

Abstract
Background:
Crystal storing histiocytosis (CSH) is a disorder characterized by local or diffuse infiltration of histiocytes containing crystalline inclusions most commonly of immunoglobulin light chain. Involvement of the central nervous system is extremely rare. CSH may be misdiagnosed as an infection or tumor. In patients with involvement of other organs, it is frequently associated with lymphoplasmacytic diseases.
Case Description:
A 20-year-old female was evaluated for 2 weeks of progressively worsening headaches. At presentation, she had no history of fevers but reported a sore throat without cough 3-4 days prior. Her past medical history was unremarkable. She denied intravenous drug use or sexually transmitted diseases but lived with an individual with a history of fungal meningitis. On examination she was afebrile, alert, and oriented with a blood pressure of 110/70 mmHg. She had no adenopathy or neurological deficits. Her white blood cell count was minimally elevated. Magnetic resonance imaging revealed a 3.5 × 1.3 × 1.9 cm contrast enhancing lesion of the left temporal lobe with a mild midline shift. Evaluation by multiple specialists suggested a differential diagnosis of an infectious or neoplastic process. Cultures for infectious agents were negative. The biopsy showed CSH. Postoperatively and at 1 month follow up, she was neurologically intact.
Conclusion:
Radiographically and intraoperatively, CSH may mimic an infectious process or neoplasm. Its recognition is critical to facilitate appropriate therapy and prompt screening for an occult lymphoplasmacytic neoplasm, plasma cell dyscrasia or other underlying disease.
Keywords: Crystals, crystal storing histiocytosis, histiocytes, pseudotumor
INTRODUCTION
Crystal storing histiocytosis (CSH) is a disorder characterized by local or diffuse accumulation of histiocytes containing crystalline inclusions with a review and classification recently proposed by Dogan et al.[1] In the majority of cases this is associated with lymphoplasmacytic diseases that produce monoclonal immunoglobulins such as plasma cell dyscrasias, multiple myeloma, or lymphoproliferative disorders such as a marginal or mantle zone B cell lymphomas. Rare cases may develop in association with a paraproteinemia such as monoclonal gammopathy of unknown significance (MGUS).[1] CSH usually occurs at sites in the head and neck, lung, bone marrow, or kidney.[1] Involvement of the central nervous system (CNS) is extremely rare. To our knowledge, only three well-documented cases involving the CNS have been reported.[2,3,4,5] We describe an atypical case of CSH and review the literature.
CASE REPORT
A 20-year-old female was evaluated at an ambulatory care center for headaches that had become progressively worse over the prior 2 weeks. She had been treated for mycoplasma infection by a primary care physician. At presentation, the headaches were reportedly severe. The patient had no history of fevers but reported a sore throat 3-4 days prior without a cough or other discomfort. Her past medical history had no notable illnesses. Regarding her social history, she lived on a farm with a friend who had been treated for fungal meningitis in the past. No tobacco, alcohol, or intravenous drug use was admitted. She denied exposure or prior treatment for sexually transmitted diseases. She had no significant past medical history. On examination she was afebrile with a blood pressure of 110/70 mmHg. She was alert and oriented with no adenopathy. A complete blood count revealed a white blood cell (WBC) of 12.4 (4.2-9.1 thousand/microliter), 87% neutrophils with no bands. Serology was negative for mycoplasma and HIV-1. Her neurological exam was normal. Magnetic resonance imaging revealed a 3.5 × 1.3 × 1.9 cm contrast enhancing lesion of the left temporal lobe with a mild midline shift [Figure 1]. Evaluation by multiple specialists suggested a differential diagnosis of a noninfectious process (favored) neoplastic process or infection. She was treated with vancomycin 1.25 g IV per 12 h and meropenem 2 g IV q 8 h and decadron.
Figure 1.
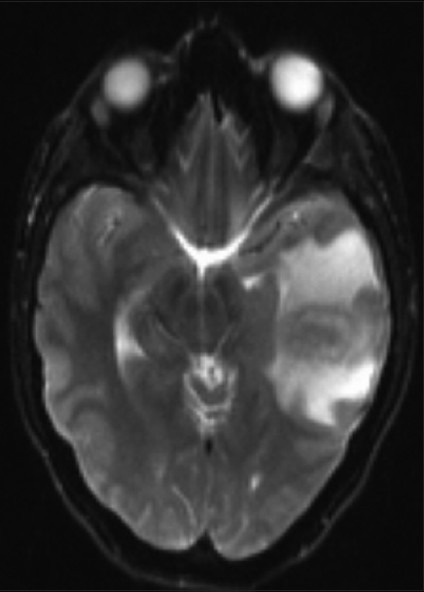
Magnetic resonance image demonstrating left temporal lobe enhancing lesion. T2-weighted image postgadolinium showing a peripherally enhancing mass with mass with edema producing mild ventricular compression.
The mass was resected and aerobic, anaerobic, mycobacterial, and fungal cultures were taken. Anaerobic and aerobic cultures as well as gram, modified acid fast bacillus, and fungal stains were negative. Brain tissue polymerase chain reaction analysis at the state Department of Public Health laboratory public was negative for infectious agents. Due to initial uncertainty about the diagnosis and anticipation of the need for multiple studies in the laboratories, she was postoperatively treated with vancomycin IV for 10 days. Postoperative scans revealed gross total resection. On postoperative day 1 and 30 she was neurologically intact.
The sections revealed a fibrotic lesion with an extensive chronic inflammatory infiltrate of CD138-immunoreactive plasma cells, CD68-immunoreactive macrophages and monocytes and S100-immunoreactive histiocytes [Figure 2a]. Plasma cells showed IgG, IgM, predominantly lambda but also some kappa immunoreactivity. There were numerous periodic acid-Schiff (PAS) staining rhomboid crystals and granular material that was predominantly intracellular but also extracellular [Figure 2b]. No polarizable material was found. Special stains for bacteria including Nocardia and Mycobacteria were negative. The Grocott's Methenamine Silver GMS stains revealed no fungal organisms. The Masson stain demonstrated extensive dense collagen deposition but no encysted amoeba. There was no Congo red staining apple green birefringent polarizable material. There were no giant cells, viral inclusions, Herpes Simple ×1 or 2, cytomegalovirus, Epstein–Barr virus, or Toxoplasma immunoreactivity. There was no Alk-1 but scattered spindle cells and blood vessels show SMA immunoreactivity. To evaluate whether immunoglobulin expression was predominantly one type, additional immunoflourescence and in situ hybridization were performed. These demonstrated overwhelmingly lambda light chain restriction [Figure 3]. Electron microscopy revealed numerous intracellular but also extracellular rhomboid and diamond-shaped crystals [Figure 4]. Collectively the findings of histiocytes with intracellular rhomboid and diamond-shaped crystals showing light chain and exclusion of other neoplastic and infectious processes were considered consistent with CSH, light chain subtype.
Figure 2.
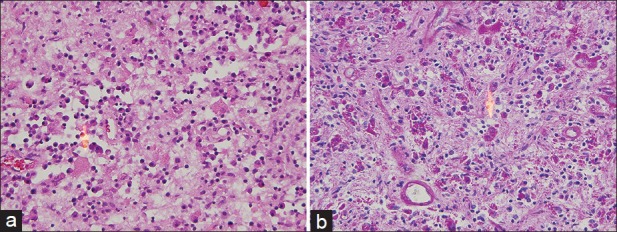
Brain lesion with crystal storing histiocytosis. (a) Numerous histiocytes contain rhomboid and needle-like kappa light chain restricted crystals are inconspicuous with standard stains (Hematoxylin and eosin, original magnification ×200). (b) Crystal deposition is apparent with periodic acid Schiff stain (PAS stain, original magnification ×200)
Figure 3.
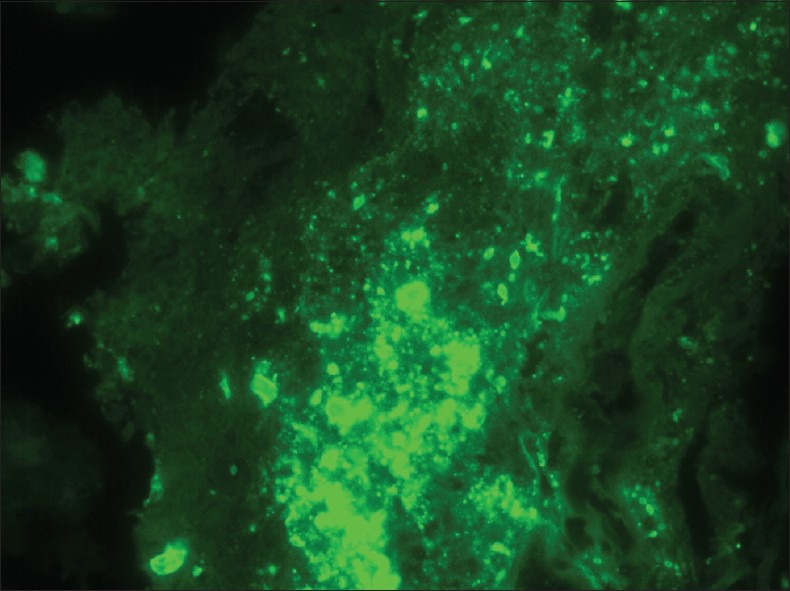
Brain lesion demonstrating lambda light chain immunofluorescence. Crystals show lambda but not kappa immunoflourescence. (Original magnification ×600)
Figure 4.
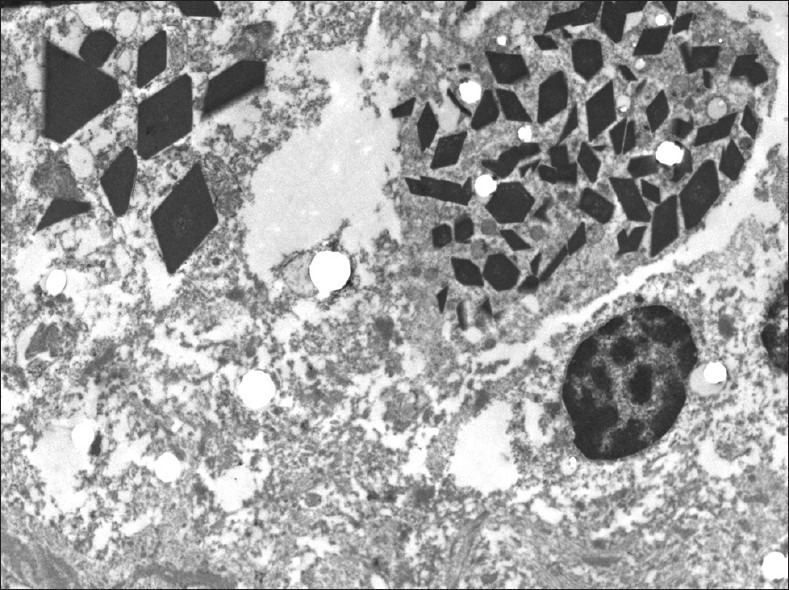
Electron microscopy. Histiocytes show intracellular rhomboid and needle-like inclusions. (Original magnification ×10,000)
DISCUSSION
Recognition of CSH is important due to the association with occult but potentially treatable lymphoplasamcytic disorders. Nonetheless, in CSH involving the CNS, the limited number of cases suggests that many may not have systemic disease. Approximately 90% of CSH occurs in a setting of plasma cell dyscrasias, myeloma, or lymphomas. However, occasionally it develops in a setting of plasma cell granulomas, atypical infections, and other inflammatory processes such as rheumatoid arthritis, mastocytosis, Crohn's disease, and hypereosinophilic syndrome.[1,2] The majority of CSH without an identified lymphoproliferative disorders occur in women[1] [Table 1]. Consequently, the diagnosis of CSH warrants careful evaluation of the underlying cause.
Table 1.
Reported cases of cerebral crystal storing histiocytosis

Systemic CSH may present as either localized or multifocal/a multiorgan disease. Most reported and the present case of CSH presented as mass lesions in the cerebral hemispheres.[2,4,5] One case presented as a serpentine enhancing lesion of the centrum semiovale.[2] The present case presented as a left temporal lobe mass.
The crystalline material in histiocytes is most commonly kappa light chain but, as in the present case, can be lambda light chain. The pathogenesis of this crystal formation remains speculative, attributed in some cases to over production or decreased clearance.[1] Nonetheless, in one case of generalized peripheral CSH, crystal formation was linked to a monoclonal gammopathy with abnormal light chain structure due to several amino acid substitutions, which were thought to promote crystallization of the light chain and/or intralysosomal degradadation.[5]
Because an associated lymphoplasmacytic disorder may not be recognized and the crystals are hard to visualize in hematoxylin and eosin-stained sections, the diagnosis may be inapparent. The differential diagnosis includes a number of entities with a lymphoplasmacytic and histiocytic infiltrate. These include the aforementioned lymphoplasmacytic disorders, amyloidoma, Erdheim–Chester and Rosai–Dorfman disease, xanthogranulomas, rare drug reactions as from Clofazimine, and infections.[1] Exclusion of other histiocytic and inflammatory entities is, of course, central to establishing the diagnosis. The sine qua non is identification of clonal, usually light chain production and crystals in histiocytes, especially in membrane bound lysosomes. The use of electron microscopy, immunofluorescence, in situ hybridization for immunoglobulins and light chains and, if available, liquid chromatography-electrospray tandem mass spectroscopy may facilitate recognition.[1,2]
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2013/4/1/112/117412
Disclaimer: The authors of this article has no conflict of interest to disclose, and has adhered to SNI's policies regarding human/animal rights, and informed consent. Advertisers in SNI did not ask for, nor did they receive access to this article prior to publication
REFERENCES
- 1.Dogan S, Barnes L, Cruz-Vetrano WP. Crystal-storing histiocytosis: Report of a case, review of the literature (80 cases) and a proposed classification. Head Neck Pathol. 2012;6:111–20. doi: 10.1007/s12105-011-0326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaminsky IA, Wang AM, Olsen J, Schechter S, Wilson J, Olson R. Central nervous system crystal-storing histiocytosis: Neuroimaging, neuropathology and literature review. Am J Neuroradiol. 2011;32:E26–8. doi: 10.3174/ajnr.A1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebeau A, Zeindl-Eberhardt E, Muller EC, Muller-Hocker J, Jungblut PR, Emmerich B, et al. Generalized crystal-storing histiocytosis associated with a monoclonal gammopathy: Molecular analysis of a disorder with rapid clinical course and review of literature. Blood. 2002;100:1817–27. [PubMed] [Google Scholar]
- 4.Orr BA, Gallia GL, Rodriquez FJ. Case 2012-10. 53rd annual diagnostic slide session. Am Assoc Neuropathol. 2012 Jun 23; [Google Scholar]
- 5.Rodriguez FJ, Gamez JD, Vrana JA, Theis JD, Giannini C, Scheithauer BW, et al. Immunoglobulin derived depositions in the nervous system: Novel mass spectrometry application for protein characterization in formalin-fixed tissue. Lab Invest. 2008;88:1024–37. doi: 10.1038/labinvest.2008.72. [DOI] [PubMed] [Google Scholar]