

Abstract
Recent studies in Aplysia have identified a new variation of synaptic plasticity in which modulatory transmitters enhance spontaneous release of glutamate, which then acts on postsynaptic receptors to recruit mechanisms of intermediate- and long-term plasticity. In this review I suggest the hypothesis that similar plasticity occurs in mammals, where it may contribute to reward, memory, and their dysfunctions in several psychiatric disorders. In Aplysia, spontaneous release is enhanced by activation of presynaptic serotonin receptors, but presynaptic D1 dopamine receptors or nicotinic acetylcholine receptors could play a similar role in mammals. Those receptors enhance spontaneous release of glutamate in hippocampus, entorhinal cortex, prefrontal cortex, ventral tegmental area, and nucleus accumbens. In all of those brain areas, glutamate can activate postsynaptic receptors to elevate Ca2+ and engage mechanisms of early-phase long-term potentiation (LTP), including AMPA receptor insertion, and of late-phase LTP, including protein synthesis and growth. Thus, presynaptic receptors and spontaneous release may contribute to postsynaptic mechanisms of plasticity in brain regions involved in reward and memory, and could play roles in disorders that affect plasticity in those regions, including addiction, Alzheimer’s disease, schizophrenia, and attention deficit hyperactivity disorder (ADHD).
Synaptic plasticity has traditionally been divided along three dimensions: (1) modulatory vs. activity-dependent forms, (2) pre- vs. postsynaptic forms, and (3) short- vs. long-term forms. In addition, these basic types of plasticity can be combined in various ways to make hybrids with different functional characteristics (Bailey et al. 2000; Antonov et al. 2003; Cassenaer and Laurent 2012; Huang et al. 2012). For example, modulatory transmitters can act on presynaptic receptors to enhance short-term plasticity (such as post-tetanic potentiation [PTP]) of the evoked release of glutamate, leading to greater recruitment of postsynaptic mechanisms and greater long-term plasticity (Puzzo et al. 2008). Recent studies in Aplysia (Jin et al. 2012a,b) have identified a new variation on this theme in which modulatory transmitters enhance spontaneous release of glutamate, which then acts on postsynaptic receptors to recruit mechanisms of intermediate- and long-term plasticity (Fig. 1A). This combination thus links presynaptic modulatory effects with postsynaptic mechanisms of “activity-dependent” plasticity in the absence of activity, and provides a new function for spontaneous release. Because of the novelty of these ideas this review focuses on spontaneous release, which in most of the studies cited was measured in the absence of presynaptic action potentials. However, as illustrated above, modulatory enhancement of presynaptic firing or evoked release can also lead to enhanced activation of glutamate receptors and recruitment of postsynaptic mechanisms of plasticity, so that many of the same arguments might apply.
Figure 1.
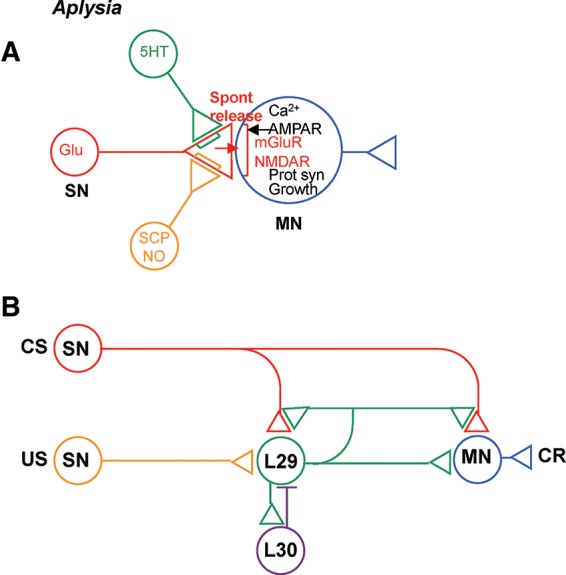
Novel synaptic (A) and circuit (B) mechanisms in Aplysia that are involved in learning and memory. (SN) Sensory neuron, (MN) motor neuron, (CS) conditioned stimulus, (US) unconditioned stimulus, (CR) conditioned response, (Glu) glutamate (red), (5HT) serotonin (green), (SCP) small cardioactive peptide, (NO) nitric oxide.
The simplicity of the Aplysia system was important for the discovery of this type of plasticity, but in this review I suggest the hypothesis that similar plasticity occurs in mammals, where it may contribute to reward, memory, and their dysfunctions in disorders including addiction, Alzheimer’s disease, schizophrenia, and attention deficit hyperactivity disorder (ADHD). These proposals are not meant to challenge the importance of other, better established mechanisms, which might play complementary roles or act cooperatively with the mechanisms described here.
In Aplysia, serotonin (5HT) acts on presynaptic receptors to produce an increase in spontaneous release of glutamate at sensory–motor neuron synapses. This increase is normally mediated by phospholipase C and protein kinase C (PKC), but it can also be produced by adenylyl cyclase and cAMP-dependent protein kinase (PKA) (Braha et al. 1990; Ghirardi et al. 1992; Jin et al. 2012a). The glutamate activates postsynaptic metabotropic glutamate (mGlu) receptors linked to the production of inositol triphosphate (IP3), leading to an increase in Ca2+ and protein synthesis-dependent intermediate-term facilitation. Many of the same postsynaptic mechanisms are recruited during an activity-dependent form of homosynaptic potentiation at these synapses (Jin and Hawkins 2003; Jin et al. 2011a). Intermediate-term facilitation is also accompanied by membrane insertion and an increase in the number of clusters of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-like glutamate receptors, which precedes increases in clusters of presynaptic proteins and presynaptic varicosities, and therefore could be an early step in synaptic growth during long-term facilitation. Consistent with that idea, spontaneous transmitter release is also required for the induction of long-term facilitation.
Recent studies in several species have shown that spontaneous release also contributes to other synaptic effects, including some cases of homeostatic scaling (Sutton et al. 2004, 2006, 2007; Frank et al. 2006, 2009; Dickman and Davis 2009; Zhang et al. 2009; Lee et al. 2010; Nosyreva et al. 2013). Scaling in those cases has additional similarities to intermediate- and long-term facilitation in Aplysia: Both types of plasticity can involve postsynaptic Ca2+, protein synthesis, AMPA receptor insertion, and modulation of the presynaptic probability of release. However, scaling has an opposite sign of action compared to facilitation (during scaling, spontaneous release acts to decrease protein synthesis and synaptic strength). One possible explanation is that a brief, large increase in spontaneous release and, consequently, postsynaptic Ca2+ produces facilitation, whereas a long, small increase produces scaling, similar to the patterns that produce long-term potentiation (LTP) and long-term depression (LTD) (Bear and Malenka 1994). Consistent with that idea, reducing background spontaneous release also tends to increase the evoked EPSP in Aplysia, suggesting that scaling and facilitation coexist at the same synapses (Jin et al. 2012a).
In Aplysia, spontaneous release is enhanced by activation of presynaptic 5HT receptors. This can also occur in mammals (Cui et al. 2012), but presynaptic D1 dopamine (DA) receptors or nicotinic acetylcholine receptors (nAChR) could play a similar role as well. Presynaptic D1 or nicotinic receptors are present on glutamatergic afferents to hippocampus, entorhinal cortex (EC), prefrontal cortex (PFC), and nucleus accumbens (NAc), as well as afferents to ventral tegmental area (VTA) dopaminergic neurons themselves (Bergson et al. 1995; Jones and Wonnacott 2004; Paspalas and Goldman-Rakic 2005; Dumartin et al. 2007). The D1 receptors act through adenylyl cyclase and PKA to enhance spontaneous release of glutamate in hippocampus, EC, PFC, NAc, and VTA (Kalivas and Duffy 1995; Bouron and Reuter 1999; Yang 1999; Wang et al. 2002, 2012). Results in NAc are mixed, and in some cases presynaptic D1 receptors instead decrease glutamatergic transmission (Nicola and Malenka 1998). Although nicotinic and D1 receptors act through very different molecular mechanisms (see the section on “Synapse-specific effects”), activation of presynaptic nicotinic receptors has the same functional outcome of enhancing spontaneous release of glutamate in hippocampus, EC, PFC, VTA, and other areas (Radcliffe and Dani 1998; Radcliffe et al. 1999; Mansvelder and McGehee 2000; Sharma and Vijayaraghavan 2003; Wang et al. 2006; Tu et al. 2009; Garduño et al. 2012). In many of these studies spontaneous release was used as an indicator of presynaptic or postsynaptic changes, but it could also recruit those changes as it does in Aplysia.
In all of those brain areas, glutamate can activate postsynaptic NMDA receptors (in addition to or instead of metabotropic glutamate receptors, as in Aplysia) to elevate Ca2+ and engage mechanisms of early-phase long-term potentiation (E-LTP) including AMPA receptor insertion (Otmakhova and Lisman 1996; Gurden et al. 2000; Mansvelder and McGehee 2000; Huang et al. 2004; Kauer and Malenka 2007; Brown et al. 2010; Luscher and Malenka 2011) and of late-phase LTP (L-LTP) including protein synthesis and growth (Figs. 2A, 3, 4; Huang and Kandel 1995; Hyman et al. 2006; Lemon and Manahan-Vaughan 2006; Navakkode et al. 2007; Bailey et al. 2008). Spontaneous EPSPs are often thought to be insufficient to activate NMDA receptors, but there is growing evidence that they can even at rest (Sutton et al. 2006, 2007; Espinosa and Kavalali 2009; Autry et al. 2011; Povysheva and Johnson 2012; Nosyreva et al. 2013). Furthermore, when spontaneous release is enhanced by a modulatory transmitter, in some cases the EPSPs can summate enough in the postsynaptic dendrites to reach threshold for firing (e.g., Sharma and Vijayaraghavan 2003). Even if the postsynaptic depolarization is subthreshold for firing, it can still be sufficient to further activate NMDA receptors and increase Ca2+ in the spines or dendrites, which is a key event in the induction of plasticity.
Figure 2.
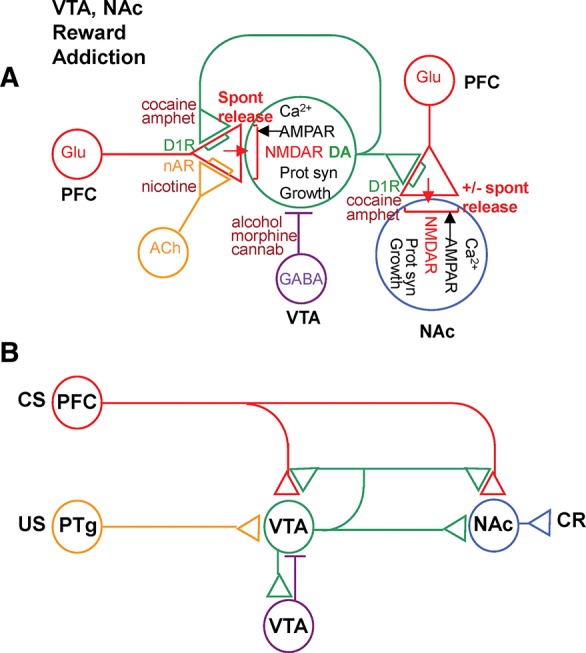
Novel synaptic (A) and circuit (B) mechanisms in ventral tegmental area (VTA) and nucleus accumbens (NAc) that could be involved in reward and its dysfunction in addiction. (PFC) Prefrontal cortex, (PTg) pedunculopontine tegmentum, (DA) dopamine (green), (ACh) acetylcholine (yellow), (D1R) D1 dopamine receptor, (nAR) nicotinic acetylcholine receptor.
Figure 3.
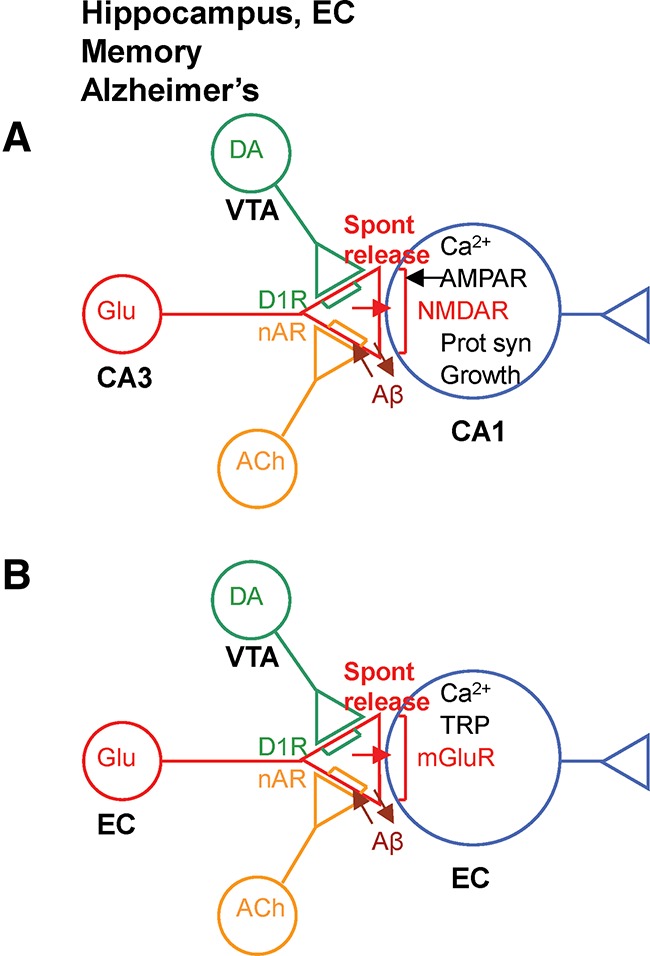
Novel synaptic (A) and intrinsic (B) mechanisms in hippocampus and entorhinal cortex (EC) that could be involved in reference memory, working memory, and their dysfunction in Alzheimer’s disease. (TRP) Transient receptor potential.
Figure 4.
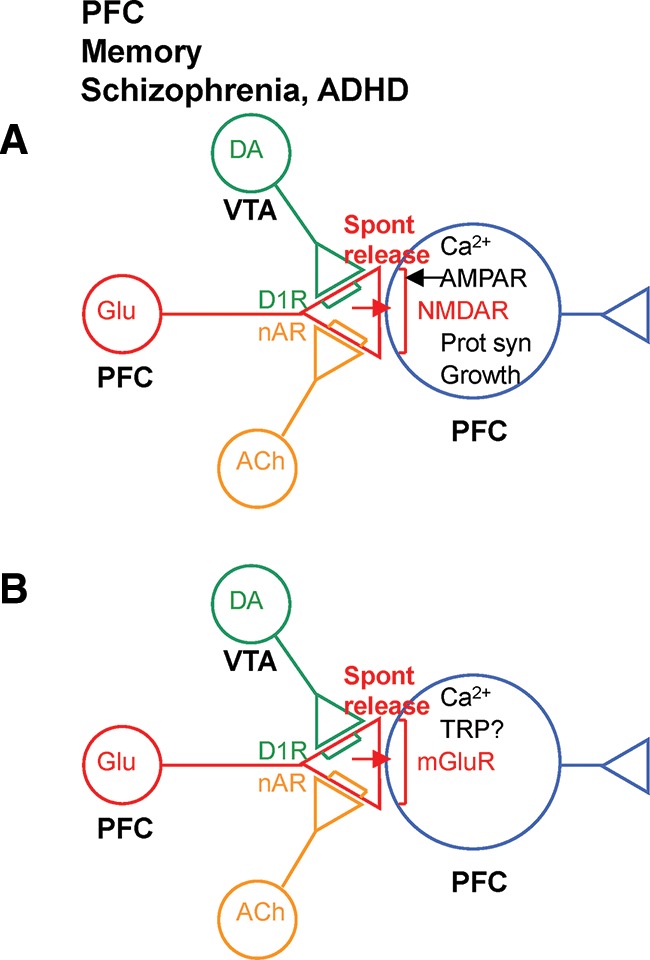
Novel synaptic (A) and intrinsic (B) mechanisms in prefrontal cortex (PFC) that could be involved in working memory and its dysfunction in schizophrenia and attention deficit hyperactivity disorder (ADHD).
Synaptic plasticity in VTA, NAc, and PFC is thought to be involved in reward, plasticity in hippocampus and EC is thought to be involved in reference memory, and plasticity in PFC and EC is thought to be involved in working memory (Goldman-Rakic 1995; Morris et al. 2003; Goldman-Rakic et al. 2004; Hyman et al. 2006; Moser et al. 2008). Thus, presynaptic receptors and spontaneous release may contribute to different aspects of learning-related synaptic plasticity in several brain regions, and could be involved in disorders that affect synaptic plasticity in those regions, including addiction (Kalivas and Duffy 1995; Mansvelder and McGehee 2000; Li et al. 2008; Xiao et al. 2009), Alzheimer’s disease (Welsby et al. 2007; Nimmrich et al. 2008; Puzzo et al. 2008; Khan et al. 2010), schizophrenia (Chen and Yang 2002; Radek et al. 2010), and ADHD (Arnsten 2009). This review summarizes some of the positive evidence in each case to illustrate the plausibility of these ideas, and is not meant to be a comprehensive survey of the literature.
Reward and addiction
Aplysia plasticity and circuitry can account for behavioral features of learning
In Aplysia the mechanisms of synaptic plasticity described above are embedded in a simple neural circuit (Fig. 1B; Hawkins 1981; Hawkins et al. 1981, 1983; Hawkins and Schacher 1989). Together with the synaptic properties, this circuit can account for many behavioral features of elementary forms of learning of the gill- and siphon-withdrawal reflex, including habituation, dishabituation, sensitization, and classical conditioning (for review, see Hawkins 1989). During conditioning, a mild conditioned stimulus (CS) to the siphon is temporally paired with a noxious unconditioned stimulus (US) to the tail. This training produces greater enhancement of the conditioned response (CR) of gill and siphon withdrawal than unpaired training or training with the US or CS alone. The CS activates sensory neurons (SNs) that make monosynaptic excitatory connections onto motor neurons (MNs) that mediate the CR. Facilitatory neurons, including a small group of L29 interneurons, also receive excitatory input from the CS as well as the US. Firing of L29 neurons activates presynaptic receptors on the CS SNs, and produces facilitation at all the terminals of those neurons, including ones onto the L29 neurons themselves. The presynaptic facilitation is activity-dependent: That is, it is greater at synapses from active SNs than from inactive SNs. Finally, the L29s receive feed forward and feedback inhibition from a small group of L30 interneurons, which act to shut off firing of the L29s. Thus, as conditioning proceeds the synapses from the CS SNs to the L29s are strengthened, causing increased firing of the L29s during the CS, increased negative feedback from the L30s, and thus decreased firing during the US. These synaptic and circuit properties embody key elements of a psychological model proposed by Rescorla and Wagner (1972), and thus can account for many higher order features of classical conditioning such as second-order conditioning and blocking (Hawkins 1989). These features of conditioning are thought to have a cognitive flavor, and therefore might form a bridge to more complex forms of learning.
Since these ideas were first formulated, a number of additional properties of synaptic plasticity and circuitry have been discovered that might account for additional features of behavioral learning (for review, see Hawkins et al. 2010). For example, the SN–MN synapses also undergo NMDA-dependent Hebbian potentiation, which is induced by nearly coincident pre- and postsynaptic activity (Lin and Glanzman 1994; Antonov et al. 2003). This happens during conditioning because the US drives the MNs (in part via the L29s, which are also excitatory interneurons) just after the CS drives the SNs. The Hebbian potentiation acts synergistically with activity-dependent presynaptic facilitation to strengthen the SN–MN synapses during conditioning (Murphy and Glanzman 1997; Antonov et al. 2003). This hybrid mechanism combines the synapse specificity of Hebbian potentiation with global instruction by motivationally significant stimuli. Furthermore, individual L29s respond to different stimuli and use different facilitatory transmitters, and therefore may serve as internal representations of different USs (Hawkins and Schacher 1989; Antonov et al. 2007). These additional synaptic and circuit properties could explain additional features of conditioning such as response specificity and post-training US exposure effects (Rescorla 1978).
As outlined above, recent studies have found that the facilitatory transmitter also enhances spontaneous release of glutamate from the SNs. The glutamate then recruits postsynaptic mechanisms of intermediate-term facilitation, and initiates a cascade of pre- and postsynaptic mechanisms that may culminate in synaptic growth during long-term facilitation (Jin et al. 2012a,b).
Mammalian plasticity and reward circuitry have many similarities to those in Aplysia
Synaptic plasticity and circuitry involved in reward in mammals have many similarities to those in Aplysia, potentially providing a mechanistic understanding of important aspects of normal reward function and its dysfunction in addiction and other disorders (Fig. 2A,B).
VTA dopamine (DA) neurons are thought to mediate reward, and are thus analogous to the L29 neurons in Aplysia (except that they are generally activated by positive USs). Like the L29 neurons, the VTA DA neurons are not homogenous and may be involved in learning about different types of USs (Lammel et al. 2011, 2012; Navratilova et al. 2012). They receive glutamatergic excitatory input from brain regions that process the CS (e.g., the PFC) and glutamatergic, cholinergic, and peptidergic excitatory input from regions that process the US (e.g., the laterodorsal and pedunculopontine tegmentum [PPTg] and lateral hypothalamus) (Floresco et al. 2003; Aston-Jones et al. 2010; Yeomans 2010; Zellner and Ranaldi 2010; Lammel et al. 2012; Kempadoo et al. 2013). During conditioning, the VTA DA neurons increase their firing to the CS and decrease their firing to the US, so that they fire in expectation of reward (Schultz et al. 1997; Schultz 2011). Their firing is thus thought to encode a reward prediction error, i.e., whether current events are better or worse than expected (Montague et al. 1996, 2004). The VTA DA neurons are also activated by rewarding brain stimulation (Wise 2005), and direct activation of those neurons with optigenetic techniques is rewarding (Tsai et al. 2009; Witten et al. 2011).
The VTA neurons release DA that stimulates presynaptic D1 receptors coupled to adenylyl cyclase on the terminals of the CS (e.g., PFC) neurons, and produces potentiation at synapses of those neurons in several brain areas, including the PFC, the NAc, and the VTA DA neurons themselves. The DA potentiation is PKA and NMDA dependent, and is also activity dependent—i.e., it is greater at synapses from active CS neurons (Gurden et al. 2000; Otani et al. 2003; Huang et al. 2004; Sun et al. 2005). Finally, the VTA DA neurons also send projections to postsynaptic neurons in several brain areas, including the PFC and NAc, and receive feed forward and feedback inhibition from GABA neurons in VTA and NAc (Hyman et al. 2006; Nugent et al. 2007; Barrot et al. 2012; Cohen et al. 2012). These properties of the VTA DA neurons are similar to those of the L29 neurons in Aplysia, and therefore might account for many of the same behavioral features of learning and reward. In particular, they might at least partially explain why the VTA DA neurons come to fire in expectation of reward during conditioning, which was initially explained by more abstract mathematical models (Montague et al. 1996, 2004; Schultz et al. 1997). These properties might also explain how firing of the VTA DA neurons helps CS neurons gain control of brain areas involved in the behavioral response (Flagel et al. 2011).
The synaptic properties and simple circuits observed in Aplysia (Fig. 1) and mammalian brain (Fig. 2) could account for many additional behavioral features of learning (Hawkins 1989; Hawkins et al. 2010), but they cannot account for some others, including learning about CS–US timing. However, the VTA DA neurons are sensitive to that timing, as demonstrated by a decrease in firing at the expected time of the US when it is omitted (Schultz et al. 1997). The behavior of the DA neurons is thus more similar to that of temporal difference models of learning (Sutton and Barto 1990), and might be explained by neural circuits that embody those models (Suri and Schultz 2001; Kobayashi and Okada 2007). In particular, if neurons that computed a temporal reward prediction signal (that is, increased firing from the time of the CS to the expected time of the US) were inserted in the CS pathway, the circuit shown in Figure 2B would embody basic elements of the temporal difference model. Such neurons have been found in a variety of brain areas that could be part of the CS pathway, including orbitofrontal cortex and PPTg (Suri and Schultz 2001; Okada et al. 2009).
Like presynaptic DA receptors, activation of presynaptic acetylcholine receptors can produce activity- and NMDA-dependent potentiation at the CS neuron synapses (Mansvelder and McGehee 2000; Welsby et al. 2006, 2007). Those synapses also undergo NMDA-dependent Hebbian potentiation, which is induced by nearly coincident pre- and postsynaptic activity driven by the CS and US, respectively. Furthermore, DA and acetylcholine enhance spontaneous release of glutamate from the CS neurons, which may act on postsynaptic receptors to engage the same mechanisms as Hebbian potentiation. As described below, these additional features of synaptic plasticity could help to explain the actions of drugs of abuse during addiction.
Synapse-specific effects
By itself, dopaminergic or cholinergic enhancement of release would produce potentiation at most or all synapses, and thus might contribute to nonassociative effects such as sensitization. If, however, dopamine or acetylcholine acts synergistically with presynaptic activity to enhance release, the resultant mechanism would be synapse specific and thus might also contribute to associative effects such as conditioning. For example, dopamine and acetylcholine can enhance not only spontaneous release but also short-term plasticity of evoked release during the CS (Young and Yang 2005; Puzzo et al. 2008), which would lead to greater stimulation of postsynaptic NMDARs and greater potentiation specifically at the CS synapses. Dopamine or acetycholine might also act synergistically with presynaptic activity to enhance long-term plasticity of release, as 5HT does in Aplysia (Schacher et al. 1997; Bailey et al. 2000).
Dopamine and acetylcholine have different presynaptic actions, and therefore might interact with presynaptic activity in different ways, possibly with different functional consequences. The nicotinic acetylcholine receptor (nAChR) is coupled to a Ca2+ permeable ionophore, so that nAChR activation and presynaptic spike activity may act synergistically to increase presynaptic Ca2+ (perhaps from intracellular stores [Welsby et al. 2006]) and thus enhance release. Recent studies have found that nAChR activation and presynaptic spike activity do act synergistically to produce LTP when spike activity comes slightly (100 msec) after the nAChR activation (Gu and Yakel 2011; Gu et al. 2012). By contrast, the D1 dopamine receptor is coupled to adenylyl cyclase, some forms of which are also Ca2+ sensitive, so that D1R activation and Ca2+ influx during spike activity may act synergistically to increase presynaptic cAMP and thus enhance release. This is the same coincidence detection mechanism that has been described in Aplysia, where spike activity shortly (500 msec) before 5HT is optimal for increasing cAMP and enhancing release (Yovell and Abrams 1992; Clark et al. 1994). The optimal timing of D1R activation and spike activity has not yet been investigated, but the prediction is that it should be similar to that of 5HT in Aplysia.
It is also not yet known in any preparation whether modulatory transmitters and presynaptic spike activity act synergistically to enhance spontaneous release. Preliminary evidence in Aplysia suggests that they do not (Eliot et al. 1994), but those experiments investigated short-term facilitation for which spontaneous release may not play an important role (Antonov et al. 2010; Jin et al. 2011a). Longer lasting forms of plasticity that involve synergistic increases in either presynaptic Ca2+ or cAMP might also involve synergistic enhancement of spontaneous release.
Drugs of abuse hijack the normal reward process
Drugs of abuse act directly or indirectly to activate the VTA DA reward system (Fig. 2A). Cocaine and amphetamine act at DA terminals to increase extracellular DA levels, whereas alcohol, morphine, and cannabinoids reduce GABA inhibitory input to the DA neurons, producing increased firing of those neurons (Kauer 2004; Hyman et al. 2006; Nugent et al. 2007; Jalabert et al. 2011; Sulzer 2011; Barrot et al. 2012; Oleson et al. 2012; Daberkow et al. 2013). The DA neurons then release DA both from their terminals in other brain areas and from their own dendrites in VTA. DA is thought to act as a signal that current events are better than expected, and drugs of abuse always produce that signal (whether or not it is true), which may explain why they have an advantage over natural rewards (Hyman et al. 2006; Schultz 2011). All of these drugs act through D1 receptors to produce NMDA-dependent E-LTP and AMPA receptor insertion at glutamatergic synapses onto the VTA DA neurons (Ungless et al. 2001; Saal et al. 2003; Hyman et al. 2006; Kauer and Malenka 2007; Sarti et al. 2007; Brown et al. 2010; Luscher and Malenka 2011). Similarly, nicotine acts through nicotinic acetylcholine receptors to produce NMDA-dependent E-LTP at glutamatergic synapses onto the VTA DA neurons (Grillner and Svensson 2000; Mansvelder and McGehee 2000; Gao et al. 2010; Jin et al. 2011b; Mao et al. 2011). The drugs also produce L-LTP, gene regulation, and synaptic growth in VTA (Licata and Pierce 2003; Hyman et al. 2006; Sarti et al. 2007; Chen et al. 2008; Flatscher-Bader et al. 2008).
NMDA-dependent Hebbian LTP at glutamatergic synapses onto the VTA DA neurons preferentially strengthens synapses from CSs that are temporally paired with the US, allowing them to activate the DA neurons themselves (Kauer and Malenka 2007; Zellner and Ranaldi 2010; Wang et al. 2011). Drugs of abuse can also produce similar potentiation by acting directly or indirectly on the same synapses. This process is thought to be involved in the initial rewarding stages of addiction when, for example, a person or animal learns positive associations with cues (CSs) connected with drug use.
Similar drug-induced plasticity occurs at synapses from the CS to other brain areas as well, with different behavioral consequences. Thus, drugs of abuse can also produce LTP In NAc and PFC (Boudreau and Wolf 2005; Boudreau et al. 2007; Huang et al. 2007; Dobi et al. 2011; Luscher and Malenka 2011; Moussawi et al. 2011; Shen et al. 2011; Pascoli et al. 2012; Gipson et al. 2013a,b). Plasticity in those areas may be involved in the early stages of addiction, such as drug sensitization and learning a behavioral response to the CS, and is also thought to be involved in later stages of addiction, such as compulsion and relapse (Bossert et al. 2011; Li et al. 2011c; Pascoli et al. 2012). However, as with D1 receptor activation (see above), the plasticity in NAc is mixed. For example, glutamatergic synapses in the shell (but not the core) of NAc undergo LTD during the early stages of cocaine withdrawal, followed by LTP during later stages of withdrawal (or with prolonged cocaine treatment) and LTD again during reinstatement (Boudreau et al. 2007; Kourrich et al. 2007; Bowers et al. 2010; Ren et al. 2010; Dobi et al. 2011).
Drugs of abuse also produce regulation of genes related to LTP in NAc, including Fos B, c-fos, NR2B and NR4A, CamKII, and dynorphin (Carlezon et al. 1998; Hyman et al. 2006; Lee et al. 2006; Brown et al. 2011; Malvaez et al. 2011; Damez-Werno et al. 2012; Pitchers et al. 2013; Robison et al. 2013; Rogge et al. 2013). That regulation involves a variety of mechanisms, including activation of CREB and epigenetic changes in histone acetylation and methylation (Carlezon et al. 1998; Brown et al. 2011; Malvaez et al. 2011; Maze et al. 2011; Damez-Werno et al. 2012; Sun et al. 2012; Taniguchi et al. 2012; Rogge et al. 2013). In addition, drugs of abuse produce synaptic growth and structural changes related to L-LTP in NAc, including increases in dendritic branching, spine density, and spine size (Lee et al. 2006; Ren et al. 2010; Dobi et al. 2011; Shen et al. 2011; Dietz et al. 2012; Dumitriu et al. 2012; Gipson et al. 2013a,b; Pitchers et al. 2013; Robison et al. 2013), as well as increases in pre- and postsynaptic proteins (Subramaniam et al. 2001; Gipson et al. 2013b). The drugs produce the same types of structural changes in PFC as well (Robinson and Kolb 1997, 1999; Robinson et al. 2001; Hamilton et al. 2010; Gourley et al. 2012).
Similar plasticity may also occur in hippocampus, which is thought to process information about the context of drug use. The hippocampus sends indirect projections to VTA and NAc (Vorel et al. 2001; Lisman and Grace 2005; Cooper et al. 2006; Fanselow and Dong 2010; Grace 2010; Luo et al. 2011; Britt et al. 2012; Keleta and Martinez 2012) and plays an important role in context conditioning and reinstatement of drug use by reexposure to the context (Meyers et al. 2003, 2006; Atkins et al. 2008; Martin-Fardon et al. 2008; Ramirez et al. 2009; Lasseter et al. 2010; Xie et al. 2010). VTA DA neurons, in turn, send projections back to hippocampus, where they are thought to enhance LTP and the formation of long-term memories about context (Fig. 3A; Rossato et al. 2009).
As in other brain areas, in vivo exposure to drugs such as cocaine enhances hippocampal LTP (Thompson et al. 2002, 2004; Del Olmo et al. 2006b; Guan et al. 2009; Perez et al. 2010), gene expression (Freeman et al. 2001; Ary and Szumlinski 2007; Krasnova et al. 2008; Perrotti et al. 2008; Tropea et al. 2008), and synaptic growth (Fole et al. 2011). The drugs also enhance synaptic transmission and/or LTP in vitro, in some cases through activation of D1 receptors (Del Olmo et al. 2006a; Stramiello and Wagner 2010; Swant et al. 2010). Likewise, activation of D1 receptors produces activity- and NMDA-dependent E-LTP and L-LTP in hippocampus (Huang and Kandel 1995; Huang et al. 1996; Otmakhova and Lisman 1996; Bach et al. 1999; Swanson-Park et al. 1999; Yang 1999; Li et al. 2003; Lemon and Manahan-Vaughan 2006; Williams et al. 2006; Navakkode et al. 2007, 2010, 2012; Stramiello and Wagner 2008, 2010), and inhibition or knock out of D1 receptors reduces LTP (Frey et al. 1991; Li et al. 2003; Morris et al. 2003; Ortiz et al. 2010).
The possible roles of presynaptic receptors and spontaneous release in reward and addiction
A question raised by these results is why the potentiation often depends on both NMDA receptors and either D1 or nACh receptors. One idea is that those receptors are all on the postsynaptic neuron and somehow interact (Kauer and Malenka 2007; Argilli et al. 2008; Sun et al. 2009; Nai et al. 2010; Luscher and Malenka 2011; Mao et al. 2011; McGranahan et al. 2011). However, another possibility is that the D1 or nACh receptors are on presynaptic terminals of glutamatergic neurons, where they enhance spontaneous as well as evoked release of glutamate that then activates postsynaptic NMDA receptors (Figs. 2A, 3A). Thus cocaine, amphetamine, alcohol, morphine, and cannabinoids act directly or indirectly to increase extracellular DA, which acts on presynaptic D1 receptors. Similarly, nicotine acts on presynaptic nACh receptors. According to this idea the D1 or nACh receptors are in series with NMDA receptors, which would explain why both are necessary.
In support of that idea, presynaptic D1 and nACh receptors are found in many brain areas, including VTA, NAc, PFC, and hippocampus. Activation of D1 receptors enhances spontaneous release of glutamate in all of those areas (Kalivas and Duffy 1995; Bouron and Reuter 1999; Yang 1999; Wang et al. 2002, 2012). Cocaine, opiates, and alcohol also act through D1 receptors to enhance spontaneous and evoked release of glutamate in several brain areas, including VTA (Kalivas and Duffy 1995; Li et al. 2008; Deng et al. 2009; Xiao et al. 2009) and NAc (Pierce et al. 1996; McFarland et al. 2003; Dobi et al. 2011; Moussawi et al. 2011; Wissman et al. 2011; Suska et al. 2013; Suto et al. 2013). Similarly, nicotine acts through nACh receptors to enhance glutamate release in VTA (Mansvelder and McGehee 2000; Nomikos et al. 2000), NAc (Gipson et al. 2013b), PFC (Wang et al. 2006), and hippocampus (Radcliffe and Dani 1998; Radcliffe et al. 1999; Liu et al. 2003; Sharma and Vijayaraghavan 2003). The glutamate could then activate postsynaptic NMDA receptors and engage mechanisms of early- and late-phase LTP. This hypothesis is very parallel to recent results from Aplysia (Fig. 1A), is consistent with much of the known mammalian data, and provides a simple explanation for why drug-induced potentiation in mammals depends on both NMDA receptors and D1 or nACh receptors.
However, other mammalian data suggest a postsynaptic action of DA and acetylcholine in VTA. Whether DA and acetylcholine have predominantly presynaptic or postsynaptic actions may depend on differences in the experimental protocols. For example, most of the evidence for presynaptic actions comes from experiments in vivo or on slices from young (P10–P20) animals (Kalivas and Duffy 1995; Mansvelder and McGehee 2000; Nomikos et al. 2000; Li et al. 2008; Deng et al. 2009), whereas most of the evidence for postsynaptic actions comes from experiments on slices from older animals (Argilli et al. 2008; Mao et al. 2011). Furthermore, presynaptic and postsynaptic actions are not mutually exclusive, and both may contribute.
Memory and Alzheimer’s disease
Presynaptic receptors, spontaneous release, and LTP
Presynaptic receptors and spontaneous release may also play important roles in mechanisms of memory in the hippocampus and entorhinal cortex, and in disruption of those mechanisms in Alzheimer’s disease (Fig. 3). LTP in both of those areas is thought to be involved in long-lasting or reference memory (Morris et al. 2003; Moser et al. 2008). Activation of D1 receptors in hippocampus produces an increase in spontaneous release of glutamate and plays an important role in LTP (see above). The D1 receptors involved in LTP have often been thought to be postsynaptic, but they might also be presynaptic. Similarly, activation of nACh receptors in hippocampus and EC produces an increase in spontaneous release of glutamate (Radcliffe and Dani 1998; Radcliffe et al. 1999; Liu et al. 2003; Sharma and Vijayaraghavan 2003) and activity-dependent LTP (Welsby et al. 2006, 2007; Kenney and Gould 2008; Lagostena et al. 2008; Tu et al. 2009; Gu and Yakel 2011; Gu et al. 2012), and blocking nAChRs reduces LTP (Yun et al. 2005). Thus presynaptic nACh receptors and spontaneous release might also contribute to LTP in those areas.
Interaction between presynaptic nACh receptors and Aβ
Alzheimer’s disease produces a devastating loss of memory that is thought to be due initially to synaptic dysfunction and later to synaptic and cell loss in hippocampus and EC (Palop and Mucke 2010). At the molecular level, these effects are due in part to toxicity caused by accumulation of β amyloid peptide (Aβ). Aβ is produced in response to activity and released in part at synaptic terminals (Kamenetz et al. 2003; Cirrito et al. 2005; Bero et al. 2011). A variety of evidence suggests that some of the effects of Aβ are caused by interactions with nACh receptors. Aβ binds to nACh receptors or associated molecules with high affinity (Wang et al. 2000; Small et al. 2007; Khan et al. 2010) and activates nACh receptors expressed in oocytes (Dineley et al. 2002). Aβ production is reduced by blocking nACh receptors, suggesting a positive feedback loop at synapses (Fig. 3A; Wei et al. 2010). In addition, intracellular accumulation of Aβ in Alzheimer’s disease may be due in part to binding of Aβ to nACh receptors and endocytosis (Nagele et al. 2002). Consistent with that idea, Aβ deposition is reduced in the entorhinal cortex of elderly people who smoke (Hellstrom-Lindahl et al. 2004; Court et al. 2005; Buckingham et al. 2009; Shimohama 2009; Srivareerat et al. 2011), perhaps due to competition of Aβ and nicotine for binding to nACh receptors. Thus the interaction of Aβ and nACh receptors may contribute to the pathology of Alzheimer’s disease in at least three ways: by modulating release of glutamate, Aβ production, and internalization of Aβ.
At very low (picomolar) concentrations Aβ acts as an agonist at nACh receptors (Khan et al. 2010) and can produce an increase in spontaneous release of glutamate (Abramov et al. 2009) and nAChR-dependent enhancements of both PTP and LTP (Puzzo et al. 2008) in hippocampus. Conversely, LTP is reduced in Aβ deficient animals, and that reduction can be rescued by nicotine (Wang et al. 2010; Puzzo et al. 2011).
At higher (nanomolar) concentrations or longer durations Aβ can produce decreases in synaptic transmission and LTP, and loss of glutamate receptors and spines. A variety of mechanisms have been proposed for these effects, including increased excitation of inhibitory interneurons, LTD of excitatory synapses, and neurotoxicity (Palop and Mucke 2010), each of which may contribute at different times and concentrations. Like the positive effects of Aβ, its negative effects depend on nACh and NMDA receptors (Welsby et al. 2007; Shankar et al. 2008; Dziewczapolski et al. 2009; Wei et al. 2010; Gu and Yakel 2011; Li et al. 2011a,b; Kessels et al. 2013). These results suggest that the negative effects may be due in part to excessive stimulation of nACh receptors and release of glutamate which, like Aβ, can produce potentiation, depression, or toxicity depending on its concentration. Consistent with that idea, the reduction in LTP by Aβ involves many of the same molecular pathways as glutamate-induced neurotoxicity (Wang and Qin 2010; Jo et al. 2011; Li et al. 2011a).
Alternatively, high Aβ can block nACh receptors (Liu et al. 2001; Chen et al. 2006; Srivareerat et al. 2011), and could thus produce reductions in spontaneous release and LTP. Consistent with that idea, nicotine or nACh agonists can “rescue” the deficits in LTP and memory produced by high Aβ (Chen et al. 2006; Welsby et al. 2007; Srivareerat et al. 2011). In either case, however, some of the negative effects of high Aβ would be due to interactions with nACh receptors.
Presynaptic receptors, spontaneous release, and persistent firing
Patients who develop Alzheimer’s disease also have earlier deficits in temporary or working memory, which is thought to involve persistent firing of neurons in EC during the delay interval of a working memory task (Suzuki et al. 1997; Young et al. 1997). Persistent firing could also contribute to the properties of grid cells in EC and the encoding of episodic memories (Hasselmo 2008; Hasselmo and Brandon 2008). Persistent firing of neurons in layers III and V of EC in vitro is due in part to activation of group I metabotropic glutamate (mGlu) receptors linked to the production of IP3, increased intracellular Ca2+, and modulation of transient receptor potential (TRP) channels (Hasselmo 2008; Yoshida et al. 2008). Similar processes also occur in CA1 pyramidal neurons (El-Hassar et al. 2011). Presynaptic receptors and spontaneous release may play important roles in these effects as well. Thus, activation of presynaptic nACh receptors enhances spontaneous release of glutamate from layer VI to layer V neurons in EC (Tu et al. 2009). As in Aplysia, the glutamate could act through group I mGlu receptors to increase postsynaptic Ca2+ and, in this case, modulate TRP channels (Fig. 3B). Binding of Aβ to the nACh receptors could interfere with these processes in Alzheimer’s disease.
Working memory, schizophrenia, and ADHD
Similarly, presynaptic receptors and spontaneous release could play an important role in persistent firing in PFC, which is thought to contribute to working memory as well (Goldman-Rakic 1995). Deficits in working memory are among the cognitive symptoms of schizophrenia and ADHD, and presynaptic nACh receptors and D1 dopamine receptors in PFC have been implicated in working memory and its dysfunction in schizophrenia and ADHD (Murphy et al. 1996; Goldman-Rakic et al. 2004; Young et al. 2007; Gamo et al. 2010; Radek et al. 2010). Because less is known about the role of nACh receptors, this review focuses on the possible role of D1 receptors.
Presynaptic D1 receptors are found in PFC (Paspalas and Goldman-Rakic 2005), where they produce a PKA-dependent enhancement of spontaneous release of glutamate (Wang et al. 2002). That enhancement might contribute to persistent firing of PFC neurons in two ways. First, D1 receptors act through PKA to produce activity- and NMDA-dependent E-LTP and L-LTP (Gurden et al. 2000; Otani et al. 2003; Huang et al. 2004; Matsuda et al. 2006), which might contribute to circuit mechanisms of persistent firing at PFC–PFC synapses (Fig. 4A; Paspalas and Goldman-Rakic 2005). Second, D1 receptors also act through PKA and mGlu receptors to modulate channels that contribute to a cell autonomous mechanism of persistent firing (Fig. 4B, Onn and Wang 2005; Witkowski et al. 2008; Sidiropoulou et al. 2009). In both cases presynaptic D1 receptors may enhance spontaneous release of glutamate, which then acts on postsynaptic NMDA or mGlu receptors. That effect would be analogous to the effect of presynaptic nACh receptors on persistent firing of neurons in EC (Fig. 3B), or of presynaptic 5HT receptors on postsynaptic mechanisms of facilitation in Aplysia (Fig. 1A).
Conclusions
Recent studies in Aplysia suggest that presynaptic receptors and spontaneous release contribute to a new form of synaptic plasticity that links presynaptic modulatory effects to postsynaptic mechanisms of activity-dependent plasticity, in the absence of activity (Jin et al. 2012a,b). This review has summarized evidence suggesting that presynaptic receptors, spontaneous release, and a similar hybrid form of plasticity may also be involved in a variety of functions in different mammalian brain areas, including reward in VTA, NAc, and PFC, reference memory in hippocampus and EC, and working memory in PFC and EC, and also in disorders that affect plasticity in those brain areas, including addiction, Alzheimer’s disease, schizophrenia, and ADHD. This novel form of plasticity could thus play widespread and important roles in mammals as well as in Aplysia.
This review is highly selective and does not include other transmitter systems, molecular pathways, and brain areas that are also important for these brain functions and disorders. In addition, a number of other, better established types of plasticity no doubt also play important roles. However, the different transmitters, pathways, brain areas, and types of plasticity are not mutually exclusive, and they could play complementary roles or act cooperatively with the mechanisms described here. For example, a large body of evidence indicates that Hebbian LTP (induced by coincident pre- and postsynaptic activity) plays an important role in reward and addiction in VTA (Kauer and Malenka 2007; Zellner and Ranaldi 2010). That mechanism could act cooperatively with the presynaptic modulatory mechanism described here, just as Hebbian LTP and activity-dependent presynaptic facilitation act cooperatively during conditioning in Aplysia (Antonov et al. 2003). Finally, although the evidence presented in this review suggests that these ideas are plausible, at the moment they are still speculative and remain to be tested experimentally. However, in so far as those tests reveal novel mechanisms of plasticity and its disruption in psychiatric disorders, they could also suggest novel treatment strategies.
Acknowledgments
I thank Ottavio Arancio, Eric Kandel, Christoph Kellendonk, Amir Levine, Eleanor Simpson, and Andrew Teich for their comments. Preparation of this article was supported by grants from the NIH (GM097502, NS083690) and Whitehall Foundation.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I 2009. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci 12: 1567–1576 [DOI] [PubMed] [Google Scholar]
- Antonov I, Antonova I, Kandel ER, Hawkins RD 2003. Activity-dependent presynaptic facilitation and Hebbian LTP are both required and interact during classical conditioning in Aplysia. Neuron 37: 135–147 [DOI] [PubMed] [Google Scholar]
- Antonov I, Ha T, Antonova I, Moroz LL, Hawkins RD 2007. Role of nitric oxide in classical conditioning of siphon withdrawal in Aplysia. J Neurosci 27: 10993–11002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonov I, Kandel ER, Hawkins RD 2010. Presynaptic and postsynaptic mechanisms of synaptic plasticity and metaplasticity during intermediate-term memory formation in Aplysia. J Neurosci 30: 5781–5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM, Bonci A 2008. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci 28: 9092–9100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF 2009. The emerging neurobiology of attention deficit hyperactivity disorder: The key role of the prefrontal association cortex. J Pediatr 154: I–S43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ary AW, Szumlinski KK 2007. Regional differences in the effects of withdrawal from repeated cocaine upon Homer and glutamate receptor expression: A two-species comparison. Brain Res 1184: 295–305 [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Smith RJ, Sartor GC, Moorman DE, Massi L, Tahsili-Fahadan P, Richardson KA 2010. Lateral hypothalamic orexin/hypocretin neurons: A role in reward-seeking and addiction. Brain Res 1314: 74–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins AL, Mashhoon Y, Kantak KM 2008. Hippocampal regulation of contextual cue-induced reinstatement of cocaine-seeking behavior. Pharmacol Biochem Behav 90: 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM 2011. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER 1999. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci 96: 5280–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Giustetto M, Huang YY, Hawkins RD, Kandel ER 2000. Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat Rev Neurosci 1: 11–20 [DOI] [PubMed] [Google Scholar]
- Bailey CH, Barco A, Hawkins RD, Kandel ER 2008. Molecular studies of learning and memory in Aplysia and hippocampus: A comparative analysis of implicit and explicit memory storage. In Learning and memory: A comprehensive reference (ed. Byrne J), pp. 11–29 Elsevier, Oxford, UK [Google Scholar]
- Barrot M, Sesack SR, Georges F, Pistis M, Hong S, Jhou TC 2012. Braking dopamine systems: A new GABA master structure for mesolimbic and nigrostriatal functions. J Neurosci 32: 14094–14101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Malenka RC 1994. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4: 389–399 [DOI] [PubMed] [Google Scholar]
- Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS 1995. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci 15: 7821–7836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci 14: 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Stern AL, Theberge FR, Cifani C, Koya E, Hope BT, Shaham Y 2011. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci 14: 420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME 2005. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25: 9144–9151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME 2007. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci 27: 10621–10635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouron A, Reuter H 1999. The D1 dopamine receptor agonist SKF-38393 stimulates the release of glutamate in the hippocampus. Neuroscience 94: 1063–1070 [DOI] [PubMed] [Google Scholar]
- Bowers MS, Chen BT, Bonci A 2010. AMPA receptor synaptic plasticity induced by psychostimulants: The past, present, and therapeutic future. Neuron 67: 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braha O, Dale N, Hochner B, Klein M, Abrams TW, Kandel ER 1990. Second messengers involved in the two processes of presynaptic facilitation that contribute to sensitization and dishabituation in Aplysia sensory neurons. Proc Natl Acad Sci 87: 2040–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A 2012. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76: 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Bellone C, Mameli M, Labouebe G, Bocklisch C, Balland B, Dahan L, Lujan R, Deisseroth K, Luscher C 2010. Drug-driven AMPA receptor redistribution mimicked by selective dopamine neuron stimulation. PloS One 5: e15870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, Lin Y, Suska A, Ishikawa M, Huang YH, et al. 2011. A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci 31: 8163–8174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham SD, Jones AK, Brown LA, Sattelle DB 2009. Nicotinic acetylcholine receptor signalling: Roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol Rev 61: 39–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ 1998. Regulation of cocaine reward by CREB. Science 282: 2272–2275 [DOI] [PubMed] [Google Scholar]
- Cassenaer S, Laurent G 2012. Conditional modulation of spike-timing-dependent plasticity for olfactory learning. Nature 482: 47–52 [DOI] [PubMed] [Google Scholar]
- Chen L, Yang CR 2002. Interaction of dopamine D1 and NMDA receptors mediates acute clozapine potentiation of glutamate EPSPs in rat prefrontal cortex. J Neurophysiol 87: 2324–2336 [DOI] [PubMed] [Google Scholar]
- Chen L, Yamada K, Nabeshima T, Sokabe M 2006. α7 Nicotinic acetylcholine receptor as a target to rescue deficit in hippocampal LTP induction in β-amyloid infused rats. Neuropharmacology 50: 254–268 [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A 2008. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48: 913–922 [DOI] [PubMed] [Google Scholar]
- Clark GA, Hawkins RD, Kandel ER 1994. Activity-dependent enhancement of presynaptic facilitation provides a cellular mechanism for the temporal specificity of classical conditioning in Aplysia. Learn Mem 1: 243–257 [PubMed] [Google Scholar]
- Cohen JY, Haesler S, Vong L, Lowell BB, Uchida N 2012. Neuron-type-specific signals for reward and punishment in the ventral tegmental area. Nature 482: 85–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, Klipec WD, Fowler MA, Ozkan ED 2006. A role for the subiculum in the brain motivation/reward circuitry. Behav Brain Res 74: 225–231 [DOI] [PubMed] [Google Scholar]
- Court JA, Johnson M, Religa D, Keverne J, Kalaria R, Jaros E, McKeith IG, Perry R, Naslund J, Perry EK 2005. Attenuation of Aβ deposition in the entorhinal cortex of normal elderly individuals associated with tobacco smoking. Neuropathol Appl Neurobiol 31: 522–535 [DOI] [PubMed] [Google Scholar]
- Cui RJ, Roberts BL, Zhao H, Zhu M, Appleyard SM 2012. Serotonin activates catecholamine neurons in the solitary tract nucleus by increasing spontaneous glutamate inputs. J Neurosci 32: 16530–16538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daberkow DP, Brown HD, Bunner KD, Kraniotis SA, Doellman MA, Ragozzino ME, Garris PA, Roitman MF 2013. Amphetamine paradoxically augments exocytotic dopamine release and phasic dopamine signals. J Neurosci 33: 452–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damez-Werno D, Laplant Q, Sun H, Scobie KN, Dietz DM, Walker IM, Koo JW, Vialou VF, Mouzon E, Russo SJ, et al. 2012. Drug experience epigenetically primes fosb gene inducibility in rat nucleus accumbens. J Neurosci 32: 10267–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Olmo N, Higuera-Matas A, Miguéns M, García-Lecumberri C, Borcel E, Solís JM, Ambrosio E 2006a. Hippocampal synaptic plasticity and water maze learning in cocaine self-administered rats. Ann N Y Acad Sci 1074: 427–437 [DOI] [PubMed] [Google Scholar]
- Del Olmo N, Miguéns M, Higuera-Matas A, Torres I, García-Lecumberri C, Solís JM, Ambrosio E 2006b. Enhancement of hippocampal long-term potentiation induced by cocaine self-administration is maintained during the extinction of this behavior. Brain Res 1116: 120–126 [DOI] [PubMed] [Google Scholar]
- Deng C, Li KY, Zhou C, Ye JH 2009. Ethanol enhances glutamate transmission by retrograde dopamine signaling in a postsynaptic neuron/synaptic bouton preparation from the ventral tegmental area. Neuropsychopharmacology 34: 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Davis GW 2009. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326: 1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz DM, Sun H, Lobo MK, Cahill ME, Chadwick B, Gao V, Koo JW, Mazei-Robison MS, Dias C, Maze I, et al. 2012. Rac1 is essential in cocaine-induced structural plasticity of nucleus accumbens neurons. Nat Neurosci 15: 891–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD 2002. β-Amyloid peptide activates α 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277: 25056–25061 [DOI] [PubMed] [Google Scholar]
- Dobi A, Seabold GK, Christensen CH, Bock R, Alvarez VA 2011. Cocaine-induced plasticity in the nucleus accumbens is cell specific and develops without prolonged withdrawal. J Neurosci 31: 1895–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin B, Doudnikoff E, Gonon F, Bloch B 2007. Differences in ultrastructural localization of dopaminergic D1 receptors between dorsal striatum and nucleus accumbens in the rat. Neurosci Lett 419: 273–277 [DOI] [PubMed] [Google Scholar]
- Dumitriu D, Laplant Q, Grossman YS, Dias C, Janssen WG, Russo SJ, Morrison JH, Nestler EJ 2012. Subregional, dendritic compartment, and spine subtype specificity in cocaine regulation of dendritic spines in the nucleus accumbens. J Neurosci 32: 6957–6966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF 2009. Deletion of the α 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. J Neurosci 29: 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Hagenston AM, D’Angelo LB, Yeckel MF 2011. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca2+ wave-dependent activation of SK and TRPC channels. J Physiol 589: 3211–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot LS, Hawkins RD, Kandel ER, Schacher S 1994. Pairing-specific, activity-dependent presynaptic facilitation at Aplysia sensory-motor neuron synapses in isolated cell culture. J Neurosci 14: 368–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET 2009. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol 101: 2290–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW 2010. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65: 7–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, Akers CA, Clinton SM, Phillips PE, Akil H 2011. A selective role for dopamine in stimulus-reward learning. Nature 469: 53–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatscher-Bader T, Zuvela N, Landis N, Wilce PA 2008. Smoking and alcoholism target genes associated with plasticity and glutamate transmission in the human ventral tegmental area. Hum Mol Genet 17: 38–51 [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA 2003. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci 6: 968–973 [DOI] [PubMed] [Google Scholar]
- Fole A, González-Martín C, Huarte C, Alguacil LF, Ambrosio E, Del Olmo N 2011. Effects of chronic cocaine administration on spatial learning and hippocampal spine density in two genetically different strains of rats. Neurobiol Learn Mem 95: 491–497 [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW 2006. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52: 663–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW 2009. A presynaptic homeostatic signaling system composed of the Eph receptor, Ephexin, Cdc42, and Cav2.1 calcium channels. Neuron 61: 556–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WM, Brebner K, Lynch WJ, Robertson DJ, Roberts DC, Vrana KE 2001. Cocaine-responsive gene expression changes in rat hippocampus. Neuroscience 108: 371–380 [DOI] [PubMed] [Google Scholar]
- Frey U, Matthies H, Reymann KG, Matthies H 1991. The effect of dopaminergic D1 receptor blockade during tetanization on the expression of long-term potentiation in the rat CA1 region in vitro. Neurosci Lett 129: 111–114 [DOI] [PubMed] [Google Scholar]
- Gamo NJ, Wang M, Arnsten AF 2010. Methylphenidate and atomoxetine enhance prefrontal function through α2-adrenergic and dopamine D1 receptors. J Am Acad Child Adolesc Psychiatry 49: 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Jin Y, Yang K, Zhang D, Lukas RJ, Wu J 2010. Mechanisms involved in systemic nicotine-induced glutamatergic synaptic plasticity on dopamine neurons in the ventral tegmental area. J Neurosci 30: 13814–13825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garduño J, Galindo-Charles L, Jiménez-Rodríguez J, Galarraga E, Tapia D, Mihailescu S, Hernandez-Lopez S 2012. Presynaptic α4β2 nicotinic acetylcholine receptors increase glutamate release and serotonin neuron excitability in the dorsal raphe nucleus. J Neurosci 32: 15148–15157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N 1992. Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron 9: 479–489 [DOI] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW 2013a. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 77: 867–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, Kalivas PW 2013b. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci 110: 9124–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS 1995. Cellular basis of working memory. Neuron 14: 477–485 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV 2004. Targeting the dopamine D1 receptor in schizophrenia: Insights for cognitive dysfunction. Psychopharmacology 174: 3–16 [DOI] [PubMed] [Google Scholar]
- Gourley SL, Olevska A, Warren MS, Taylor JR, Koleske AJ 2012. Arg kinase regulates prefrontal dendritic spine refinement and cocaine-induced plasticity. J Neurosci 32: 2314–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA 2010. Dopamine system dysregulation by the ventral subiculum as the common pathophysiological basis for schizophrenia psychosis, psychostimulant abuse, and stress. Neurotoxicol Res 18: 367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner P, Svensson TH 2000. Nicotine-induced excitation of midbrain dopamine neurons in vitro involves ionotropic glutamate receptor activation. Synapse 38: 1–9 [DOI] [PubMed] [Google Scholar]
- Gu Z, Yakel JL 2011. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron 71: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, Yakel JL 2012. Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J Neurosci 32: 12337–12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Zhang R, Xu Y, Li S 2009. Cocaine withdrawal enhances long-term potentiation in rat hippocampus via changing the activity of corticotropin-releasing factor receptor subtype 2. Neuroscience 161: 665–670 [DOI] [PubMed] [Google Scholar]
- Gurden H, Takita M, Jay TM 2000. Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J Neurosci 20: RC106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton GF, Whitcher LT, Klintsova AY 2010. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC layer II/III pyramidal neurons. Synapse 64: 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME 2008. Grid cell mechanisms and function: Contributions of entorhinal persistent spiking and phase resetting. Hippocampus 18: 1213–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Brandon MP 2008. Linking cellular mechanisms to behavior: Entorhinal persistent spiking and membrane potential oscillations may underlie path integration, grid cell firing, and episodic memory. Neural Plast 2008: 658323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD 1981. Interneurons involved in mediation and modulation of gill-withdrawal reflex in Aplysia. III. Identified facilitating neurons increase Ca2+ current in sensory neurons. J Neurophysiol 45: 327–339 [DOI] [PubMed] [Google Scholar]
- Hawkins RD 1989. A biologically based computational model for several simple forms of learning in Aplysia. Psychol Learn Motiv 23: 65–108 [Google Scholar]
- Hawkins RD, Schacher S 1989. Identified facilitator neurons L29 and L28 are excited by cutaneous stimuli used in dishabituation, sensitization, and classical conditioning of Aplysia. J Neurosci 9: 4236–4245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Castellucc VF, Kandel ER 1981. Interneurons involved in mediation and modulation of gill-withdrawal reflex in Aplysia. I. Identification and characterization. J Neurophysiol 45: 304–314 [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Abrams TW, Carew TJ, Kandel ER 1983. A cellular mechanism of classical conditioning in Aplysia: Activity-dependent amplification of presynaptic facilitation. Science 219: 400–405 [DOI] [PubMed] [Google Scholar]
- Hawkins RD, Bailey CH, Kandel ER 2010. The neuronal circuit for simple forms of learning in Aplysia. In Handbook of brain microcircuits (ed. Shepherd G, Grillner S), pp. 485–502 Oxford University Press, New York [Google Scholar]
- Hellstrom-Lindahl E, Mousavi M, Ravid R, Nordberg A 2004. Reduced levels of αβ 40 and Aβ 42 in brains of smoking controls and Alzheimer’s patients. Neurobiol Dis 15: 351–360 [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER 1995. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci 92: 2446–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Bach ME, Lipp HP, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind JM, Mulligan R, et al. 1996. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci 93: 8699–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER 2004. Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci 101: 3236–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lin HJ, Hsu KS 2007. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex 17: 1877–1888 [DOI] [PubMed] [Google Scholar]
- Huang S, Trevino M, He K, Ardiles A, Pasquale R, Guo Y, Palacios A, Huganir R, Kirkwood A 2012. Pull-push neuromodulation of LTP and LTD enables bidirectional experience-induced synaptic scaling in visual cortex. Neuron 73: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ 2006. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598 [DOI] [PubMed] [Google Scholar]
- Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, Georges F 2011. Neuronal circuits underlying acute morphine action on dopamine neurons. Proc Natl Acad Sci 108: 16446–16450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Hawkins RD 2003. Presynaptic and postsynaptic mechanisms of a novel form of homosynaptic potentiation at Aplysia sensory-motor neuron synapses. J Neurosci 23: 7288–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Kandel ER, Hawkins RD 2011a. Whereas short-term facilitation is presynaptic, intermediate-term facilitation involves both presynaptic and postsynaptic protein kinases and protein synthesis. Learn Mem 18: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Yang K, Wang H, Wu J 2011b. Exposure of nicotine to ventral tegmental area slices induces glutamatergic synaptic plasticity on dopamine neurons. Synapse 65: 332–338 [DOI] [PubMed] [Google Scholar]
- Jin I, Puthanveettil S, Udo H, Karl K, Kandel ER, Hawkins RD 2012a. Spontaneous transmitter release is critical for the induction of long-term and intermediate-term facilitation in Aplysia. Proc Natl Acad Sci 109: 9131–9136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin I, Udo H, Rayman JB, Puthanveettil S, Kandel ER, Hawkins RD 2012b. Spontaneous transmitter release recruits postsynaptic mechanisms of long-term and intermediate-term facilitation in Aplysia. Proc Natl Acad Sci 109: 9137–9142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Whitcomb DJ, Olsen KM, Kerrigan TL, Lo SC, Bru-Mercier G, Dickinson B, Scullion S, Sheng M, Collingridge G, et al. 2011. Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat Neurosci 14: 545–547 [DOI] [PubMed] [Google Scholar]
- Jones IW, Wonnacott S 2004. Precise localization of α7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci 24: 11244–11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P 1995. D1 receptors modulate glutamate transmission in the ventral tegmental area. J Neurosci 15: 5379–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R 2003. APP processing and synaptic function. Neuron 37: 925–937 [DOI] [PubMed] [Google Scholar]
- Kauer JA 2004. Learning mechanisms in addiction: Synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol 66: 447–475 [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC 2007. Synaptic plasticity and addiction. Nat Rev Neurosci 8: 844–858 [DOI] [PubMed] [Google Scholar]
- Keleta YB, Martinez JL 2012. Brain circuits of methamphetamine place reinforcement learning: The role of the hippocampus–VTA loop. Brain Behav 2: 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempadoo KA, Tourino C, Cho SL, Magnani F, Leinninger GM, Stuber GD, Zhang F, Myers MG, Deisseroth K, de Lecea L, et al. 2013. Hypothalamic neurotensin projections promote reward by enhancing glutamate transmission in the VTA. J Neurosci 33: 7618–7626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ 2008. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol 38: 101–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Nabav S, Malinow R 2013. Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc Natl Acad Sci 110: 4033–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan GM, Tong M, Jhun M, Arora K, Nichols RA 2010. β-Amyloid activates presynaptic α7 nicotinic acetylcholine receptors reconstituted into a model nerve cell system: Involvement of lipid rafts. Eur J Neurosci 31: 788–796 [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Okada K 2007. Reward prediction error computation in the pedunculopontine tegmental nucleus neurons. Ann N Y Acad Sci 1104: 310–323 [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ 2007. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27: 7921–7928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Li SM, Wood WH, McCoy MT, Prabhu VV, Becker KG, Katz JL, Cadet JL 2008. Transcriptional responses to reinforcing effects of cocaine in the rat hippocampus and cortex. Genes Brain Behav 7: 193–202 [DOI] [PubMed] [Google Scholar]
- Lagostena L, Trocme-Thibierge C, Morain P, Cherubini E 2008. The partial α7 nicotine acetylcholine receptor agonist S 24795 enhances long-term potentiation at CA3–CA1 synapses in the adult mouse hippocampus. Neuropharmacology 54: 676–685 [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC 2011. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70: 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC 2012. Input-specific control of reward and aversion in the ventral tegmental area. Nature 491: 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasseter HC, Xie X, Ramirez DR, Fuchs RA 2010. Sub-region specific contribution of the ventral hippocampus to drug context-induced reinstatement of cocaine-seeking behavior in rats. Neuroscience 171: 830–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P 2006. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci 103: 3399–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-C, Yasuda R, Ehlers MD 2010. Metaplasticity at single glutamatergic synapses. Neuron 66: 859–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon N, Manahan-Vaughan D 2006. Dopamine D1/D5 receptors gate the acquisition of novel information through hippocampal long-term potentiation and long-term depression. J Neurosci 26: 7723–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Cullen WK, Anwyl R, Rowan MJ 2003. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nat Neurosci 6: 526–531 [DOI] [PubMed] [Google Scholar]
- Li KY, Xiao C, Xiong M, Delphin E, Ye JH 2008. Nanomolar propofol stimulates glutamate transmission to dopamine neurons: A possible mechanism of abuse potential? J Pharmacol Exp Ther 325: 165–174 [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ 2011a. Soluble αβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31: 6627–6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SF, Wu MN, Wang XH, Yuan L, Yang D, Qi JS 2011b. Requirement of α7 nicotinic acetylcholine receptors for amyloid β protein-induced depression of hippocampal long-term potentiation in CA1 region of rats in vivo. Synapse 65: 1136–1143 [DOI] [PubMed] [Google Scholar]
- Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, Sacktor TC, Shaham Y, Lu L 2011c. Inhibition of PKMζ in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci 31: 5436–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata SC, Pierce RC 2003. The roles of calcium/calmodulin-dependent and Ras/mitogen-activated protein kinases in the development of psychostimulant-induced behavioral sensitization. J Neurochem 85: 14–22 [DOI] [PubMed] [Google Scholar]
- Lin XY, Glanzman DL 1994. Hebbian induction of long-term potentiation of Aplysia sensorimotor synapses: Partial requirement for activation of an NMDA-related receptor. Proc Biol Sci 255: 215–221 [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA 2005. The hippocampal–VTA loop: Controlling the entry of information into long-term memory. Neuron 46: 703–713 [DOI] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK 2001. β-Amyloid peptide blocks the response of α 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci 98: 4734–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Yang S, Zhang YX, Liu CH 2003. Presynaptic α-7 nicotinic acetylcholine receptors modulate excitatory synaptic transmission in hippocampal neurons. Sheng Li Xue Bao 55: 731–735 [PubMed] [Google Scholar]
- Luo AH, Tahsili-Fahadan P, Wise RA, Lupica CR, Aston-Jones G 2011. Linking context with reward: A functional circuit from hippocampal CA3 to ventral tegmental area. Science 333: 353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Malenka RC 2011. Drug-evoked synaptic plasticity in addiction: From molecular changes to circuit remodeling. Neuron 69: 650–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M, Mhillaj E, Matheos DP, Palmery M, Wood MA 2011. CBP in the nucleus accumbens regulates cocaine-induced histone acetylation and is critical for cocaine-associated behaviors. J Neurosci 31: 16941–16948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS 2000. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27: 349–357 [DOI] [PubMed] [Google Scholar]
- Mao D, Gallagher K, McGehee DS 2011. Nicotine potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J Neurosci 31: 6710–6720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fardon R, Ciccocioppo R, Aujla H, Weiss F 2008. The dorsal subiculum mediates the acquisition of conditioned reinstatement of cocaine-seeking. Neuropsychopharmacology 33: 1827–1834 [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Marzo A, Otani S 2006. The presence of background dopamine signal converts long-term synaptic depression to potentiation in rat prefrontal cortex. J Neurosci 26: 4803–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ 2011. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci 108: 3035–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW 2003. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan TM, Patzlaff NE, Grady SR, Heinemann SF, Booker TK 2011. α4β2 nicotinic acetylcholine receptors on dopaminergic neurons mediate nicotine reward and anxiety relief. J Neurosci 31: 10891–10902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Neisewander JL 2003. Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. Neuroreport 14: 2127–2131 [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Speer CM, Neisewander JL 2006. Dorsal hippocampus inhibition disrupts acquisition and expression, but not consolidation, of cocaine conditioned place preference. Behav Neurosci 120: 401–412 [DOI] [PubMed] [Google Scholar]
- Montague PR, Dayan P, Sejnowski TJ 1996. A framework for mesencephalic dopamine systems based on predictive Hebbian learning. J Neurosci 16: 1936–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague PR, Hyman SE, Cohen JD 2004. Computational roles for dopamine in behavioural control. Nature 431: 760–767 [DOI] [PubMed] [Google Scholar]
- Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O’Carroll C 2003. Elements of a neurobiological theory of the hippocampus: The role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci 358: 773–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Kropff E, Moser MB 2008. Place cells, grid cells, and the brain’s spatial representation system. Annu Rev Neurosci 31: 69–89 [DOI] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW 2011. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci 108: 385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GG, Glanzman DL 1997. Mediation of classical conditioning in Aplysia californica by long-term potentiation of sensorimotor synapses. Science 278: 467–471 [DOI] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AF, Goldman-Rakic PS, Roth RH 1996. Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Natl Acad Sci 93: 1325–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele RG, D’Andrea MR, Anderson WJ, Wang HY 2002. Intracellular accumulation of β-amyloid(1-42) in neurons is facilitated by the α 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110: 199–211 [DOI] [PubMed] [Google Scholar]
- Nai Q, Li S, Wang SH, Liu J, Lee FJ, Frankland PW, Liu F 2010. Uncoupling the D1-N-methyl-D-aspartate (NMDA) receptor complex promotes NMDA-dependent long-term potentiation and working memory. Biol Psychiatry 67: 246–254 [DOI] [PubMed] [Google Scholar]
- Navakkode S, Sajikumar S, Frey JU 2007. Synergistic requirements for the induction of dopaminergic D1/D5-receptor-mediated LTP in hippocampal slices of rat CA1 in vitro. Neuropharmacology 52: 1547–1554 [DOI] [PubMed] [Google Scholar]
- Navakkode S, Sajikumar S, Sacktor TC, Frey JU 2010. Protein kinase Mζ is essential for the induction and maintenance of dopamine-induced long-term potentiation in apical CA1 dendrites. Learn Mem 17: 605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navakkode S, Sajikumar S, Korte M, Soong TW 2012. Dopamine induces LTP differentially in apical and basal dendrites through BDNF and voltage-dependent calcium channels. Learn Mem 19: 294–299 [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, Ossipov MH, King T, Fields HL, Porreca F 2012. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci 109: 20709–20713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC 1998. Modulation of synaptic transmission by dopamine and norepinephrine in ventral but not dorsal striatum. J Neurophysiol 79: 1768–1776 [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C 2008. Amyloid β oligomers (A β(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 28: 788–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomikos GG, Schilstrom B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH 2000. Role of α7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res 113: 97–103 [DOI] [PubMed] [Google Scholar]
- Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET 2013. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci 33: 6990–7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA 2007. Opioids block long-term potentiation of inhibitory synapses. Nature 446: 1086–1090 [DOI] [PubMed] [Google Scholar]
- Okada K, Toyama K, Inoue Y, Isa T, Kobayashi Y 2009. Different pedunculopontine tegmental neurons signal predicted and actual task rewards. J Neurosci 29: 4858–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson EB, Beckert MV, Morra JT, Lansink CS, Cachope R, Abdullah RA, Loriaux AL, Schetters D, Pattij T, Roitman MF, et al. 2012. Endocannabinoids shape accumbal encoding of cue-motivated behavior via CB1 receptor activation in the ventral tegmentum. Neuron 73: 360–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onn SP, Wang XB 2005. Differential modulation of anterior cingulate cortical activity by afferents from ventral tegmental area and mediodorsal thalamus. Eur J Neurosci 21: 2975–2992 [DOI] [PubMed] [Google Scholar]
- Ortiz O, Delgado-Garcia JM, Espadas I, Bahi A, Trullas R, Dreyer JL, Gruart A, Moratalla R 2010. Associative learning and CA3-CA1 synaptic plasticity are impaired in D1R null, Drd1a–/– mice and in hippocampal siRNA silenced Drd1a mice. J Neurosci 30: 12288–12300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani S, Daniel H, Roisin MP, Crepel F 2003. Dopaminergic modulation of long-term synaptic plasticity in rat prefrontal neurons. Cereb Cortex 13: 1251–1256 [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE 1996. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci 16: 7478–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L 2010. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 13: 812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Luscher C 2012. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481: 71–75 [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Goldman-Rakic PS 2005. Presynaptic D1 dopamine receptors in primate prefrontal cortex: Target-specific expression in the glutamatergic synapse. J Neurosci 25: 1260–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MF, Gabach LA, Almiron RS, Carlini VP, De Barioglio SR, Ramirez OA 2010. Different chronic cocaine administration protocols induce changes on dentate gyrus plasticity and hippocampal dependent behavior. Synapse 64: 742–753 [DOI] [PubMed] [Google Scholar]
- Perrotti LI, Weaver RR, Robison B, Renthal W, Maze I, Yazdani S, Elmore RG, Knapp DJ, Selley DE, Martin BR, et al. 2008. Distinct patterns of ΔFosB induction in brain by drugs of abuse. Synapse 62: 358–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW 1996. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchers KK, Vialou V, Nestler EJ, Laviolette SR, Lehman MN, Coolen LM 2013. Natural and drug rewards act on common neural plasticity mechanisms with ΔFosB as a key mediator. J Neurosci 33: 3434–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povysheva NV, Johnson JW 2012. Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol 107: 2232–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O 2008. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28: 14537–14545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, et al. 2011. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann Neurol 69: 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA 1998. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci 18: 7075–7083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA 1999. Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci 868: 591–610 [DOI] [PubMed] [Google Scholar]
- Radek RJ, Kohlhaas KL, Rueter LE, Mohler EG 2010. Treating the cognitive deficits of schizophrenia with α4β2 neuronal nicotinic receptor agonists. Curr Pharm Des 16: 309–322 [DOI] [PubMed] [Google Scholar]
- Ramirez DR, Bell GH, Lasseter HC, Xie X, Traina SA, Fuchs RA 2009. Dorsal hippocampal regulation of memory reconsolidation processes that facilitate drug context-induced cocaine-seeking behavior in rats. Eur J Neurosci 30: 901–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Sun WL, Jiao H, Zhang D, Kong H, Wang X, Xu M 2010. Dopamine D1 and N-methyl-D-aspartate receptors and extracellular signal-regulated kinase mediate neuronal morphological changes induced by repeated cocaine administration. Neuroscience 168: 48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA 1978. Some implications of a cognitive perspective on Pavlovian conditioning. In Cognitive processes in animal behavior (ed. Hulse SH, et al. ), pp. 15–50 Erlbaum, Hillsdale, NJ [Google Scholar]
- Rescorla RA, Wagner AR 1972. A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and non-reinforcement. In Classical conditioning II: Current research and theory (ed. Black AH, Prokasy WF), pp. 64–99 Appleton-Century-Crofts, New York [Google Scholar]
- Robinson TE, Kolb B 1997. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci 17: 8491–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B 1999. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci 11: 1598–1604 [DOI] [PubMed] [Google Scholar]
- Robinson TE, Gorny G, Mitton E, Kolb B 2001. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse 39: 257–266 [DOI] [PubMed] [Google Scholar]
- Robison AJ, Vialou V, Mazei-Robison M, Feng J, Kourrich S, Collins M, Wee S, Koob G, Turecki G, Neve R, et al. 2013. Behavioral and structural responses to chronic cocaine require a feedforward loop involving ΔFosB and calcium/calmodulin-dependent protein kinase II in the nucleus accumbens shell. J Neurosci 33: 4295–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogge GA, Singh H, Dang R, Wood MA 2013. HDAC3 is a negative regulator of cocaine-context-associated memory formation. J Neurosci 33: 6623–6632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M 2009. Dopamine controls persistence of long-term memory storage. Science 325: 1017–1020 [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC 2003. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582 [DOI] [PubMed] [Google Scholar]
- Sarti F, Borgland SL, Kharazia VN, Bonci A 2007. Acute cocaine exposure alters spine density and long-term potentiation in the ventral tegmental area. Eur J Neurosci 26: 749–756 [DOI] [PubMed] [Google Scholar]
- Schacher S, Wu F, Sun ZY 1997. Pathway-specific synaptic plasticity: Activity-dependent enhancement and suppression of long-term heterosynaptic facilitation at converging inputs on a single target. J Neurosci 17: 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W 2011. Potential vulnerabilities of neuronal reward, risk, and decision mechanisms to addictive drugs. Neuron 69: 603–617 [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR 1997. A neural substrate of prediction and reward. Science 275: 1593–1599 [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. 2008. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14: 837–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S 2003. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38: 929–939 [DOI] [PubMed] [Google Scholar]
- Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW 2011. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci 108: 19407–19412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S 2009. Nicotinic receptor-mediated neuroprotection in neurodegenerative disease models. Biol Pharm Bull 32: 332–336 [DOI] [PubMed] [Google Scholar]
- Sidiropoulou K, Lu FM, Fowler MA, Xiao R, Phillips C, Ozkan ED, Zhu MX, White FJ, Cooper DC 2009. Dopamine modulates an mGluR5-mediated depolarization underlying prefrontal persistent activity. Nat Neurosci 12: 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Maksel D, Kerr ML, Ng J, Hou X, Chu C, Mehrani H, Unabia S, Azari MF, Loiacono R, et al. 2007. The β-amyloid protein of Alzheimer’s disease binds to membrane lipids but does not bind to the α7 nicotinic acetylcholine receptor. J Neurochem 101: 1527–1538 [DOI] [PubMed] [Google Scholar]
- Srivareerat M, Tran TT, Salim S, Aleisa AM, Alkadhi KA 2011. Chronic nicotine restores normal Aβ levels and prevents short-term memory and E-LTP impairment in Aβ rat model of Alzheimer’s disease. Neurobiol Aging 32: 834–844 [DOI] [PubMed] [Google Scholar]
- Stramiello M, Wagner JJ 2008. D1/5 receptor-mediated enhancement of LTP requires PKA, Src family kinases, and NR2B-containing NMDARs. Neuropharmacology 55: 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stramiello M, Wagner JJ 2010. Cocaine enhancement of long-term potentiation in the CA1 region of rat hippocampus: Lamina-specific mechanisms of action. Synapse 64: 644–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Marcotte ER, Srivastava LK 2001. Differential changes in synaptic terminal protein expression between nucleus accumbens core and shell in the amphetamine-sensitized rat. Brain Res 901: 175–183 [DOI] [PubMed] [Google Scholar]
- Sulzer D 2011. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69: 628–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME 2005. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci 25: 7342–7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zhou L, Quinones-Jenab V, Jenab S 2009. Cocaine effects on dopamine and NMDA receptors interactions in the striatum of Fischer rats. Brain Res Bull 80: 377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D, Neve RL, Zachariou V, Shen L, Nestler EJ 2012. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci 32: 17454–17464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri RE, Schultz W 2001. Temporal difference model reproduces anticipatory neural activity. Neural Comput 13: 841–862 [DOI] [PubMed] [Google Scholar]
- Suska A, Lee BR, Huang YH, Dong Y, Schlüter OM 2013. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci 110: 713–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto N, Elmer GI, Wang B, You ZB, Wise RA 2013. Bidirectional modulation of cocaine expectancy by phasic glutamate fluctuations in the nucleus accumbens. J Neurosci 33: 9050–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RS, Barto AG 1990. Time derivative models of Pavlovian reinforcement. In Learning and computational neuroscience: Foundations of adaptive networks (ed. Gabriel M, Moore J), pp. 539–602 MIT Press, Cambridge, MA [Google Scholar]
- Sutton MA, Wall NR, Aakalu G, Schuman EM 2004. Regulation of dendritic protein synthesis by miniature synaptic events. Science 304: 1979–1983 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM 2006. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125: 785–799 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM 2007. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron 55: 648–661 [DOI] [PubMed] [Google Scholar]
- Suzuki WA, Miller EK, Desimone R 1997. Object and place memory in the macaque entorhinal cortex. J Neurophysiol 78: 1062–1081 [DOI] [PubMed] [Google Scholar]
- Swanson-Park JL, Coussens CM, Mason-Parker SE, Raymond CR, Hargreaves EL, Dragunow M, Cohen AS, Abraham WC 1999. A double dissociation within the hippocampus of dopamine D1/D5 receptor and β-adrenergic receptor contributions to the persistence of long-term potentiation. Neuroscience 92: 485–497 [DOI] [PubMed] [Google Scholar]
- Swant J, Chirwa S, Stanwood G, Khoshbouei H 2010. Methamphetamine reduces LTP and increases baseline synaptic transmission in the CA1 region of mouse hippocampus. PLoS One 5: e11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW 2012. Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron 73: 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AM, Gosnell BA, Wagner JJ 2002. Enhancement of long-term potentiation in the rat hippocampus following cocaine exposure. Neuropharmacology 42: 1039–1042 [DOI] [PubMed] [Google Scholar]
- Thompson AM, Swant J, Gosnell BA, Wagner JJ 2004. Modulation of long-term potentiation in the rat hippocampus following cocaine self-administration. Neuroscience 127: 177–185 [DOI] [PubMed] [Google Scholar]
- Tropea TF, Kosofsky BE, Rajadhyaksha AM 2008. Enhanced CREB and DARPP-32 phosphorylation in the nucleus accumbens and CREB, ERK, and GluR1 phosphorylation in the dorsal hippocampus is associated with cocaine-conditioned place preference behavior. J Neurochem 106: 1780–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K 2009. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science 324: 1080–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B, Gu Z, Shen JX, Lamb PW, Yakel JL 2009. Characterization of a nicotine-sensitive neuronal population in rat entorhinal cortex. J Neurosci 29: 10436–10448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A 2001. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587 [DOI] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL 2001. Relapse to cocaine-seeking after hippocampal θ burst stimulation. Science 292: 1175–1178 [DOI] [PubMed] [Google Scholar]
- Wang Y, Qin ZH 2010. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 15: 1382–1402 [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB 2000. β-Amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem 275: 5626–5632 [DOI] [PubMed] [Google Scholar]
- Wang Z, Feng XQ, Zheng P 2002. Activation of presynaptic D1 dopamine receptors by dopamine increases the frequency of spontaneous excitatory postsynaptic currents through protein kinase A and protein kinase C in pyramidal cells of rat prelimbic cortex. Neuroscience 112: 499–508 [DOI] [PubMed] [Google Scholar]
- Wang BW, Liao WN, Chang CT, Wang SJ 2006. Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse 59: 491–501 [DOI] [PubMed] [Google Scholar]
- Wang H, Song L, Lee A, Laird F, Wong PC, Lee HK 2010. Mossy fiber long-term potentiation deficits in BACE1 knock-outs can be rescued by activation of α7 nicotinic acetylcholine receptors. J Neurosci 30: 13808–13813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LP, Li F, Wang D, Xie K, Wang D, Shen X, Tsien JZ 2011. NMDA receptors in dopaminergic neurons are crucial for habit learning. Neuron 72: 1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, Bamford NS 2012. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. J Physiol 590: 3743–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R 2010. Amyloid β from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 13: 190–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby P, Rowan M, Anwyl R 2006. Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. Eur J Neurosci 24: 3109–3118 [DOI] [PubMed] [Google Scholar]
- Welsby PJ, Rowan MJ, Anwyl R 2007. β-Amyloid blocks high frequency stimulation induced LTP but not nicotine enhanced LTP. Neuropharmacology 53: 188–195 [DOI] [PubMed] [Google Scholar]
- Williams S, Mmbaga N, Chirwa S 2006. Dopaminergic D1 receptor agonist SKF 38393 induces GAP-43 expression and long-term potentiation in hippocampus in vivo. Neurosci Lett 402: 46–50 [DOI] [PubMed] [Google Scholar]
- Wise RA 2005. Forebrain substrates of reward and motivation. J Comp Neurol 493: 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissman AM, McCollum AF, Huang GZ, Nikrodhanond AA, Woolley CS 2011. Sex differences and effects of cocaine on excitatory synapses in the nucleus accumbens. Neuropharmacology 61: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkowski G, Szulczyk B, Rola R, Szulczyk P 2008. D(1) dopaminergic control of G protein-dependent inward rectifier K+ (GIRK)-like channel current in pyramidal neurons of the medial prefrontal cortex. Neuroscience 155: 53–63 [DOI] [PubMed] [Google Scholar]
- Witten IB, Steinberg EE, Lee SY, Davidson TJ, Zalocusky KA, Brodsky M, Yizhar O, Cho SL, Gong S, Ramakrishnan C, et al. 2011. Recombinase-driver rat lines: Tools, techniques, and optogenetic application to dopamine-mediated reinforcement. Neuron 72: 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC III, Li KY, Krnjevic K, Zhou C, Ye JH 2009. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology 34: 307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Ramirez DR, Lasseter HC, Fuchs RA 2010. Effects of mGluR1 antagonism in the dorsal hippocampus on drug context-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology 208: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SN 1999. Presynaptic involvement of nitric oxide in dopamine D1/D5 receptor-induced sustained enhancement of synaptic currents mediated by ionotropic glutamate receptors in the rat hippocampus. Neurosci Lett 270: 87–90 [DOI] [PubMed] [Google Scholar]
- Yeomans J. 2010. Rewarding brain stimulation. In Encyclopedia of Behavioral Neuroscience (ed. Koob GF, et al. ), pp. 154–160 Academic Press, Oxford, UK [Google Scholar]
- Yoshida M, Fransen E, Hasselmo ME 2008. mGluR-dependent persistent firing in entorhinal cortex layer III neurons. Eur J Neurosci 28: 1116–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CE, Yang CR 2005. Dopamine D1-like receptor modulates layer- and frequency-specific short-term synaptic plasticity in rat prefrontal cortical neurons. Eur J Neurosci 21: 3310–3320 [DOI] [PubMed] [Google Scholar]
- Young BJ, Otto T, Fox GD, Eichenbaum H 1997. Memory representation within the parahippocampal region. J Neurosci 17: 5183–5195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey J 2007. Impaired attention is central to the cognitive deficits observed in α 7 deficient mice. Eur Neuropsychopharmacol 17: 145–155 [DOI] [PubMed] [Google Scholar]
- Yovell Y, Abrams TW 1992. Temporal asymmetry in activation of Aplysia adenylyl cyclase by calcium and transmitter may explain temporal requirements of conditioning. Proc Natl Acad Sci 89: 6526–6530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SH, Park KA, Sullivan P, Pasternak JF, Ladu MJ, Trommer BL 2005. Blockade of nicotinic acetylcholine receptors suppresses hippocampal long-term potentiation in wild-type but not ApoE4 targeted replacement mice. J Neurosci Res 82: 771–777 [DOI] [PubMed] [Google Scholar]
- Zellner MR, Ranaldi R 2010. How conditioned stimuli acquire the ability to activate VTA dopamine cells: A proposed neurobiological component of reward-related learning. Neurosci Biobehav Rev 34: 769–780 [DOI] [PubMed] [Google Scholar]
- Zhang S-Y, Xu M, Miao Q-L, Poo M-M, Zhang X-H 2009. Endocannabinoid-dependent homeostatic regulation of inhibitory synapses by miniature excitatory synaptic activities. J Neurosci 29: 13222–13231 [DOI] [PMC free article] [PubMed] [Google Scholar]