

Abstract
Most eukaryotic lineages belong to one of a few major groups. However, several protistan lineages have not yet been robustly placed in any of these groups. Both the breviates and apusomonads are two such lineages that appear to be related to the Amoebozoa and Opisthokonta (i.e. the ‘unikonts’ or Amorphea); however, their precise phylogenetic positions remain unclear. Here, we describe a novel microaerophilic breviate, Pygsuia biforma gen. nov. sp. nov., isolated from a hypoxic estuarine sediment. Ultrastructurally, this species resembles the breviate genera Breviata and Subulatomonas but has two cell morphologies, adherent and swimming. Phylogenetic analyses of the small sub-unit rRNA gene show that Pygsuia is the sister to the other breviates. We constructed a 159-protein supermatrix, including orthologues identified in RNA-seq data from Pygsuia. Phylogenomic analyses of this dataset show that breviates, apusomonads and Opisthokonta form a strongly supported major eukaryotic grouping we name the Obazoa. Although some phylogenetic methods disagree, the balance of evidence suggests that the breviate lineage forms the deepest branch within Obazoa. We also found transcripts encoding a nearly complete integrin adhesome from Pygsuia, indicating that this protein complex involved in metazoan multicellularity may have evolved earlier in eukaryote evolution than previously thought.
Keywords: eukaryote evolution, protist, mitochondria, integrin, animal–fungal clade
1. Introduction
Our understanding of the evolutionary history of eukaryotes has been revolutionized by phylogenomic approaches that use hundreds of proteins to infer phylogenies (reviewed in [1]). Most eukaryotes may be placed into one of only three major high-level groups: Amorphea, Diaphoretickes and Excavata [2]. The Amorphea (or ‘unikonts’ [3]) is composed of the well-supported supergroups Amoebozoa, containing amoeboid protists and Opisthokonta, which unites the Fungi and Metazoa (animals) [2]. It also includes a number of protists whose precise phylogenetic positions remain unclear. These taxa include the breviate amoeboid flagellates (Breviatea [4]) and a heterogeneous collection of flagellate taxa such as the apusomonads (Apusomonadida [5–7]), ancyromonads (Ancyromonadida/Planomonadida [8,9]) and miscellaneous other flagellate genera [10]. Determining the composition of the Amorphea and the phylogeny of its constituent lineages is pivotal for understanding major events that occurred early in the evolutionary history of eukaryotes. For instance, some evidence suggests that the root of the eukaryotic tree lies within the Amorphea [3,6] or is just outside of this group as, for example, suggested by the ‘unikont/bikont hypothesis’ [11,12]. Furthermore, the Amorphea contains at least five lineages—Metazoa, Fungi, dictyostelids, Fonticula and Copromyxa—in which a multicellular lifestyle has evolved independently [2].
The breviate amoebae, exemplified by Breviata anathema, are small uniflagellate heterotrophic amoeboid organisms inhabiting hypoxic environments [13]. Their phylogenetic position is unclear, since they are placed in various positions in the eukaryotic tree based on different data and analyses [4,13–17]. Phylogenomic analyses of 78 or 124 proteins indicate that B. anathema is sister to, or in, Amoebozoa [16,17], or is sister to the Opisthokonta [17]. However, these analyses included large amounts of missing data for B. anathema (42% [16] and 61% [17], missing sites, respectively) and lacked the apusomonads, a group of flagellates that were suggested relatives of breviates [13] and appeared to branch within the Amorphea clan [7,18,19]. A breviate + apusomonad (BA) clade was recovered in a 16-protein phylogenetic analysis, however the group was only weakly supported [20], a result that is perhaps unsurprising given that little data from apusomonads were included (only one to six genes were sampled). Analyses of a 159-protein supermatrix that included both B. anathema and the apusomonad Thecamonas trahens weakly grouped Breviata with Amoebozoa, not apusomonads [19], but again, these analyses were compromised by the large amount of missing data (approx. 80% of sites missing) for B. anathema. Another study that used an eight-protein matrix weakly inferred that B. anathema is sister to the Amoebozoa with the ancyromonads sister to Opisthokonta + apusomonads (OA) [21], but these results were again compromised by missing data for B. anathema. Most recently, a 30-protein phylogeny showed a BA clade with weak support that could not be conclusively placed on an overall tree of eukaryotes [22].
The deep phylogenetic relationships within Amorphea are important for understanding the origins of multicellularity [2]. Recent studies show that many genes and signalling pathways related to multicellularity in animals actually predate the origin of Metazoa and even Opisthokonta [23–28]. One of the most notable examples is that of integrin-mediated adhesion complex (IMAC), recently found in the apusomonad Thecamonas trahens [26]. In animal cells, integrins form heterodimeric transmembrane receptors made up of α-integrin (ITA) and β-integrin (ITB) subunits that play key roles in adhesion to the extracellular matrix, cell signalling and cell motility [29]. IMACs are able to transduce extracellular signals from the extracellular matrix or adjacent cells to intracellular proteins that regulate actin polymerization [29]. In metazoans, the extracellular integrin ligand affinities can be mediated by interactions of intracellular proteins with the cytoplasmic tail of ITB [30]. The discovery of IMAC proteins in Thecamonas indicates that the origin of the complex substantially predates the origin of Metazoa and suggests that a number of opisthokont lineages, including choanoflagellates and Fungi, have secondarily lost parts of this system. Pinpointing the origins and ancestral functions of this complex and other protein systems associated with multicellularity within Amorphea is important in understanding the origins of the multicellular lifestyle.
Here, we describe a novel amoeboid flagellate, Pygsuia biforma gen. nov. sp. nov., isolated from hypoxic estuarine sediments. Molecular phylogenetic analyses of small sub-unit (SSU) rDNA and morphological observations indicate that P. biforma is a member of the Breviatea [4]. Phylogenomic analyses of a 159-protein dataset suggest that the breviates are a sister lineage to a strongly supported OA clade [7]. Consistent with this position, we identified a nearly complete metazoan-like IMAC similar in content and architecture to that found in the apusomonad Thecamonas trahens [26].
2. Material and methods
Details of experimental methods for P. biforma culturing, nucleic acid extraction, SSU rDNA analyses, cDNA construction, Illumina sequencing, cluster assembly, model cross validation, topology testing using the phylogenomic dataset, further phylogenetic methods, homology searches for IMAC transcripts and electron microscopic methods are described in the electronic supplementary material.
(a). Phylogenomic dataset construction
Our 159-protein phylogenomic dataset was constructed using seed protein alignments [19]. The RNA-seq clusters of P. biforma were screened for orthologues in the seed dataset using TBlastN with sequences from a reference protein dataset (RefDat) (electronic supplementary material, table S1). The TBlastN hits were then translated to amino acid residues, using Blastmonkey from the barrel_of_monkeys toolkit (http://rogerlab.biochemistryandmolecularbiology.dal.ca/Software/). BlastP was used to screen homologues from P. biforma against the OrthoMCL v. 4 database (www.orthomcl.org) and the output for each gene was compared against a dictionary of orthologous OrthoMCL IDs (see the electronic supplementary material, table S2); those that did not match were designated as paralogues and removed. The remaining protein sequences were added to the seed protein alignments using Mafft [31]. Ambiguously aligned positions from all 159 single protein alignments were identified and removed by visual inspection.
Maximum-likelihood (ML) trees were inferred for each single-gene alignment in RAxML v7.2.6 [32] using the LG + Γ distribution with four rate categories (LG + Γ4) [33], with 10 ML tree searches and 100 ML bootstrap replicates. To test undetected paralogues or contaminants, we constructed a consensus tree (ConTree) representing phylogenetic groupings of well-established eukaryotic clades [15–17,19,34–38], allowing P. biforma + B. anathema to branch in any place (see the electronic supplementary material, figure S1). Individual protein trees that placed taxa in conflicting positions relative to ConTree with more than 70% bootstrap support and trees with zero-length or extremely long branches were checked by eye. Orthology was assessed using reciprocal BlastP against Oryza sativa homologues on NCBI. All problematic sequences identified using these methods were removed. The resulting alignments (see the electronic supplementary material, file) were then concatenated into a 43 615 amino acid position supermatrix. Sequence data from P. biforma were deposited in GenBank under BioProject PRJNA185780. Data sources, details on gene sampling and information on missing data are in the electronic supplementary material, table S1.
(b). Phylogenomic analyses
Bayesian inferences (BIs) were made in Phylobayes-mpi v. 1.2e [39] under the best-fitting CAT-GTR + Γ4 model, and four independent Markov chain Monte Carlo chains were run for 30 000 generations sampling every two generations. For Phylobayes analyses, constant sites were removed to decrease computational time. Convergence was achieved for three of the chains at 3000 generations, with the largest discrepancy in posterior probabilities (PPs) (maxdiff) less than 0.16 and the effective size of continuous model parameters were in the range of acceptable values (more than 50). PPs of post-burn in bipartitions (three chains, 12 000 trees, sampling every 10 trees) are mapped on to the consensus BI topology. The chain that did not converge on the same posterior distribution yielded the same topology for the taxa of interest (see the electronic supplementary material, figure S2).
ML trees were estimated from 60 independent searches (see the electronic supplementary material, figure S3) using RAxML under LG + Γ4 and empirical amino acid frequencies (LG + Γ4 + F) model, selected by the Akaike information criterion. Topological support was assessed by 500 RAxML bootstrap replicates. BIs under the LG + Γ4 + F model were based on two chains run for 10 000 generations, sampled every two generations (see the electronic supplementary material, figure S4). Convergence was achieved at 2000 generations (maxdiff < 0.17) and an acceptable effective size of model parameters.
(c). Fast-evolving site removal
Site rates of the phylogenomic dataset were estimated using dist_est [40] with the LG + Γ model, using discrete gamma probability estimation [40]. The sites were removed from the dataset from the highest rate to the lowest rate in 1000 site increments. Each of these datasets was analysed using 100 RAxML bootstrap replicates with the PROTCAT + LG + F model. A bootstrapped ML tree (see the electronic supplementary material, figure S5) was inferred under the LG + Γ4 + F model on the dataset that contained 25 615 of the original sites (approx. 59%), as this site-removal step showed a dramatic change in bootstrap values.
3. Results and discussion
(a). Pygsuia is a novel breviate
Pygsuia biforma is a small heterotrophic, amoeboid flagellate with two cell forms: (i) adherent cells with a conspicuous apical flagellum and a shorter posterior flagellum that attaches to the cell body (figure 1a) and (ii) cells with two conspicuous long flagella (‘swimming cells’; figure 1c). Swimming cells predominate in cultures that are passed every 5–7 days. Adherent cells appeared in culture after 1.5–2 weeks.
Figure 1.

Micrographs of P. biforma. (a) Differential interference contrast micrograph (DIC) of an adherent amoeboid cell with a long apical flagellum, af, and prominent filose pseudopodia, f, (arrowheads) projecting from one side. (b) DIC of an adherent amoeboid cell showing the inconspicuous posteriorly directed flagellum, pf, running along the cell surface. (c) DIC of a swimming cell with two long opposed flagella. (d) Scanning election micrograph of an adherent cell with a filose pseudopodium. (e) Transmission electron micrograph (TEM) of a whole cell. (f) TEM showing the flagellar apparatus of the cell shown in (c), four serial sections deeper into the cell. Note the microtubular dorsal fan, d, closely associated with the anterior basal body, ab. (g) TEM of the pf projecting from the posterior basal body, pb. Same cell as shown in (c,d), three sections deeper than (c). (h) TEM of the mitochondrion-related organelle (m) closely associated with, d. n, nucleus; s, starch-like body; fv, food vacuole; g, Golgi apparatus. Scale bars: (b) 10 µm, (c) 10 µm (a scaled to c), (d) 2.5 µm, (e) 2 µm, (f,g) 0.5 µm, (h) 0.5 µm.
Adherent cells are similar to B. anathema and Subulatomonas tetraspora (figure 1a,b). Cells are typically pear-shaped (8.5–18.5 µm long and 5–8 µm broad (n = 15)) with the apical flagellum approximately 8–30 µm long. The (usually) shorter posteriorly directed flagellum is difficult to observe clearly under light microscopy (figure 1b). It inserts subapically, is usually tightly associated with the cell surface, and is less than the length of the cell (figure 1b,d). In actively gliding adherent cells, filose pseudopodia form near the anterior end of the cell and move along the cell body in a ‘conveyer belt’ fashion as the cell progresses forwards. Similar pseudopodial activity is seen in Breviata and Subulatomonas, as well as in the apusomonad Amastigomonas filosa [41]. Cells can form posterior attachments to the substratum that elongate as the cell moves away from the point of attachment (electronic supplementary material, figures S6 and S7; they are also seen in swimming cells when these adhere—see below). These attachments may branch but do not appear to anastomose.
The ‘swimming cell’ forms have a rounded to elongate cell body and are 8.5–13 µm long and 3.7–5.3 µm broad (n = 17). The anterior flagellum (8.5–28 µm long) inserts apically. The posterior flagellum is directed posteriorly, but is typically free of the cell body, and is often approximately 50% longer than the apical flagellum (16–37 µm long; figure 1c). Both flagella beat with rapid undulatory motions during swimming, and the entire cell vibrates. Swimming cells sometimes attach to surfaces, and can then glide on their long posterior flagellum (see the electronic supplementary material, figure S7A); the cell outline remains smooth, or sometimes forms short finger-like pseudopodia or elongate posterior attachments (see above). This gliding is similar to apusomonads, among other taxa [10].
Examination of P. biforma using transmission electron microscopy (TEM) reveals a number of ultrastructural similarities to B. anathema [42] (figure 1e–h). The two flagellar basal bodies are at an obtuse angle, and a fan of microtubules surrounds the dorsum of the anterior basal body (figures 1e and electronic supplementary material, S8–S10). Both basal bodies bore flagella in all examined cells (see the electronic supplementary material, figure 1f,g). There is usually only a single mitochondrion-related organelle (MRO), which is tubular in shape, with one end closely associated with the basal bodies (figure 1e,h and serial sections in electronic supplementary material, figures S8–S10). The MRO has an electron-dense matrix and is bound by a double membrane, but lacks obvious cristae (figure 1e,h; electronic supplementary material, S8–S10). This organelle is similar to the putative hydrogenosome-like organelle found in B. anathema [13]. Cells often contain ingested bacteria and/or many starch-like granules (electronic supplementary material, figure S8–S10). Unlike apusomonads, P. biforma and the other breviates possess neither an organic theca nor a proboscis [41].
Phylogenetic analyses of SSU rDNA sequences illustrate that P. biforma is clearly related to, but distinct from, B. anathema and S. tetraspora (see the electronic supplementary material, figure S11). Pygsuia biforma branches with moderate support (92%/0.88) as sister to the named breviates and several environmental SSU rDNA sequences that fall within the breviate clade [20]. Of the named breviate genera, Pygsuia is the deepest branch, with near maximal bootstrap and PP support recovered for the Breviata and Subulatomonas clade to exclusion of Pygsuia (see the electronic supplementary material, figure S11). Although these results confirm that P. biforma is a breviate, the placement of the group in the global SSU rDNA phylogeny of eukaryotes is not well resolved (see the electronic supplementary material, figure S11) [4,13,43].
(b). Phylogenomic evidence for the Obazoa, a group containing Opisthokonta, breviates and apusomonads
To determine the phylogenetic placement of breviates within eukaryotes, we obtained RNA-seq data from P. biforma. We included these data into a 159-protein phylogenomic supermatrix (43 615 amino acid positions, 37 043 of which were represented in 152 orthologues from P. biforma) [19] that includes representatives from each of the proposed eukaryotic supergroups [2]. Phylogenetic analyses using both BI under the site-heterogeneous CAT-GTR + Γ4 model and ML using the site-homogeneous LG + Γ4 + F model revealed that the breviates robustly group in a large clade with the apusomonads (T. trahens and Manchomonas bermudiensis) and the supergroup Opisthokonta (OBA) (1.0/100%, BI PP/ML-bootstrap (BS); figure 2).
Figure 2.

Phylogenetic tree estimated from the 159-protein dataset, inferred by Phylobayes-mpi under the CAT-GTR + Γ4 model. Posterior probabilities (PPs) are shown under branches. ML bootstrap support was also estimated under the LG + Γ4 + F model (upper value). Black dots indicate 100% bootstrap support and PPs of 1.0. Topologies not recovered in the ML analyses are denoted by asterisks (*).
The exact position of the breviates within the OBA clade depended strongly on the method used for phylogenetic inference: the breviates are either sister to the OA (BI, figure 2) or sister to the apusomonads (ML, electronic supplementary material, figure S3). In each of these analyses, the phylogenetic position of the breviates was strongly supported by the method, receiving a PP of 1.0 and 98% BS. We suspected that the differences in the topologies recovered by these analyses stem from the different models used in the BI versus the ML analyses. This was confirmed by BI analyses under the LG + Γ4 + F model, which recovered the same BA clade as in the ML analysis (see the electronic supplementary material, figure S4) (the reciprocal experiment could not be carried out because the CAT-GTR model is not implemented in any ML software). To further examine support for various placements of the breviates in eukaryote phylogeny, we used ‘approximately unbiased’ (AU) topology tests [44]. Under the LG + Γ4 + F model, these tests reject the placement of breviates as sister to Amoebozoa, Collodictyon or Malawimonas (electronic supplementary material, table S1). However, the AU tests could not reject that breviates are sister to an OA clade, the topology that is observed in the CAT-GTR + Γ4 analyses.
To investigate the sensitivity of these results to taxon sampling, we conducted systematic fast-evolving taxon removal analyses. The deletion series were investigated using ML (LG + Γ4 + F), with which, support for the OBA clade remained strong (greater than 99% BS) as did support for the BA grouping (see the electronic supplementary material, figure S12). Investigation of the 56-taxon and last deletion (46-taxon) datasets with Phylobayes CAT-Poisson + Γ4 model showed support for the OBA clade, with the breviates branching outside of the OA clade (PP = 0.94 and 0.78, respectively; electronic supplementary material, figures S13 and S14). To see whether the topology within Amorphea is affected by other taxa in the tree, we also analysed a 23-taxon dataset that contained only Amoebozoa and OBA taxa and again, the same two conflicting placements of breviates were obtained in ML (LG + Γ4 + F) versus Phylobayes CAT-GTR + Γ4 analyses (see the electronic supplementary material, figure S15), showing that this feature of our analyses is insensitive to this substantial difference in taxon sampling.
Cross-validation analyses of the full dataset and the 23-taxon datasets in Phylobayes show that the CAT-GTR + Γ4 model fits the data much better than LG + Γ4 + F (average lnL difference of 9664.8 [σ ± 223] and 1735.02 [σ ± 83] in favour of CAT-GTR + Γ4 for respective datasets). Therefore, we suspected that the BA clade recovered in ML and BI analyses under LG + Γ4 + F may be caused by systematic error arising from failure to model site-specific substitution dynamics under the site-homogeneous LG + Γ4 + F model, as has been previously observed [45]. To further test this possibility, we assessed the likelihood of the two alternative topologies recovered in the analyses of the 23-taxon dataset that differed only in the position of the breviates using a 10 component mixture model (10cF) that, like CAT-GTR, is designed to capture site-specific substitution dynamics implemented in QMM-RAxML [46,47]. This mixture model fits the dataset significantly better than LG + Γ4 + F (ΔlnL = −8812.05, likelihood-ratio-test p-value = 0) and prefers the Phylobayes CAT-GTR + Γ4 topology with breviates splitting off before the OA clade.
We also evaluated the impact of removing the fastest evolving sites from the 43 615 amino acid supermatrix, since these are expected to be the ‘noisiest’ and contribute the most to systematic error in the analysis [45,48]. Sites were removed in a stepwise fashion from fastest to slowest in 1000 site increments and ML-bootstrap analyses were performed under LG + Γ4 + F for resulting alignments. This illustrates that support for the monophyly of BA rapidly decreases as fast-evolving sites are removed, whereas support for the larger clade of OBA remains very stable (figure 3). As support for the BA decreased, support for the alternative OA clade increased in agreement with the CAT-GTR + Γ4 BI analyses. After 18 000 sites were removed, the latter topology was optimal (see the electronic supplementary material, figure S5). Therefore, the topology observed in the ML and BI analyses with LG + Γ4 + F is probably an artefact resulting from model misspecification, which can be overcome either through the use of a more realistic phylogenetic model like site-heterogeneous models such as CAT-GTR + Γ4 [39,49] and the 10cF model [46,47] or through deletion of fast-evolving sites. Manipulations of other model features, including amino acid frequencies in ML analyses (i.e. LG + Γ4 versus LG + Γ4 + F), removal of constant sites (data not shown) or accounting for heterotachy under LG + Γ4 + F using the Pahadist ML distance method (see the electronic supplementary material, figure S16), still showed an apusomonad + breviate clade, but with weaker support (65%). In the absence of heterotachy modelling, this clade remained strongly supported despite sequential removal of fast-evolving genes (see the electronic supplementary material, figure S17).
Figure 3.
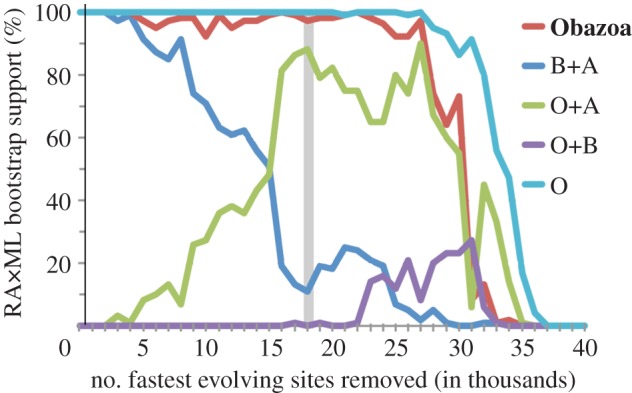
Sites were sorted based on their rates of evolution and removed from the dataset from highest to lowest rate. The bootstrap values for each bipartition of interest are plotted. The dataset with 18 000 sites removed (grey bar) was analysed further; see the electronic supplementary material, figure S5. O, Opisthokonta; B, Breviatea; A, Apusomonadida.
Regardless of the precise position of the breviates, the uniformly strong support for an OBA clade in all analyses under all models argues strongly against previous inferences of an Amoebozoa affinity for breviates, based on more limited site coverage and taxon sampling [16]. This clade is also robust to long branching taxa and gene removal experiments (see the electronic supplementary material, figure S12–S16). The OBA clade is a very large and important eukaryote group that has no name at present. We propose the name ‘Obazoa’ for this clade (see below).
One of the proposed molecular synapomorphies suggested to unite the ‘bikont’ lineages (i.e. Diaphoretickes plus Excavata [2]) as a eukaryotic mega-clade is a dihydrofolate reductase-thymidylate synthase (DHFR-TS) gene fusion. This fusion is lacking in prokaryotes, Amoebozoa and Opisthokonta, which all have separate DHFR and TS genes, when present. However, the DHFR-TS gene fusion is also present in the apusomonad, Thecamonas trahens [11], which casts doubt on its robustness as a phylogenetic character, given that the apusomonads are closely related to the Opisthokonta (figure 2) [7,18,50]. We searched the P. biforma RNA-seq data for DHFR and TS transcripts, and found sequences encoding separate DHFR and TS proteins (see the electronic supplementary material, data), such as opisthokonts and amoebozoans, but unlike apusomonads. The precise history of the DHFR-TS gene fusion in the ‘bikonts’ and apusomonads is currently unclear. Another molecular synapomorphy proposed to support an Amoebozoa and Opisthokonta clade is a unique glycine insertion in myosin class II paralogues in the motif FIDFGLDL [51]. Both Thecamonas and P. biforma have myosin-II proteins with this glycine insertion (electronic supplementary material, data). However, this character also has a complex history, as a myosin-II with this glycine insertion is also found in Naegleria gruberi [52].
(c). Pygsuia has a nearly complete integrin-mediated adhesion complex
Recent evidence indicates that some protein complexes important for metazoan multicellularity originated much earlier in evolution than true animals, even before the divergence of Metazoa from Fungi [26]. One of these systems, the IMAC, apparently predates the OA split, as genes encoding its core components were found in the genome of the apusomonad Thecamonas [26]. Given the relationship of the breviates to the opisthokonts and apusomonads described earlier, we sought transcripts encoding IMAC proteins in the RNA-seq data from Pygsuia. We identified a similar set of IMAC proteins to that described in Thecamonas [26]. Specifically, in P. biforma, we identified one ITA and one ITB. Both subunits are significantly longer than their metazoan homologues, in both the expansion is in the extracellular stalk region of the protein (figure 4a,b). The stalk region of ITA is dramatically extended in P. biforma, resulting in a protein that is three times as long as its metazoan orthologues, even though it lacks the ITA leg domain that is found only in metazoan ITAs (figure 4a). Canonical metazoan ITBs have a stalk made up of a single extracellular epidermal growth factor (EGF) domain with three to four cysteine-rich stalk repeat (CRSR) motifs. However, the ITB homologue in P. biforma has 29 EGF domains and 27 CRSR (figure 4b). Many integrin-interacting scaffolding and signalling cytoplasmic proteins are also present in P. biforma (figure 4c). Scaffolding proteins, such as paxilin, talin, α-actinin and pinch, are present, as well as an integrin-linked kinase (ILK; see the electronic supplementary material, data). We were unable to identify a focal adhesion kinase (FAK) or a c-src kinase in the data, which is consistent with their absence in Thecamonas [26] and suggests that they may have appeared later during opisthokont evolution. We were also unable to identify two actin binding proteins, parvin and vinculin (figure 4c). The absence of a vinculin gene is further supported by the fact that the talin protein of P. biforma conspicuously lacks a vinculin-binding domain, which is otherwise present in talin homologues found throughout Amorphea (see the electronic supplementary material, figure S18). Tentatively, these data indicate that parvin originated after the divergence of OA from the breviates (figure 4c) and vinculin originated early in the Amorphea, but were secondarily lost in P. biforma.
Figure 4.

(a) Domain architecture of ITA from P. biforma (Pb) compared to a canonical ITA of Homo sapiens (Hs) (ITGA5, GenBank NP_002196) and the ITA in Thecamonas trahens (Tt) [26]. (b) Domain architecture of ITB of Pb compared to a canonical ITB of Hs (ITGB1, GenBank NP_391988) and the ITB in Tt [26]. SP, signal peptide (SignalP-NN(euk)); ITA head, integrin α, β-propeller (IPR013519); FG, FG-gap (PS51470); ITA leg, integrin α-2 (IPR013649); TM, transmembrane domain; ITB head, integrin β chain (PF00362); EGF, epidermal growth factor; extracellular (IPR013111); BT, integrin-B tail (PF07965); BC, integrin-B cytoplasmic region (PF08725). Vertical purple lines represent cysteine-rich repeats (CxCxxCxC) (PS00243). The number of EGF and CxCxxCxC are indicated. (c) Phylogenetic distribution of IMAC. The inset depicts IMAC coloured in accordance with the distributions shown in the tree. Circle, innovation/gain; bar, loss. Abbreviations: vin, vinculin; pax, paxillin; tal, talin; par, parvin; pin, particularly interesting new cysteine-histidine-rich protein; FAK, focal adhesion kinase; ILK, integrin-linked kinase; αA, alpha-actinin; α, α-integrin; β, β-integrin; IP, Interpro; PF, Pfam; PS, ProSite.
The discovery of IMAC components in Pygsuia, originally thought to be specific to Metazoa, pushes back the origin for the integrin machinery to before the origin of the Obazoa. As has been previously described, several cytoplasmic signalling proteins that function with integrins such as pinch, paxillin, talin and vinculin appear to be Amorphea-specific innovations, as they are also present in amoebozoans [26]. Many of these proteins were subsequently lost in the Fungi, in particular the Dikaryomycetes [26]. The loss of the IMAC machinery was gradual in Fungi, as some signalling proteins, and ILK, are present in basal Fungi (figure 4c) [26]. The full suite of proteins in the integrin-related adhesion machinery as characterized in animals (i.e. including FAK and c-src) appears to be a holozoan innovation, with integrins secondarily lost in choanoflagellates (figure 4c). The similar IMAC machineries of Thecamonas and Pygsuia possibly reflect the ancestral architecture of the system. The functions of integrins in these protists are unknown at present, but could be related to motility, predation or a sexual cycle. In metazoans, the IMAC functions in the recruitment of actin polymerization in protrusion of new pseudopodia and could have an analogous function in protistan cells, especially given that the breviates, apusomonads and Capsaspora all produce pseudopodia.
(d). Conclusion
With the inclusion of the RNA-seq data presented here, we definitively show that the breviates are related to the opisthokonts and apusomonads and do not emerge as sister to, or within, the Amoebozoa as previously reported [16,21]. We suspect that previous analyses were not robust because of taxon sampling issues, particularly the lack of apusomonad taxa as well as the relatively few genes that were available from B. anathema [16,17]. The Obazoa clade is very well supported and stable in our analyses (figure 3). The placement of breviates as sister to the opisthokonts and apusomonads is consistent with an emerging scheme of morphological evolution for Opisthokonta [21,53]. For example, like Pygsuia and the apusomonads, the last common ancestor of the Obazoa is very likely to have been a biflagellate. Moreover, flagellated organisms in the Obazoa clade appear to have a bicentriolar flagellar apparatus (kinetid), some with a secondarily reduced kinetid with a non-flagellated secondary basal body as in some breviates and nearly all flagellated opisthokonts (some Neocallimastigaceae fungi secondarily have a unicentriolar flagellar apparatus [2,54]). The ancestral organism was probably capable of forming filose pseudopodia-like breviates, some apusomonads, the nucleariid amoebae, many holozoans and many metazoan cell types (also see [10]).
The nearly complete IMAC in P. biforma and Thecamonas trahens suggests that the bulk of these proteins are an ancestral innovation in the Obazoa clade (figure 4) and were secondarily lost in various opisthokont groups [26]. We expect data from more unicellular relatives of Opisthokonta will have significant implications for understanding the underlying molecular mechanisms and ancestral genetic repertoire that enabled the opisthokonts to evolve into the diversity of multicellular life forms alive today.
Although the analyses presented here clarify our understanding of deep-level eukaryotic evolution in the major group Amorphea, there are several outstanding issues that still need to be addressed. Most importantly, the other organisms considered apusozoans or recently sulcozoans [3,10], such as Ancyromonas, Planomonas, Micronuclearia, Rigifila [55] and Mantamonas [56] should be examined using phylogenomics. Although small-scale analyses weakly suggest that these are distantly related to the breviates, apusomonads or Opisthokonta (see the electronic supplementary material, figure S11) [21,55], their inclusion in phylogenomic datasets is of key importance to elucidating the evolutionary history of Amorphea.
4. Taxonomic summary and description
Amorphea Adl et al. 2012
Obazoa Brown et al.: the least inclusive clade containing Homo sapiens Linnaeus 1758 (Opisthokonta), Neurospora crassa Shear & Dodge 1927 (Opisthokonta), P. biforma Brown et al. (Breviatea), and Thecamonas trahens Larsen & Patterson 1990 (Apusomonadida). This is a node-based definition in which all of the specifiers are extant; it is intended to apply to a crown clade.
Etymology: the term Obazoa is based on an acronym of Opisthokonta, Breviatea and Apusomonadida, plus ‘zóa’ (pertaining to ‘life’ in Greek).
Breviatea Cavalier-Smith 2004
Pygsuia Brown et al. gen. nov.
Small, pear-shaped but amoeboid flagellates, with two flagella, one apical, one directed posteriorly. In adherent cells, the posterior flagellum usually attaches to the cell; the cells do not swim, but locomote along substrates, typically forming filose pseudopodia at the anterior end, which seem to move along the cell body in a ‘conveyer belt’ fashion. Swimming cells have a free and long posterior flagellum, swim with vibration and rotation, or glide on the posterior flagellum. Cells often form elongate posterior attachments that may branch. Mitochondrion-like organelles elongate, usually singular and lacking obvious cristae. Microaerophilic and bacterivorous. Cysts not observed.
Type species: Pygsuia biforma nov. sp.
Pygsuia biforma Brown et al. nov. sp.
Species with characteristics of the genus. Flagellates of two cell types. Adherent cells 8.5–18.5 µm long and 5–8 µm broad. Swimming cells 8.5–13 µm long and 3.7–5.3 µm broad. Anterior flagellum 8.5–28 µm. Posterior flagellum 16–37 µm in length, in swimming cells, less than the cell length in adherent cells. Fine pseudopodia typically 2–5 µm. Posterior filaments may reach ca 4 µm.
Habitat: this species was isolated from brackish estuarine sediment collected just below the waterline from Prince Cove, Marstons Mills, MA, USA (41.641681° N, –70.413421° W).
Reference material: a fixed and embedded resin TEM block of this culture was deposited in the Smithsonian Museum. This permanent physical specimen is considered the hapantotype (name-bearing type) of the species (see Art. 73.3 of the International Code for Zoological Nomenclature, 4th Edition).
Gene sequence data: the nearly complete SSU rRNA gene of the type isolate (PCbi66) is deposited in GenBank under accession no. KC433554.
Etymology: the genus name is derived from part of the University of Arkansas Razorback's sports cheer ‘Woo Pig Sooie’, because the row of filose pseudopodia resembles the dorsal bristles of razorbacks (feral pigs). ‘Pyg’ replaces ‘pig’ as a play on Pygmaeī (Latin) (‘a mythical race of pygmies’) referring to their small size and ‘sui’ replaces ‘sooie’ for brevity and to refer to the animal family to which suids belong (Suidae). Consequently, the genus name also means ‘little pig’ in mock Latin. The species name is derived from the presence of two distinct cell forms (adherent and swimming) (‘bi-forma’) (Latin) that are observed in the life cycle.
Acknowledgements
We thank the Broad Institute (http://www.broadinstitute.org/annotation/genome/multicellularity_project/) and the Baylor College of Medicine (http://www.hgsc.bcm.tmc.edu/microbial-detail.xsp?project_id=163) for access to genome data. We thank Dr Martin Kolisko for bioinformatics help. Thanks to Dr Ping Li for electron microscopy support. Also, thanks to Dr Nicolas Lartillot for use of Phylobayes-mpi before its public release.
Funding statement
Computing resources were provided by the University of Toronto's SciNet Supercomputing facility, of Compute/Calcul Canada. M.W.B. was supported by a postdoctoral fellowship from the Tula Foundation. This work was supported by a grant no. MOP-62809, awarded to A.J.R. and A.G.B.S. by the Canadian Institutes for Health Research. This research was partly supported by the Natural Sciences and Engineering Research Council of Canada Discovery grants no. 298366-09 (A.G.B.S.) and 194560-201 (B.F.L.). J.D.S. acknowledges support from the Arkansas Biosciences Institute.
References
- 1.Walker G, Dorrell RG, Schlacht A, Dacks JB. 2011. Eukaryotic systematics: a user's guide for cell biologists and parasitologists. Parasitology 138, 1638–1663 (doi:10.1017/S0031182010001708) [DOI] [PubMed] [Google Scholar]
- 2.Adl SM, et al. 2012. The revised classification of eukaryotes. J. Eukaryot. Microbiol. 59, 429–514 (doi:10.1111/j.1550-7408.2012.00644.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavalier-Smith T. 2002. The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa. Int. J. Syst. Evol. Microbiol. 52, 297–354 [DOI] [PubMed] [Google Scholar]
- 4.Cavalier-Smith T, Chao EEY, Oates B. 2004. Molecular phylogeny of Amoebozoa and the evolutionary significance of the unikont Phalansterium. Eur. J. Protistol. 40, 21–48 (doi:10.1016/J.Ejop.2003.10.001) [Google Scholar]
- 5.Karpov SA, Mylnikov AP. 1989. Biology and ultrastructure of colorless flagellates Apusomonadida Ord N. Zool. Zh. 68, 5–17 [Google Scholar]
- 6.Derelle R, Lang F. 2012. Rooting the eukaryotic tree with mitochondrial and bacterial proteins. Mol. Biol. Evol. 29, 1277–1289 (doi:10.1093/Molbev/Msr295) [DOI] [PubMed] [Google Scholar]
- 7.Torruella G, Derelle R, Paps J, Lang BF, Roger AJ, Shalchian-Tabrizi K, Ruiz-Trillo I. 2012. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 29, 531–544 (doi:10.1093/Molbev/Msr185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalier-Smith T, Chao EE, Stechmann A, Oates B, Nikolaev S. 2008. Planomonadida ord. nov. (Apusozoa): ultrastructural affinity with Micronuclearia podoventralis and deep divergences within Planomonas gen. nov. Protist 159, 535–562 (doi:10.1016/j.protis.2008.06.002) [DOI] [PubMed] [Google Scholar]
- 9.Heiss AA, Walker G, Simpson AG. 2010. Clarifying the taxonomic identity of a phylogenetically important group of eukaryotes: Planomonas is a junior synonym of Ancyromonas. J. Eukaryot. Microbiol. 57, 285–293 (doi:10.1111/j.1550-7408.2010.00477.x) [DOI] [PubMed] [Google Scholar]
- 10.Cavalier-Smith T. 2012. Early evolution of eukaryote feeding modes, cell structural diversity, and classification of the protozoan phyla Loukozoa, Sulcozoa, and Choanozoa. Eur. J. Protistol. 49, 115–178 (doi:10.1016/j.ejop.2012.06.001) [DOI] [PubMed] [Google Scholar]
- 11.Stechmann A, Cavalier-Smith T. 2002. Rooting the eukaryote tree by using a derived gene fusion. Science 297, 89–91 (doi:10.1126/science.1071196) [DOI] [PubMed] [Google Scholar]
- 12.Stechmann A, Cavalier-Smith T. 2003. The root of the eukaryote tree pinpointed. Curr. Biol. 13, R665–666 (doi:10.1016/S0960-9822(03)00602-X) [DOI] [PubMed] [Google Scholar]
- 13.Walker G, Dacks JB, Martin Embley T. 2006. Ultrastructural description of Breviata anathema, n. gen., n. sp., the organism previously studied as ‘Mastigamoeba invertens’. J. Eukaryot. Microbiol. 53, 65–78 (doi:10.1111/j.1550-7408.2005.00087.x) [DOI] [PubMed] [Google Scholar]
- 14.Parfrey LW, Grant J, Tekle YI, Lasek-Nesselquist E, Morrison HG, Sogin ML, Patterson DJ, Katz LA. 2010. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst. Biol. 59, 518–533 (doi:10.1093/Sysbio/Syq037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burki F, Shalchian-Tabrizi K, Minge M, Skjaeveland A, Nikolaev SI, Jakobsen KS, Pawlowski J. 2007. Phylogenomics reshuffles the eukaryotic supergroups. PLoS ONE 2, e790 (doi:10.1371/journal.pone.0000790) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minge MA, Silberman JD, Orr RJ, Cavalier-Smith T, Shalchian-Tabrizi K, Burki F, Skjaeveland A, Jakobsen KS. 2009. Evolutionary position of breviate amoebae and the primary eukaryote divergence. Proc. R. Soc. B 276, 597–604 (doi:10.1098/rspb.2008.1358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao S, Burki F, Bråte J, Keeling PJ, Klaveness D, Shalchian-Tabrizi K. 2012. Collodictyon: an ancient lineage in the tree of eukaryotes. Mol. Biol. Evol. 29, 1557–1568 (doi:10.1093/molbev/mss001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim E, Simpson AG, Graham LE. 2006. Evolutionary relationships of apusomonads inferred from taxon-rich analyses of 6 nuclear encoded genes. Mol. Biol. Evol. 23, 2455–2466 (doi:10.1093/molbev/msl120) [DOI] [PubMed] [Google Scholar]
- 19.Brown MW, Kolisko M, Silberman JD, Roger AJ. 2012. Aggregative multicellularity evolved independently in the eukaryotic supergroup Rhizaria. Curr. Biol. 22, 1123–1127 (doi:10.1016/J.Cub.2012.04.021) [DOI] [PubMed] [Google Scholar]
- 20.Katz LA, Grant J, Parfrey LW, Gant A, O'Kelly CJ, Anderson OR, Molestina RE, Nerad T. 2011. Subulatomonas tetraspora nov gen. nov. sp. is a member of a previously unrecognized major clade of eukaryotes. Protist 162, 762–773 (doi:10.1016/J.Protis.2011.05.002) [DOI] [PubMed] [Google Scholar]
- 21.Paps J, Medina-Chacón LA, Marshall W, Suga H, Ruiz-Trillo I. 2012. Molecular phylogeny of unikonts: new insights into the position of apusomonads and ancyromonads and the internal relationships of opisthokonts. Protist. 164, 2–12 (doi:10.1016/j.protis.2012.09.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant JR, Lahr DJG, Rey F, Burleigh JG, Gordon J, Knight R, Molestina R, Katz L. 2012. Gene discovery from a pilot study of the transcriptomes from three diverse microbial eukaryotes: Corallomyxa tenera, Chilodonella uncinata, and Subulatomonas tetraspora. Protist Genomics 1, 3–18 (doi:10.2478/prge-2012-0002) [Google Scholar]
- 23.Suga H, Dacre M, de Mendoza A, Shalchian-Tabrizi K, Manning G, Ruiz-Trillo I. 2012. Genomic survey of premetazoans shows deep conservation of cytoplasmic tyrosine kinases and multiple radiations of receptor tyrosine kinases. Sci. Signal. 5, ra35 (doi:10.1126/scisignal.2002733) [DOI] [PubMed] [Google Scholar]
- 24.King N, et al. 2008. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788 (doi:10.1038/nature06617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King N. 2004. The unicellular ancestry of animal development. Dev. Cell 7, 313–325 (doi:10.1016/j.devcel.2004.08.010) [DOI] [PubMed] [Google Scholar]
- 26.Sebe-Pedros A, Roger AJ, Lang FB, King N, Ruiz-Trillo I. 2010. Ancient origin of the integrin-mediated adhesion and signaling machinery. Proc. Natl Acad. Sci. USA 107, 10 142–10 147 (doi:10.1073/pnas.1002257107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai X, Clapham DE. 2012. Ancestral Ca2+ signaling machinery in early animal and fungal evolution. Mol. Biol. Evol. 29, 91–100 (doi:10.1093/molbev/msr149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai X. 2012. Ancient origin of four-domain voltage-gated Na+ channels predates the divergence of animals and fungi. J. Membr. Biol. 245, 117–123 (doi:10.1007/s00232-012-9415-9) [DOI] [PubMed] [Google Scholar]
- 29.Vicente-Manzanares M, Choi CK, Horwitz AR. 2009. Integrins in cell migration: the actin connection. J. Cell Sci. 122, 199–206 (doi:10.1242/jcs.018564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harburger DS, Calderwood DA. 2009. Integrin signalling at a glance. J. Cell Sci. 122, 159–163 (doi:10.1242/Jcs.018093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518 (doi:10.1093/Nar/Gki198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 (doi:10.1080/10635150802429642) [DOI] [PubMed] [Google Scholar]
- 33.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320 (doi:10.1093/molbev/msn067) [DOI] [PubMed] [Google Scholar]
- 34.Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, Roger AJ. 2009. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic ‘supergroups’. Proc. Natl Acad. Sci. USA 106, 3859–3864 (doi:10.1073/pnas.0807880106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burki F, Okamoto N, Pombert JF, Keeling PJ. 2012. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc. R. Soc. B 279, 2246–2254 (doi:10.1098/rspb.2011.2301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burki F, et al. 2009. Large-scale phylogenomic analyses reveal that two enigmatic protist lineages, Telonemia and Centroheliozoa, are related to photosynthetic chromalveolates. Genome Biol. Evol. 1, 231–238 (doi:10.1093/gbe/evp022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burki F, Shalchian-Tabrizi K, Pawlowski J. 2008. Phylogenomics reveals a new ‘megagroup’ including most photosynthetic eukaryotes. Biol. Lett. 4, 366–369 (doi:10.1098/rsbl.2008.0224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burki F, Kudryavtsev A, Matz MV, Aglyamova GV, Bulman S, Fiers M, Keeling PJ, Pawlowski J. 2010. Evolution of Rhizaria: new insights from phylogenomic analysis of uncultivated protists. BMC Evol. Biol. 10, 377 (doi:10.1186/1471-2148-10-377) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lartillot N, Lepage T, Blanquart S. 2009. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (doi:10.1093/bioinformatics/btp368) [DOI] [PubMed] [Google Scholar]
- 40.Susko E, Field C, Blouin C, Roger AJ. 2003. Estimation of rates-across-sites distributions in phylogenetic substitution models. Syst. Biol. 52, 594–603 (doi:10.1080/10635150390235395) [DOI] [PubMed] [Google Scholar]
- 41.Larsen J, Patterson DJ. 1990. Some flagellates (Protista) from tropical marine-sediments. J. Nat. Hist. 24, 801–937 (doi:10.1080/00222939000770571) [Google Scholar]
- 42.Heiss AA, Walker G, Simpson AG. 2013. The flagellar apparatus of Breviata anathema, a eukaryote without a clear supergroup affinity. Eur. J. Protistol. 49, 354–372 (doi:10.1016/j.ejop.2013.01.001) [DOI] [PubMed] [Google Scholar]
- 43.Edgcomb VP, Simpson AG, Zettler LA, Nerad TA, Patterson DJ, Holder ME, Sogin ML. 2002. Pelobionts are degenerate protists: insights from molecules and morphology. Mol. Biol. Evol. 19, 978–982 (doi:10.1093/oxfordjournals.molbev.a004157) [DOI] [PubMed] [Google Scholar]
- 44.Shimodaira H, Hasegawa M. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247 (doi:10.1093/bioinformatics/17.12.1246) [DOI] [PubMed] [Google Scholar]
- 45.Lartillot N, Philippe H. 2008. Improvement of molecular phylogenetic inference and the phylogeny of Bilateria. Proc. R. Soc. B 363, 1463–1472 (doi:10.1098/rstb.2007.2236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang HC, Li K, Susko E, Roger AJ. 2008. A class frequency mixture model that adjusts for site-specific amino acid frequencies and improves inference of protein phylogeny. BMC Evol. Biol. 8, 331 (doi:10.1186/1471-2148-8-331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gaston D, Susko E, Roger AJ. 2011. A phylogenetic mixture model for the identification of functionally divergent protein residues. Bioinformatics 27, 2655–2663 (doi:10.1093/bioinformatics/btr470) [DOI] [PubMed] [Google Scholar]
- 48.Olsen GJ. 1987. Earliest phylogenetic branchings: comparing rRNA-based evolutionary trees inferred with various techniques. Cold Spring Harb. Symp. Quant. Biol. 52, 825–837 (doi:10.1101/SQB.1987.052.01.090) [DOI] [PubMed] [Google Scholar]
- 49.Quang LS, Gascuel O, Lartillot N. 2008. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 24, 2317–2323 (doi:10.1093/Bioinformatics/Btn445) [DOI] [PubMed] [Google Scholar]
- 50.Roger AJ, Simpson AG. 2009. Evolution: revisiting the root of the eukaryote tree. Curr. Biol. 19, R165–R167 (doi:10.1016/j.cub.2008.12.032) [DOI] [PubMed] [Google Scholar]
- 51.Richards TA, Cavalier-Smith T. 2005. Myosin domain evolution and the primary divergence of eukaryotes. Nature 436, 1113–1118 (doi:10.1038/nature03949) [DOI] [PubMed] [Google Scholar]
- 52.Odronitz F, Kollmar M. 2007. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 8, R196 (doi:10.1186/Gb-2007-8-9-R196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown MW, Spiegel FW, Silberman JD. 2009. Phylogeny of the ‘forgotten’ cellular slime mold, Fonticula alba, reveals a key evolutionary branch within Opisthokonta. Mol. Biol. Evol. 26, 2699–2709 (doi:10.1093/Molbev/Msp185) [DOI] [PubMed] [Google Scholar]
- 54.James TY, Letcher PM, Longcore JE, Mozley-Standridge SE, Porter D, Powell MJ, Griffith GW, Vilgalys R. 2006. A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98, 860–871 (doi:10.3852/mycologia.98.6.860) [DOI] [PubMed] [Google Scholar]
- 55.Yabuki A, Ishida KI, Cavalier-Smith T. 2012. Rigifila ramosa n. gen., n. sp., a filose apusozoan with a distinctive pellicle, is related to Micronuclearia. Protist 164, 75–88 (doi:10.1016/j.protis.2012.04.005) [DOI] [PubMed] [Google Scholar]
- 56.Glücksman E, Snell EA, Berney C, Chao EE, Bass D, Cavalier-Smith T. 2011. The novel marine gliding zooflagellate genus Mantamonas (Mantamonadida ord. n.: Apusozoa). Protist 162, 207–221 (doi:10.1016/j.protis.2010.06.004) [DOI] [PubMed] [Google Scholar]