

Abstract
Understanding drivers of genetic diversity at the major histocompatibility complex (MHC) is vitally important for predicting how vertebrate immune defence might respond to future selection pressures and for preserving immunogenetic diversity in declining populations. Parasite-mediated selection is believed to be the major selective force generating MHC polymorphism, and while MHC-based mating preferences also exist for multiple species including humans, the general importance of mate choice is debated. To investigate the contributions of parasitism and sexual selection in explaining among-species variation in MHC diversity, we applied comparative methods and meta-analysis across 112 mammal species, including carnivores, bats, primates, rodents and ungulates. We tested whether MHC diversity increased with parasite richness and relative testes size (as an indicator of the potential for mate choice), while controlling for phylogenetic autocorrelation, neutral mutation rate and confounding ecological variables. We found that MHC nucleotide diversity increased with parasite richness for bats and ungulates but decreased with parasite richness for carnivores. By contrast, nucleotide diversity increased with relative testes size for all taxa. This study provides support for both parasite-mediated and sexual selection in shaping functional MHC polymorphism across mammals, and importantly, suggests that sexual selection could have a more general role than previously thought.
Keywords: major histocompatibility complex, mate choice, parasite-mediated selection, parasite richness, phylogenetic comparative methods, meta-analysis
1. Introduction
A significant fraction of the mammal genome is dedicated to immune defence, and immune genes are well known for their genetic variability [1,2]. Parasites have long been viewed as a major selective force in shaping host genetic diversity [3,4], and the rate of adaptive evolution for genes that interact most directly with pathogens can be exceptionally high [5]. Sexual selection can also influence immunogenetic variation; in particular, the ‘good genes’ hypothesis for resistance to parasites has been invoked to explain why some animals hold mating preferences in the absence of direct benefits of being choosy [6]. Thus, direct effects of parasites on host fitness, combined with sexual selection for mates that might confer beneficial genes to progeny, are the two most likely forces shaping immunogenetic diversity in animals.
The major histocompatibility complex (MHC) is an ideal candidate for identifying factors that determine immune gene diversity, because it plays a crucial role in immune defence for virtually all vertebrates and can mediate mate choice in a variety of species, including humans [7,8]. The MHC encodes glycoproteins that bind to foreign antigens and present them to T-cells, initiating an immune response [9]. There are two major groups of MHC genes: class I responds to intracellular pathogens and class II interacts with extracellular pathogens [10]. In particular, the class II DRB locus has been extensively studied because of its high allelic diversity, and both diversity and specific alleles at this locus predict parasite resistance in animals [7]. The DRB exon 2 region encodes the functionally important antigen-binding sites (ABS) that recognize pathogen peptides, with evidence of intense positive selection at codons along the sequence [10]. Because different ABS bind to different pathogen proteins, multiple alleles are required to confer resistance to diverse pathogen strains and species [9].
Past work showed that even endangered species (which otherwise harbour extremely low diversity based on selectively neutral loci) can display high MHC genotypic diversity [11–13] with such observations attributed to strong balancing selection operating on MHC loci [3,7]. Despite the potential for universally strong selection on MHC genes across vertebrate taxa, species do differ in their levels of MHC variability [14]. Some of this variation can be explained by differential parasite pressure across species [15], but mechanisms underlying among-species variation in MHC have rarely been studied in a comparative sense (but see [15–17]).
Theory suggests that disassortative mating could also preserve allelic diversity across MHC loci [18], and numerous species, including rodents, fishes, birds and humans can discern MHC genotypes based on olfactory and other cues [19–21], and prefer scents of mates with complementary or dissimilar MHC genotypes [22,23]. A key challenge facing researchers studying mate choice and MHC is that ecological and demographic factors influence the opportunity for and benefits of being choosy [24]. As a result, most studies showing MHC-based mating preferences within species are based on laboratory or captive experiments, and studies conducted on wild populations have shown mixed results [25,26].
Here, we use a comparative approach to investigate the relative influence of the two proposed main selective forces on MHC polymorphism: parasite-mediated selection and sexual selection. Our analysis focuses on mammals from five orders, as mammals have been relatively well studied for MHC variation, parasites and infectious diseases, and traits associated with sexual selection and reproductive skew. Key questions motivating this study were: (i) how does MHC diversity vary across mammal groups? (ii) are measures of allelic and nucleotide diversity elevated in species with higher potential for mate choice? (iii) is MHC diversity also elevated in species with greater parasite richness? and (iv) is there an association between parasite richness and degree of sexual selection, and how might this interaction shape the relationship with MHC polymorphism? We estimated the potential for sexual selection using relative testes size as a proxy for competition among males to produce offspring, as past work showed this measure is greater in species with promiscuous or polygynous (as opposed to monogamous) mating systems [27–29]. To infer selection pressures exerted by diverse parasite communities, we augmented existing data on parasites and pathogens (including viruses, bacteria, protozoa, helminths and arthropods) from free-living mammal populations [30–32]. We used phylogenetically informed analyses to test key predictors of immune genes diversity across species, controlling for the potential effects of host phylogeny, ecological traits and uneven sampling effort. We also used meta-analyses to compare effect sizes across taxa and to better support the generality of the findings. To our knowledge, this work represents the first test of the importance of sexual selection for explaining immunogenetic variation across a wide range of mammals.
2. Material and methods
(a). Major histocompatibility complex data
Sequence data for 112 mammal species were compiled from GenBank using Geneious v. 5.6.3. We first performed a preliminary search with the key term ‘MHC class II DRB’, recovered all mammal sequences and retained sequences including exon 2 of the DRB locus. We also searched on Web of Science and Biosis using each previously identified species Latin binomial and MHC as key terms. Sequences from subspecies were combined at the species level, and we followed the taxonomy of Wilson & Reeder [33]. Primate taxonomy followed the nomenclature from the Global Mammal Parasite Database [30] and the dataset from Garamszegi & Nunn [15] to correspond with parasite data. For each species, we recorded the number of animals sampled at the DRB locus because more alleles tend to be discovered as more individuals are sequenced.
Sequences were grouped according to Order (Carnivora, Chiroptera, Primate, Rodentia and Artiodactyla), imported into Mega v. 5 [34] and aligned by MUSCLE [35]. Because sequences differed in length, we trimmed all exon 2 sequences to 171 bp to estimate substitution rates. We removed pseudogenes and alleles with nucleotide insertions or deletions that might represent non-functioning alleles. We also removed DRB6 alleles from primates, as this locus is thought to be non-functional [36]. We checked for duplicates within species and removed non-unique sequences. Final numbers of sequences were recorded as numbers of alleles per species. For analyses of allelic richness, we used residuals from a regression analysis of log(number of alleles) on log(number of animals sampled) to control for uneven sampling across species.
Rates of selection for functional variation is a biologically important measure of diversity, especially for sites that encode proteins responsible for binding to foreign peptides (ABS [10]). To estimate substitution rates, we used the most commonly used method [37] with correction for multiple substitutions at the same site [38]. Mega v. 5 was used to compute within-species averages for amino acid changing non-synonymous substitutions (dN) at 15 ABS based on [39]. We repeated this process for synonymous substitutions (dS) at ABS to provide a baseline for neutral substitution rates. We avoided using the ratio dN : dS at ABS as in [15], as correlations with ratios may be more difficult to interpret, being influenced by both the nominator and denominator. However, we also ran all analyses using this ratio, and results were generally consistent (see the electronic supplementary material, figures S1 and S2, though power to detect significant associations was reduced owing to some species having no synonymous substitutions at the ABS). We considered using alternative ABS sites determined using the consensus of codon-based maximum-likelihood methods [40] applied to each main order sequence set (e.g. carnivore, bat, primate, rodent and ungulate). However, these predicted sites were strongly and significantly correlated with the 15 peptide-binding region residues determined based on protein crystallography (Pearson's correlation ranged from r(29) = 0.49, p < 0.01, primates, to r(12) = 0.93, p < 0.001, rodents). In addition, these 15 sites are known to be involved with antigen binding and have been shown to be under positive selection across a diverse set of taxa (carnivores, rodents and primates [41]; bats [42]). Therefore, we focus analyses on these documented 15 ABS sites, though analyses with putative taxon-specific ABS sites show overall consistent results (see the electronic supplementary material, figure S3). The number of MHC alleles varies by allelic lineage [43] and by the number of duplicated DRB loci [44]. Because most species in our dataset are non-model organisms and because no information is available on the specific DRB lineage or gene copy number, we could not include these variables in our analysis. We compiled the number of recorded DRB loci for each of 61 mammal species for follow-up analyses.
(b). Parasite data
Hosts exposed to a more diverse parasite assemblage could experience selection for greater genetic diversity for resistance [16]. Because MHC class II genes recognize extracellular parasites, there might be stronger relationships between parasites with prominent extracellular stages (such as helminths, arthropods and some microbes) and measures of DRB diversity. We compiled parasite richness data for each host species using the Global Mammal Parasite Database (www.mammalparasites.org), the most comprehensive collection of published records of parasitic organisms from free-living mammals [30]. For each host species, we recorded parasite richness as the total number of viruses, bacteria, protozoa, helminths and parasitic arthropods, as defined at the species-level based on current taxonomic schemes. We also recorded separately the numbers of helminths (thought to have strong coevolutionary relationships with their hosts), microparasites (viruses, bacteria and protozoa) and macroparasites (helminths and arthropods) to test whether some groups were more strongly associated with MHC class II diversity than others. We could not examine all parasite subgroups (e.g. viruses and bacteria) individually owing to low numbers for some host taxa.
Parasite richness estimates depend strongly on research effort [45]; better-studied host species tend to have more parasites reported to infect them. We therefore controlled for uneven sampling effort among hosts using the total number of citations for each host species (using Latin binomials and common taxonomic variants) from Web of Science as an indicator of scientific effort per host. Following previous studies [15,31,46,47], we used citation counts to control for research effort instead of the cumulative number of individual hosts sampled across all studies, as some studies did not publish the number of animals sampled, and other studies had high sample sizes despite testing for only a single parasite. We used residuals from a regression analysis of log(parasite richness) against log(citation count) (R2 = 0.45, F1,96 = 78.29, p < 0.001) to estimate corrected parasite richness per host.
(c). Estimates of sexual selection and ecological traits
Genetic mating system, and specifically the potential for female mate choice (as females tend to be the choosier sex [48]), is expected to influence the strength of sexual selection on the MHC. Females in monogamous or polygynous mating systems are likely to be more constrained in their choice for mates that can provide direct or indirect benefits [49]. By contrast, females in promiscuous or polyandrous mating systems are expected to have greater opportunity to select among potential mates. Relative testes size (testes mass/body mass) was used as a proxy for female promiscuity and opportunities for mate choice, as this measure has been shown to predict sexual selection and mating system across mammals (e.g. primates [27]; rodents [28]; carnivores [29]) and it is available for a large number of species. We compiled testes mass data and male body mass from the literature (see the electronic supplementary material, dataset S1). In some instances, only testes length, circumference or volume measurements were available, and in those cases, we converted these data to mass using the method of [27]. We then used the residuals from a regression analysis of log(testes mass) against log(male body mass) to obtain relative testes size per species and performed this separately for each taxonomic group.
For each mammal species, we also compiled data on two variables that could strongly influence parasitism, mating behaviour, and/or MHC diversity and evolution. First, effective population size (Ne) can impact genetic diversity by affecting the realized mutation rate, strength of selection and the amount of genetic drift experienced by a population [50]. Ne can also influence parasite richness measures as larger populations can theoretically retain more parasites than smaller populations [51,52]. In addition, species with greater population density might harbour more parasites with density-dependent transmission [53], and species with larger geographical range sizes could encounter a greater diversity of parasites [31,32]. We used census population size as a proxy for effective population size, with the caveat that the ratio of Ne/N is approximately 0.1 on average [54]. Population size was estimated by multiplying average population density (individuals per km2) from the PanTHERIA database [55] by the species geographical range size (km2) extracted from spatial data provided by the 2010 International Union for Conservation of Nature Red List (http://www.iucnredlist.org/technical-documents/spatial-data#mammals, last accessed on 6 July 2012). Previous studies have shown that this measure of population size is a significant predictor of both Ne [15,56] and parasite richness [52]. Second, body mass is known to scale with many life-history traits, including population size, reproductive rate, evolutionary rate [57] and parasite diversity [31,32,52], and thus was included as a covariate. Body mass (g) data were extracted from PanTHERIA [55], or when not available, the primary literature. Our full comparative dataset can be found in the electronic supplementary material, S1. Variables were log transformed or square root arcsin transformed (dS rate data; [58]) when necessary to meet normality assumptions, and no predictor variables showed strong collinearity (r < 0.07) [59].
(d). Comparative analyses
We tested whether log MHC allelic diversity and rates of positive selection (dN) at functional sites (as two separate dependent variables) were associated with parasite richness and estimates of sexual selection. Because closely related species are more likely to share genetic and life-history traits [60], we used phylogenetic least-squares (PGLS) regressions that handle phylogenetic structure through a variance–covariance matrix [61]. We assessed the importance of each predictor through a stepwise model selection procedure, in which the full models included the following: corrected parasite richness, relative testes size, log body mass, log population size, and the interaction between corrected parasite richness and relative testes size, while also including dS at ABS as a covariate to control for underlying substitution rate. We then simplified the starting model using Akaike information criteria corrected for smaller sample sizes (AICc) following [62] and removed variables that did not improve model fit (ΔAICc > 4). To avoid problems associated with missing values in AIC-based model comparison where sample size changes as terms are removed, we removed any species from our dataset that did not have complete coverage for all of the predictor variables of interest in the starting full model. To test for associations between relative testes size (as a response variable) and corrected parasite richness (predictor variable), we ran PGLS models controlling for the effects of taxonomic group, log population size and male body mass.
The PGLS regression was conducted using the caper package in R [63] using Pagel's λ to adjust models for the amount of phylogenetic signal observed in each variable. The phylogeny was constructed using the mammalian supertree [64], and polytomies were randomly resolved (by adding branches of length equal to zero) using the multi2di function in the ape R package [65]. Species in the dataset but missing from the supertree assumed the names of the closest relatives (i.e. Papio cynocephalus was changed to Papio hamadryas, and Zalophus wollebaeki was changed to Zalophus californianus).
To test the robustness of our results, we estimated taxon-specific effect sizes in the form of correlation coefficients [66] and ran meta-analyses to compare the overall effect sizes for predictor variables across taxa in explaining MHC variation. We estimated grand classwise effect sizes after considering differences in sample size among orders, and also examined differences among taxonomic groups. Meta-analysis was performed using the metafor package [67] in R [68].
3. Results
Our final dataset comprised 112 mammal species (26 carnivores; 14 chiropterans; 37 primates; 16 rodents and 19 ungulates), 2454 sequences and 2665 host–parasite species combinations (list of species with full trait and genetic datasets are provided in the electronic supplementary material, dataset S1 and S2). We tested for relationships between parasitism (using total parasite richness, helminth richness, and micro- and macroparasite richness) and sexual selection (using relative testes size as an indicator of mating system) on the rate of positive selection (dN at ABS) and MHC allelic richness across mammals. Analyses controlled for mammal phylogeny, the rate of neutral substitutions (dS at ABS), measures of sampling effort and two host traits known to be important for parasitism and/or genetic diversity based on previous studies (body mass and population size; electronic supplementary material, tables S1–S3). All predictor and response variables except relative allelic richness and parasite richness showed strong phylogenetic signal and were more similar among closer relatives (see the electronic supplementary material, table S2).
Multivariate models showed that the strength of selection at ABS increased with relative testes size across all five mammal orders tested here (figure 1a and the electronic supplementary material, table S1). Effect sizes differed among orders with approximately 64% of the variability attributed to heterogeneity among the true effects [67] (Q = 8.43, p = 0.04, I2 = 63.84%), possibly owing to biological differences among mammal groups or the methodology used in different studies. When corrected parasite richness was included as a predictor in the full model with relative testes size, its main effect and interaction with relative testes size was non-significant. However, removing the relative testes size variable (and including 21 additional species that were missing testes data) showed that the strength of selection at ABS increased with total parasite richness (corrected for sampling effort) for bats and ungulates and decreased with parasite richness for carnivores (figure 1b and the electronic supplementary material, table S1).
Figure 1.
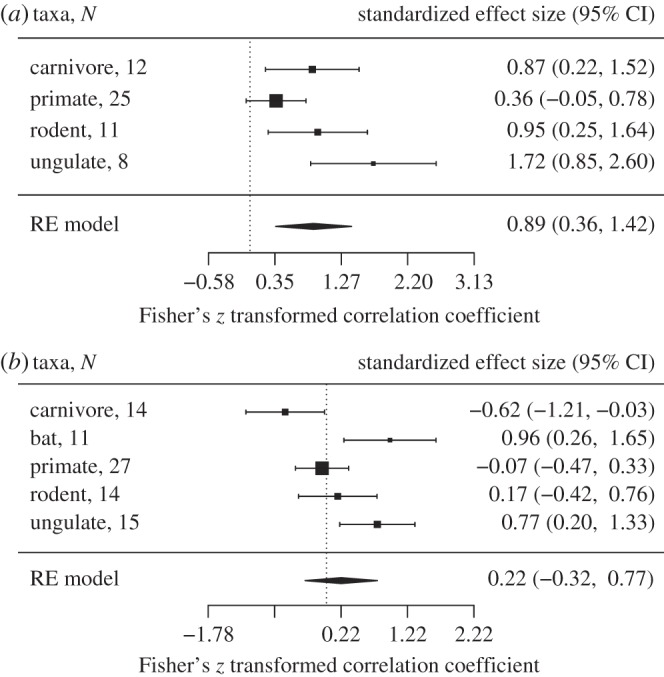
Forest plots showing predictors of the rate of non-synonymous substitutions (dN) at ABS. PGLS models show the effect of (a) relative testes size and (b) parasite richness, run separately for each mammal group. The vertical dotted line is positioned at zero and error bars denote 95% CIs. N refers to the number of species included; RE model, random effects model. Heterogeneity test for relative testes size: Q = 8.433, p = 0.038, I2 = 63.84%; parasite richness: Q = 17.408, p = 0.002, I2 = 79.06%.
Tests using data from parasite subgroups, including microparasites, macroparasites and helminths, showed that selection on ABS decreased with helminth and macroparasite richness for carnivores, and also decreased with microparasite richness for primates (figure 2; electronic supplementary material, figure S2). Ratios of dN : dS increased with macroparasite and microparasite richness for carnivores and ungulates (see the electronic supplementary material, figure S2). Neutral substitution rate (dS at ABS) was a positive predictor of dN at ABS (see the electronic supplementary material, figure S4) but only in models without relative testes size (and larger sample sizes). Taxonomic group was also a significant predictor of allelic substitution at ABS, with both dN and dS being greatest for bats and primates and lowest for carnivores and ungulates (figure 3c).
Figure 2.
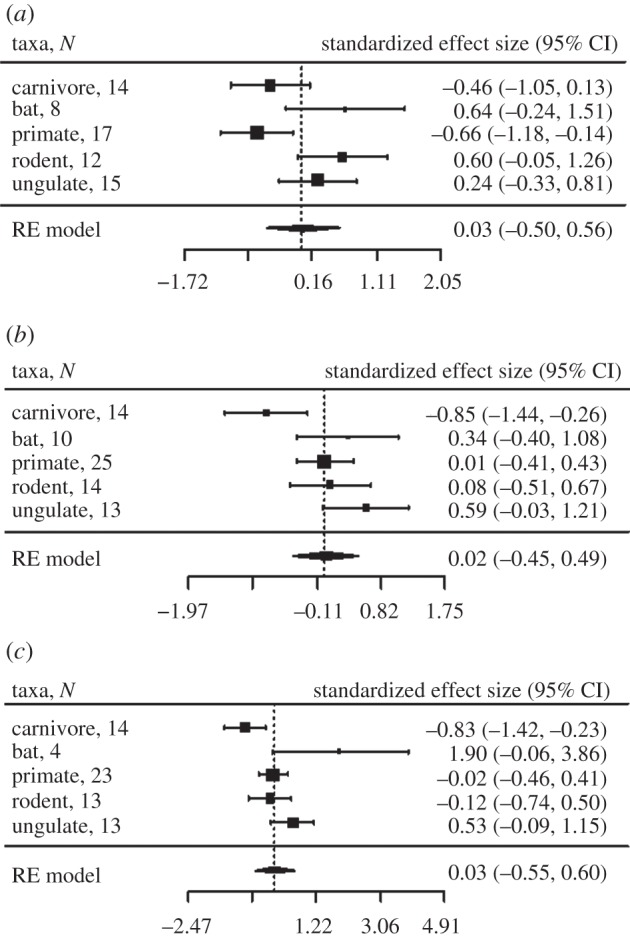
Forest plots showing predictors of the rate of non-synonymous substitutions at ABS. The results of PGLS models showing the effect of (a) microparasite richness, (b) macroparasite richness and (c) helminth richness, run separately for each host and parasite group. No meta-analysis random effects model was found to be significant for the parasite predictor variables (p > 0.1). The vertical dotted line is positioned at zero for Fisher's z transformed correlation coefficient. N refers to number of species; RE model, random effects model. Heterogeneity test for microparasite richness: Q = 5.186, p = 0.159, I2 = 43.24%; macroparasite richness: Q = 6.506, p = 0.089, I2 = 55.08%; helminth richness: Q = 4.329, p = 0.228, I2 = 0.00%.
Figure 3.
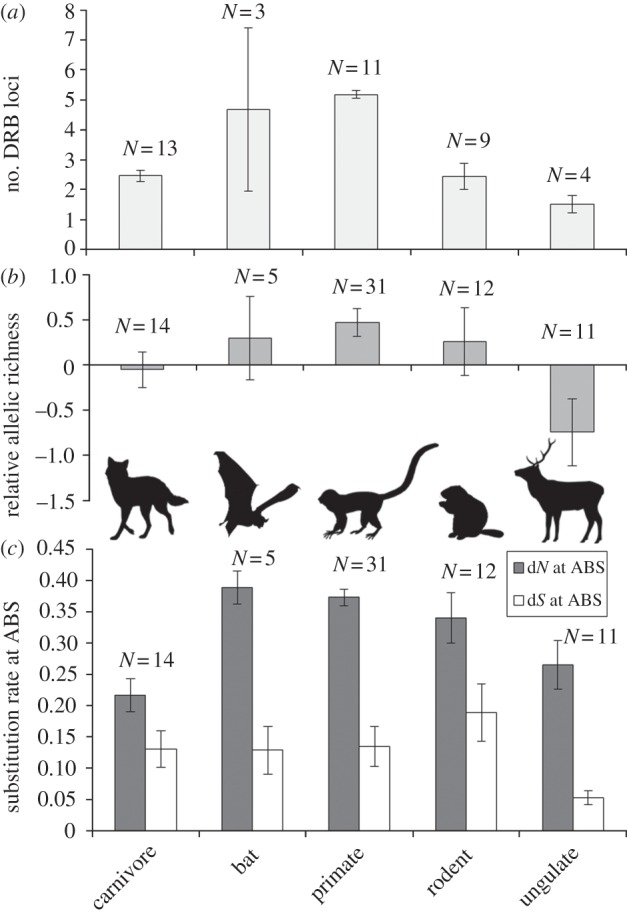
Genetic diversity at the MHC by mammal group. (a) The number of DRB loci, (b) relative allelic richness and (c) the rate of non-synonymous (dN) and synonymous substitutions (dS) at the ABS for carnivores, bats, primates, rodents and ungulates. Error bars denote 95% CIs. N refers to the number of species with data.
We tested allelic richness as a separate measure of selection on ABS. This measure differed among mammal taxonomic groups and increased with population size for ungulates (electronic supplementary material, tables S1 and S3e) but did not depend on measures of parasite richness or testes size. Allelic richness was significantly lower for ungulates than any other mammal group (figure 3b). A post hoc ANOVA revealed that ungulates also had significantly fewer duplicated DRB loci than other mammal orders, whereas primates had the most (F1,39 = 8.359, p = 0.006; figure 3a).
Finally, we tested for a relationship between relative testes size and corrected parasite richness, to ask whether the strength of sexual selection might covary positively with parasite pressure. Across all orders, we found a weak negative association between relative testes size and total parasite richness (p = 0.09; electronic supplementary material, figure S5a) with homogeneous effect sizes across taxa (Q = 1.079, p = 0.18, I2 = 0.00%), although other components of parasite richness showed no trend (p > 0.1; electronic supplementary material, figure S5).
4. Discussion
We found evidence that pressure from a diverse parasite fauna (represented by corrected parasite richness at the host species level) was associated with positive selection at the MHC DRB locus in bats and ungulates only. Species in these two groups that harboured greater parasite richness also showed higher rates of functionally significant evolutionary change within the MHC. By comparison, greater potential for sexual selection (represented by relative testes size as an indicator of mating system) predicted greater positive selection on functionally important MHC sites across all five orders of mammals examined here.
Very few studies have considered predictors of cross-species variation in MHC polymorphism, and those conducted to date focused on the relationship between parasite richness and MHC allelic richness [15–17]. In spite of the extensive intraspecific empirical evidence for MHC–parasite associations (reviewed in [7,69]), there is surprisingly weak support for parasites driving variation in MHC diversity among species. One study found that allelic richness increased with helminth richness across 10 rodent species [16], and another study found that the rate of positive selection at ABS (but not allelic richness) was positively related to nematode richness (but not total parasite richness) across 27 primates [15]. Our study differed from prior studies by using genetic data from multiple populations per species, by including broader taxonomic groups in parasite richness estimates, by not delineating alleles by their functional lineages, and by not separately analysing nematode richness (for which insufficient data were available from all orders).
The negative relationship between parasite richness and measures of the strength of selection on MHC in carnivores ran counter to our expectations. One reason for this pattern could be due to interactions between threat status and infectious disease risk, such that carnivores might be more vulnerable to population bottlenecks and genetic drift that concurrently reduce their genetic diversity and increases their susceptibility to parasitism. Many high-profile threatened carnivores have depleted MHC diversity (wild dogs [70]; Ethiopian wolves [71]; cheetahs [72]) and have simultaneously experienced declines from introduced infectious diseases such as rabies, canine distemper and sarcoptic mange [73,74]; these carnivore species might be exceptionally well studied and better represented in our dataset. One way to examine this issue further might be to include estimates of effective population size in comparative analyses and to distinguish between native versus introduced parasites and pathogens. An alternative explanation is that different taxa are in different stages of the coevolutionary arms race. If parasites lead the game, evidence might support parasitism as a driver of MHC polymorphism (leading to a positive relationship). However, if hosts lead the game, greater MHC diversity might reduce parasite pressure (leading to a negative relationship).
In contrast to the taxon-specific evidence of parasite-mediated selection, we found that relative testes size, as an indicator of sperm competition and the potential for sexual selection to operate at the species level [27], was positively associated with the rate of evolution at ABS across all mammal groups in our study. This finding provides evidence that species with high potential for mate choice tend to have higher MHC nucleotide diversity at functionally important sites. There are several non-exclusive explanations for this result. First, species with greater relative testes size and sperm competition might have faster reproductive rates, increasing the speed of selection for new variants. Indeed, Sommer et al. [75] found higher levels of MHC variation in a fast-reproducing and promiscuous rodent relative to a monogamous and slower reproducing relative, and hypothesized that slow reproduction might constrain MHC polymorphism. A second hypothesis is that greater sperm competition indicates greater promiscuity and increased exposure to sexually or socially transmitted diseases, which could enhance selection on immune defences [76,77]. As a third possibility, females with more potential mates might select genetically complementary or non-related mates and by doing so, serve to increase MHC variability. A fourth hypothesis is that relative testes size is correlated with androgen levels [78], which can suppress immune function or mediate male behaviour and increase exposure to and selection by parasites [79,80]. Our finding of a weak negative relationship between parasite richness and relative testes size, however, is not consistent with this hypothesis. Importantly, each of these mechanisms predicts that greater promiscuity will lead to greater genetic diversity, a result already observed for MHC and neutral genetic diversity across passerine birds [81]. Our analysis did not support an interactive effect between parasite richness and relative testes size, as might be expected if both high parasite pressure and the potential for mate choice were necessary to drive high MHC diversity.
Our study strongly supported taxonomic group as an important predictor for MHC allelic and sequence diversity. Specifically, ungulates had significantly lower allelic richness than any other order, possibly owing to fewer duplicated DRB loci (figure 2a). Primates, in comparison, had significantly greater allelic richness and more duplicated DRB loci. Average nucleotide divergence (π) is positively associated with the number of duplicated DRB loci in rodents [44] and this could be an important mechanism providing baseline genetic variation. Life-history traits or ecological conditions that affect the likelihood of MHC gene duplication events might therefore help predict MHC polymorphism in natural populations.
Overall, our study extends previous comparative work on MHC evolution by showing that both parasite-mediated selection and sexual selection can operate as independent forces maintaining differences in MHC diversity across mammal species. Evidence that parasites served as agents of selection was only found for bats and ungulates, but support for sexual selection was universal across mammal groups tested here. Potential explanations for this pattern include greater selection on immune genes driven by higher pressure from socially or sexually transmitted disease, and greater opportunities for mate choice leading to faster rates of substitution. Importantly, our analyses emphasize that comparative studies can contribute to knowledge on MHC ecology and evolution. We expect results of this study will encourage more work on the influence of sexual selection on MHC variability in wild populations, with great relevance for conservation genetics and predicting species responses to future disease risk.
Acknowledgements
We thank B. Han and C. Nunn for access to parasite data, J. Wares, J. Carroll, J. Moore, M. Yabsley, V. Ezenwa, J. Rushmore, D. Streicker, the Altizer Laboratory group, M. MacManes and an anonymous reviewer for discussion and comments that greatly improved the previous manuscript. J.C.W., L.Z.G. and S.A. designed the research; J.C.W., S.G.M., S.H. and P.R.S. compiled data; J.C.W. and L.Z.G. analysed data; and J.C.W., L.Z.G., S.H., P.R.S. and S.A. wrote the paper.
Funding statement
The AnthroTree workshop (supported by NSF grant nos. BCS-0923791 and EF-0905606) provided guidance on phylogenetic comparative analyses. The Odum School of Ecology, National Geographic Society, Animal Behavior Society, Association for Women in Science and the Philosophical Society provided financial support. J.C.W. was supported by a Graduate School Assistantship from the University of Georgia and a T-32 Training grant from the National Institutes of Health. L.Z.G. was supported by the ‘Plan Nacional’ program of the Spanish government (ref. no. CGL2012-40026 and CGL2012-38262). S.A. was supported by funding from the NSF (DEB-0643831).
References
- 1.Chinwalla AT, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (doi:10.1038/nature01262) [DOI] [PubMed] [Google Scholar]
- 2.Trowsdale J, Parham P. 2004. Mini-review: defense strategies and immunity-related genes. Eur. J. Immunol. 34, 7–17 (doi:10.1002/eji.200324693) [DOI] [PubMed] [Google Scholar]
- 3.Sommer S. 2005. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2, 1742–1760 (doi:10.1186/1742-9994-2-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barreiro LB, Quintana-Murci L. 2010. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 11, 17–30 (http://www.nature.com/nrg/journal/v11/n1/suppinfo/nrg2698_S1.html) [DOI] [PubMed] [Google Scholar]
- 5.Huang H, et al. 2004. Evolutionary conservation and selection of human disease gene orthologs in the rat and mouse genomes. Genome Biol. 5, 234 (doi:10.1186/gb-2004-5-7-234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton WD, Zuk M. 1982. Heritable true fitness and bright birds: a role for parasites. Science 218, 384–387 (doi:10.1126/science.7123238) [DOI] [PubMed] [Google Scholar]
- 7.Bernatchez L, Landry C. 2003. MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J. Evol. Biol. 16, 363–377 (doi:10.1046/j.1420-9101.2003.00531.x) [DOI] [PubMed] [Google Scholar]
- 8.Potts WK, Wakeland EK. 1990. Evolution of diversity at the major histocompatibility complex. Trends Ecol. Evol. 5, 181–187 (doi:10.1016/0169-5347(90)90207-T) [DOI] [PubMed] [Google Scholar]
- 9.Klein J. 1986. Natural history of the major histocompatibility complex. New York, NY: Wiley and Sons [Google Scholar]
- 10.Hughes AL, Yeager M. 1998. Natural selection at major histocompatibility complex loci of vertebrates. Annu. Rev. Genet. 32, 415 (doi:10.1146/annurev.genet.32.1.415) [DOI] [PubMed] [Google Scholar]
- 11.Garrigan D, Hedrick PW. 2003. Perspective: detecting adaptive molecular polymorphism: lessons from the MHC. Evolution 57, 1707–1722 (doi:10.1111/j.0014-3820.2003.tb00580.x) [DOI] [PubMed] [Google Scholar]
- 12.Gutierrez-Espeleta GA, Hedrick PW, Kalinowski ST, Garrigan D, Boyce WM. 2001. Is the decline of desert bighorn sheep from infectious disease the result of low MHC variation? Heredity 86, 439–450 (doi:10.1046/j.1365-2540.2001.00853.x) [DOI] [PubMed] [Google Scholar]
- 13.Aguilar A, Roemer G, Debenham S, Binns M, Garcelon D, Wayne RK. 2004. High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl Acad. Sci. USA 101, 3490–3494 (doi:10.1073/pnas.0306582101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikko S, Røed K, Schmutz S, Andersson L. 1999. Monomorphism and polymorphism at MHC DRB loci in domestic and wild ruminants. Immunol. Rev. 167, 169–178 (doi:10.1111/j.1600-065X.1999.tb01390.x) [DOI] [PubMed] [Google Scholar]
- 15.Garamszegi LZ, Nunn CL. 2011. Parasite-mediated evolution of the functional part of the MHC in primates. J. Evol. Biol. 24, 184–195 (doi:10.1111/j.1420-9101.2010.02156.x) [DOI] [PubMed] [Google Scholar]
- 16.de Bellocq JG, Charbonnel N, Morand S. 2008. Coevolutionary relationship between helminth diversity and MHC class II polymorphism in rodents. J. Evol. Biol. 21, 1144–1150 (doi:10.1111/j.1420-9101.2008.01538.x) [DOI] [PubMed] [Google Scholar]
- 17.Simkova A, Ottova E, Morand S. 2006. MHC variability, life-traits and parasite diversity of European cyprinid fish. Evol. Ecol. 20, 465–477 [Google Scholar]
- 18.Hedrick PW. 1992. Female choice and variation in the major histocompatibility complex. Genetics 132, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki K, Yamaguchi M, Andrews PW, Peake B, Boyse EA. 1978. Mating preferences of F2 segregants of crosses between MHC-congenic mouse strains. Immunogenetics 6, 253–259 (doi:10.1007/BF01563915) [Google Scholar]
- 20.Penn DJ. 2002. The scent of genetic compatibility: sexual selection and the major histocompatibility complex. Ethology 108, 1–21 (doi:10.1046/j.1439-0310.2002.00768.x) [Google Scholar]
- 21.Milinski M, Griffiths S, Wegner KM, Reusch TBH, Haas-Assenbaum A, Boehm T. 2005. Mate choice decisions of stickleback females predictably modified by MHC peptide ligands. Proc. Natl Acad. Sci. USA 102, 4414–4418 (doi:10.1073/pnas.0408264102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wedekind C, Furi S. 1997. Body odour preferences in men and women: do they aim for specific MHC combinations or simply heterozygosity? Proc. R. Soc. Lond. B 264, 1471–1479 (doi:10.1098/rspb.1997.0204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cutrera AP, Fanjul MS, Zenuto RR. 2012. Females prefer good genes: MHC-associated mate choice in wild and captive tuco-tucos. Anim. Behav. 83, 847–856 (doi:10.1016/j.anbehav.2012.01.006) [Google Scholar]
- 24.Roberts SC. 2009. Complexity and context of MHC-correlated mating preferences in wild populations. Mol. Ecol. 18, 3121–3123 (doi:10.1111/j.1365-294X.2009.04244.x) [DOI] [PubMed] [Google Scholar]
- 25.Setchell JM, Huchard E. 2010. The hidden benefits of sex: evidence for MHC-associated mate choice in primate societies. Bioessays 32, 940–948 (doi:10.1002/bies.201000066) [DOI] [PubMed] [Google Scholar]
- 26.Paterson S, Pemberton JM. 1997. No evidence for major histocompatibility complex-dependent mating patterns in a free-living ruminant population. Proc. R. Soc. Lond. B 264, 1813–1819 (doi:10.1098/rspb.1997.0250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harcourt AH, Harvey PH, Larson SG, Short RV. 1981. Testis weight, body weight and breeding system in primates. Nature 293, 55–57 (doi:10.1038/293055a0) [DOI] [PubMed] [Google Scholar]
- 28.Kenagy GJ, Trombulak SC. 1986. Size and function of mammalian testes in relation to body size. J. Mammal. 67, 1–22 (doi:10.2307/1380997) [Google Scholar]
- 29.Iossa G, Soulsbury CD, Baker PJ, Harris S. 2008. Sperm competition and the evolution of testes size in terrestrial mammalian carnivores. Funct. Ecol. 22, 655–662 (doi:10.1111/j.1365-2435.2008.01409.x) [Google Scholar]
- 30.Nunn CL, Altizer SM. 2005. The global mammal parasite database: an online resource for infectious disease records in wild primates. Evol. Anthropol. Issues News Rev. 14, 1–2 (doi:10.1002/evan.20041) [Google Scholar]
- 31.Lindenfors P, Nunn CL, Jones KE, Cunningham AA, Sechrest W, Gittleman JL. 2007. Parasite species richness in carnivores: effects of host body mass, latitude, geographical range and population density. Glob. Ecol. Biogeogr. 16, 496–509 (doi:10.1111/j.1466-8238.2006.00301.x) [Google Scholar]
- 32.Ezenwa VO, Price SA, Altizer S, Vitone ND, Cook KC. 2006. Host traits and parasite species richness in even and odd-toed hoofed mammals, Artiodactyla and Perissodactyla. Oikos 115, 526–536 (doi:10.1111/j.2006.0030-1299.15186.x) [Google Scholar]
- 33.Wilson DE, Reeder DAM. 2005. Mammal species of the world: a taxonomic and geographic reference. Baltimore, MD: Johns Hopkins University Press [Google Scholar]
- 34.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. Mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (doi:10.1093/molbev/msr121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bodmer JG, et al. 1992. Nomenclature for factors of the HLA system, 1991. Immunogenetics 36, 135–148 (doi:10.1007/bf00661090) [PubMed] [Google Scholar]
- 37.Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 [DOI] [PubMed] [Google Scholar]
- 38.Jukes TH, Cantor CR. 1969. Evolution of protein molecules. In Mammalian protein metabolism (ed. Munro HN.), pp. 21–132 New York, NY: Academic Press [Google Scholar]
- 39.Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. 1993. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364, 33–39 (doi:10.1038/364033a0) [DOI] [PubMed] [Google Scholar]
- 40.Kosakovsky Pond SL, Frost SDW. 2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (doi:10.1093/molbev/msi105) [DOI] [PubMed] [Google Scholar]
- 41.Furlong R, Yang Z. 2008. Diversifying and purifying selection in the peptide binding region of DRB in mammals. J. Mol. Evol. 66, 384–394 (doi:10.1007/s00239-008-9092-6) [DOI] [PubMed] [Google Scholar]
- 42.Schad J, Voigt CC, Greiner S, Dechmann DK, Sommer S. 2012. Independent evolution of functional MHC class II DRB genes in New World bat species. Immunogenetics 64, 535–547 (doi:10.1007/s00251-012-0609-1) [DOI] [PubMed] [Google Scholar]
- 43.Garamszegi L, de Groot N, Bontrop R. 2009. Correlated evolution of nucleotide substitution rates and allelic variation in MHC–DRB lineages of primates. BMC Evol. Biol. 9, 73 (doi:10.1186/1471-2148-9-73) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winternitz JC, Wares JP. 2013. Duplication and population dynamics shape historic patterns of selection and genetic variation at the major histocompatibility complex in rodents. Ecol. Evol. 3, 1552–1568 (doi:10.1002/ece3.567) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walther B, Cotgreave P, Price R, Gregory R, Clayton D. 1995. Sampling effort and parasite species richness. Parasitol. Today 11, 306–310 (doi:10.1016/0169-4758(95)80047-6) [DOI] [PubMed] [Google Scholar]
- 46.Altizer S, Nunn CL, Thrall PH, Gittleman JL, Antonovics J, Cunningham AA, Dobson AP, Ezenwa V, Jones KE. 2003. Social organization and parasite risk in mammals: integrating theory and empirical studies. Annu. Rev. Ecol. Evol. Syst. 34, 517–547 (doi:10.1146/annurev.ecolsys.34.030102.151725) [Google Scholar]
- 47.Sol D, Duncan RP, Blackburn TM, Cassey P, Lefebvre L. 2005. Big brains, enhanced cognition, and response of birds to novel environments. Proc. Natl Acad. Sci. USA 102, 5460–5465 (doi:10.1073/pnas.0408145102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tregenza T, Wedell N. 2000. Genetic compatibility, mate choice and patterns of parentage: invited review. Mol. Ecol. 9, 1013–1027 (doi:10.1046/j.1365-294x.2000.00964.x) [DOI] [PubMed] [Google Scholar]
- 49.Clutton-Brock TH, Parker GA. 1995. Sexual coercion in animal societies. Anim. Behav. 49, 1345–1365 (doi:10.1006/anbe.1995.0166) [Google Scholar]
- 50.Hartl DL, Clark AG. 1997. Principles of population genetics. Sunderland, MA: Sinauer Associates [Google Scholar]
- 51.Anderson RM, May RM. 1978. Regulation and stability of host–parasite population interactions. 1. Regulatory processes. J. Anim. Ecol. 47, 219–247 (doi:10.2307/3933) [Google Scholar]
- 52.Nunn CL, Altizer S, Jones KE, Sechrest W. 2003. Comparative tests of parasite species richness in primates. Am. Nat. 162, 597–614 (doi:10.1086/378721) [DOI] [PubMed] [Google Scholar]
- 53.Arneberg P. 2002. Host population density and body mass as determinants of species richness in parasite communities: comparative analyses of directly transmitted nematodes of mammals. Ecography 25, 88–94 (doi:10.1034/j.1600-0587.2002.250110.x) [Google Scholar]
- 54.Frankham R. 1995. Effective population size/adult population size ratios in wildlife: a review. Genet. Res. 66, 95–107 (doi:10.1017/S0016672300034455) [DOI] [PubMed] [Google Scholar]
- 55.Jones KE, et al. 2009. PanTHERIA: a species-level database of life history, ecology, and geography of extant and recently extinct mammals. Ecology 90, 2648 (doi:10.1890/08-1494.1) [Google Scholar]
- 56.Møller A, Garamszegi L, Spottiswoode C. 2008. Genetic similarity, breeding distribution range and sexual selection. J. Evol. Biol. 21, 213–225 (doi:10.1111/j.1420-9101.2008.01556.x) [DOI] [PubMed] [Google Scholar]
- 57.Brown JH. 1995. Macroecology. Chicago, IL: University of Chicago Press [Google Scholar]
- 58.Sokal RR, Rohlf FJ. 1995. Biometry: the principles and practice of statistics in biological research. New York, NY: WH Freeman [Google Scholar]
- 59.Dormann CF, et al. 2012. Collinearity: a review of methods to deal with it and a simulation study evaluating their performance. Ecography 36, 27–46 (doi:10.1111/j.1600-0587.2012.07348.x) [Google Scholar]
- 60.Harvey PH, Pagel MD. 1991. The comparative method in evolutionary biology. Oxford, UK: Oxford University Press [Google Scholar]
- 61.Pagel MD. 1992. A method for the analysis of comparative data. J. Theor. Biol. 156, 431–442 (doi:10.1016/S0022-5193(05)80637-X) [Google Scholar]
- 62.Crawley MJ. 2002. Statistical computing: an introduction to data analysis using S-plus, p. 761 Chichester, UK: Wiley [Google Scholar]
- 63.Orme D. 2011. The caper package: comparative analysis of phylogenetics and evolution in R. See http://cran.r-project.org/web/packages/caper/vignettes/caper.pdf
- 64.Bininda-Emonds ORP, et al. 2007. The delayed rise of present-day mammals. Nature 446, 507–512 (doi:10.1038/nature05634) [DOI] [PubMed] [Google Scholar]
- 65.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (doi:10.1093/bioinformatics/btg412) [DOI] [PubMed] [Google Scholar]
- 66.Nakagawa S, Cuthill IC. 2007. Effect size, confidence interval and statistical significance: a practical guide for biologists. Biol. Rev. 82, 591–605 (doi:10.1111/j.1469-185X.2007.00027.x) [DOI] [PubMed] [Google Scholar]
- 67.Viechtbauer W. 2010. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 [Google Scholar]
- 68.Team RDC. 2012. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- 69.Spurgin LG, Richardson DS. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. R. Soc. B 277, 979–988 (doi:10.1098/rspb.2009.2084) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marsden CD, Mable BK, Woodroffe R, Rasmussen GSA, Cleaveland S, McNutt JW, Emmanuel M, Thomas R, Kennedy LJ. 2009. Highly endangered African wild dogs (Lycaon pictus) lack variation at the major histocompatibility complex. J. Hered. 100(Suppl. 1), S54–S65 (doi:10.1093/jhered/esp031) [Google Scholar]
- 71.Kennedy LJ, et al. 2011. Major histocompatibility complex diversity in the endangered Ethiopian wolf (Canis simensis). Tissue Antigens 77, 118–125 (doi:10.1111/j.1399-0039.2010.01591.x) [DOI] [PubMed] [Google Scholar]
- 72.Castro-Prieto A, Wachter B, Sommer S. 2011. Cheetah paradigm revisited: MHC diversity in the world's largest free-ranging population. Mol. Biol. Evol. 28, 1455–1468 (doi:10.1093/molbev/msq330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O'Brien SJ, Evermann JF. 1988. Interactive influence of infectious-disease and genetic diversity in natural-populations. Trends Ecol. Evol. 3, 254–259 (doi:10.1016/0169-5347(88)90058-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pedersen AB, Jones KE, Nunn CL, Altizer S. 2007. Infectious diseases and extinction risk in wild mammals. Conserv. Biol. 21, 1269–1279 (doi:10.1111/j.1523-1739.2007.00776.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sommer S, Schwab D, Ganzhorn JU. 2002. MHC diversity of endemic Malagasy rodents in relation to geographic range and social system. Behav. Ecol. Sociobiol. 51, 214–221 (doi:10.1007/s00265-001-0432-4) [Google Scholar]
- 76.MacManes MD, Lacey EA. 2012. Is promiscuity associated with enhanced selection on MHC-DQα in mice (genus Peromyscus)? PLoS ONE 7, e37562 (doi:10.1371/journal.pone.0037562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nunn CL, Gittleman JL, Antonovics J. 2000. Promiscuity and the primate immune system. Science 290, 1168–1170 (doi:10.1126/science.290.5494.1168) [DOI] [PubMed] [Google Scholar]
- 78.Parapanov R, Nusslé S, Crausaz M, Senn A, Hausser J, Vogel P. 2009. Testis size, sperm characteristics and testosterone concentrations in four species of shrews (Mammalia, Soricidae). Anim. Reprod. Sci. 114, 269–278 (doi:10.1016/j.anireprosci.2008.09.013) [DOI] [PubMed] [Google Scholar]
- 79.Folstad I, Karter AJ. 1992. Parasites, bright males, and the immunocompetence handicap. Am. Nat. 139, 603–622 (doi:10.1086/285346) [Google Scholar]
- 80.Veiga JP, Salvador A, Merino S, Puerta M. 1998. Reproductive effort affects immune response and parasite infection in a lizard: a phenotypic manipulation using testosterone. Oikos 82, 313–318 (doi:10.2307/3546971) [Google Scholar]
- 81.Gohli J, Anmarkrud JA, Johnsen A, Kleven O, Borge T, Lifjeld JT. 2012. Female promiscuity is positively associated with neutral and selected genetic diversity in passerine birds. Evolution 67, 1406–1419 [DOI] [PubMed] [Google Scholar]