investigator Award in Cancer Prevention and Control from the Cancer Training Branch of the National Cancer Institute (K05 CA136975); the South Carolina Cancer Disparities Community Network from the National Cancer Institute’s Center to Reduce Cancer Health Disparities (Community Networks Program Center) (U54 CA153461, JR Hébert, P.I.); and the South Carolina Cancer Prevention and Control Research Network (U48 DP001936, JR Hébert, P.I.) from the Centers for Disease Prevention and Control.
Footnotes
Disclaimer:The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the VA or the United States government.
Note:This manuscript was the result of PhD dissertation project of Dr. Gowtham A. Rao. The dissertation committee was chaired by Dr. James R. Hébert.
References
- 1.Sipahi I, Debanne S, Rowland D, Simon D, Fang J. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. The lancet oncology. 2010 doi: 10.1016/S1470-2045(10)70106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bangalore S, Kumar S, Kjeldsen SE, et al. Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324?168 participants from randomised trials. The lancet oncology. 2011;12(1):65–82. doi: 10.1016/S1470-2045(10)70260-6. [DOI] [PubMed] [Google Scholar]
- 3.Pasternak B, Svanstrom H, Callreus T, Melbye M, Hviid A. Use of Angiotensin Receptor Blockers and the Risk of Cancer. Circulation. 2011 Apr 26;123(16):1729–1736. doi: 10.1161/CIRCULATIONAHA.110.007336. 2011; [DOI] [PubMed] [Google Scholar]
- 4.Bhaskaran K, Douglas I, Evans S, Staa Tv, Smeeth L. Angiotensin receptor blockers and risk of cancer: cohort study among people receiving antihypertensive drugs in UK General Practice Research Database. BMJ. 2012;344 doi: 10.1136/bmj.e2697. 2012-04-24 00:00:00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collaboration TAT. Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138 769 individuals. Journal of Hypertension. 2011;29(4):623–635. doi: 10.1097/HJH.0b013e328344a7de. 610.1097/HJH.1090b1013e328344a328347de. [DOI] [PubMed] [Google Scholar]
- 6.Hallas J, dePont Christensen R, Andersen M, Friis S, Bjerrum L. Long-term use of drugs affecting the renin-angiotensin system and the risk of cancer. A population-based case-control study. British Journal of Clinical Pharmacology. 2012 doi: 10.1111/j.1365-2125.2012.04170.x. no-no. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshino K, Ishiguro H, Teranishi Ji, et al. Regulation of androgen receptor expression through angiotensin II type 1 receptor in prostate cancer cells. The Prostate. 2011;71(9):964–975. doi: 10.1002/pros.21312. [DOI] [PubMed] [Google Scholar]
- 8.Uemura H, Ishiguro H, Nakaigawa N, et al. Angiotensin II receptor blocker shows antiproliferative activity in prostate cancer cells: A possibility of tyrosine kinase inhibitor of growth factor. Molecular Cancer Therapeutics. 2003 Nov 1;2(11):1139–1147. 2003; [PubMed] [Google Scholar]
- 9.Uemura H, Ishiguro H, Nagashima Y, et al. Antiproliferative activity of angiotensin II receptor blocker through cross-talk between stromal and epithelial prostate cancer cells. Molecular Cancer Therapeutics. 2005 Nov 1;4(11):1699–1709. doi: 10.1158/1535-7163.MCT-04-0295. 2005; [DOI] [PubMed] [Google Scholar]
- 10.Uemura H, Hasumi H, Kawahara T, et al. Pilot study of angiotensin II receptor blocker in advanced hormone-refractory prostate cancer. Int J Clin Oncol. 2005 Dec;10(6):405–410. doi: 10.1007/s10147-005-0520-y. [DOI] [PubMed] [Google Scholar]
- 11.Uemura H, Ishiguro H, Kubota Y. Pharmacology and new perspectives of angiotensin II receptor blocker in prostate cancer treatment. International Journal of Urology. 2008;15(1):19–26. doi: 10.1111/j.1442-2042.2007.01937.x. [DOI] [PubMed] [Google Scholar]
- 12.Hernan MA, Robins JM. Estimating causal effects from epidemiological data. J Epidemiol Community Health. 2006 Jul;60(7):578–586. doi: 10.1136/jech.2004.029496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.VA Information Resource Center. 2nd ed. Hines, IL: U.S. Dept. of Veterans Affairs, Health Services Research and Development Service, VA Information Resource Center; 2009. Sep 1, VIReC Research User Guide: VHA Decision Support System Clinical National Data Extracts. wwwvirecresearchvagov/References/RUG/RUG-DSS-2nd-Ed-erpdf. [Google Scholar]
- 14.McGinnis KA, Brandt CA, Skanderson M, et al. Validating Smoking Data From the Veteran’s Affairs Health Factors Dataset, an Electronic Data Source. Nicotine & Tobacco Research. 2011 Sep 12; doi: 10.1093/ntr/ntr206. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Department of Veterans Affiars (VA) VA Information Resource Center (VIReC) Web site. [Accessed March 2nd, 2012];2012 www.virec.research.va.gov.
- 16.Hernan MA, Alonso A, Logan R, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008 Nov;19(6):766–779. doi: 10.1097/EDE.0b013e3181875e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D'Agostino RB., Jr Propensity score methods for bias reduction in the comparison of a treatment to a non-randomized control group. Stat Med. 1998 Oct 15;17(19):2265–2281. doi: 10.1002/(sici)1097-0258(19981015)17:19<2265::aid-sim918>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 18.Austin PC. The performance of different propensity-score methods for estimating differences in proportions (risk differences or absolute risk reductions) in observational studies. Stat Med. 2010 Sep 10;29(20):2137–2148. doi: 10.1002/sim.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson ML, Crown W, Martin BC, Dormuth CR, Siebert U. Good Research Practices for Comparative Effectiveness Research: Analytic Methods to Improve Causal Inference from Nonrandomized Studies of Treatment Effects Using Secondary Data Sources: The ISPOR Good Research Practices for Retrospective Database Analysis Task Force Report—Part III. Value in Health. 2009;12(8):1062–1073. doi: 10.1111/j.1524-4733.2009.00602.x. [DOI] [PubMed] [Google Scholar]
- 20.Thompson IM, Goodman PJ, Tangen CM, et al. The Influence of Finasteride on the Development of Prostate Cancer. New England Journal of Medicine. 2003;349(3):215–224. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 21.Ager EI, Neo J, Christophi C. The renin–angiotensin system and malignancy. Carcinogenesis. 2008;29(9):1675–1684. doi: 10.1093/carcin/bgn171. [DOI] [PubMed] [Google Scholar]
- 22.Cheng ZJ, Vapaatalo H, Mervaala E. Angiotensin II and vascular inflammation. Medical science monitor : international medical journal of experimental and clinical research. 2005;11(6):RA194–RA205. 06/ [PubMed] [Google Scholar]
- 23.Rosenthal T, Gavras I. Angiotensin inhibition and malignancies: a review. Journal of human hypertension. 2009;23(10):623–635. doi: 10.1038/jhh.2009.21. [DOI] [PubMed] [Google Scholar]
- 24.Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor-&kgr; B through AT1 and AT2 receptors1. Kidney international. 2002;61(6):1986–1995. doi: 10.1046/j.1523-1755.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- 25.Sarlos S, Rizkalla B, Moravski CJ, Cao Z, Cooper ME, Wilkinson-Berka JL. Retinal Angiogenesis Is Mediated by an Interaction between the Angiotensin Type 2 Receptor, VEGF, and Angiopoietin. The American Journal of Pathology. 2003;163(3):879–887. doi: 10.1016/S0002-9440(10)63448-7. 9// [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christian JB, Lapane KL, Hume AL, Eaton CB, Weinstock MA. Association of ACE inhibitors and angiotensin receptor blockers with keratinocyte cancer prevention in the randomized VATTC trial. J Natl Cancer Inst. 2008 Sep 3;100(17):1223–1232. doi: 10.1093/jnci/djn262. [DOI] [PubMed] [Google Scholar]
- 27.Miyajima A, Kosaka T, Asano T, et al. Angiotensin II Type I Antagonist Prevents Pulmonary Metastasis of Murine Renal Cancer by Inhibiting Tumor Angiogenesis. Cancer Research. 2002 Aug 1;62(15):4176–4179. 2002; [PubMed] [Google Scholar]
- 28.Wilop S, von Hobe S, Crysandt M, Esser A, Osieka R, Jost E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. Journal of Cancer Research and Clinical Oncology. 2009;135(10):1429–1435. doi: 10.1007/s00432-009-0587-3. [DOI] [PubMed] [Google Scholar]
- 29.George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. Nov;10(11):745–759. doi: 10.1038/nrc2945. [DOI] [PubMed] [Google Scholar]
- 30.FDA Drug Safety Communication. No increase in risk of cancer with certain blood pressure drugs--Angiotensin Receptor Blockers (ARBs) [on May 31st 2012]; Retrieved from www.fda.gov.