

Abstract
The development of a bovine papillomavirus (BPV) vaccine is an outstanding challenge. BPV protein L1 gene transfection in the Drosophila melanogaster S2 cell expression system failed to produce L1 protein notwithstanding correct L1 gene insertion. Severe genetic inbalance in the host cell line, including cytogenetic alterations, may account for the lack of protein expression.
Keywords: Bovine Papillomavirus, BPV4, S2 expression cells.
Bovine papillomaviruses (BPV) are known to be etiologically related to bovine papillomatosis, esophagic and bladder cancer (endemic diseases that lead to serious economic losses). Superficial epithelial lesions enable viruses to infect the basal layer cells. The virus completes its full cycle in the stratified squamous epithelia of the skin and mucous membranes. The BPV non-enveloped 8 kb double-stranded circular DNA genome is packed in a capsid assembled with 360 copies of the L1 55 kDa protein, organized in 72 capsomeres, and approximately 12 copies of the L2 39 kDa protein (3,4). The benign lesions, that originated the term “papillomatosis”, can spontaneously or under the influence of cofactors progress to cancer (3). Prophylactic or therapeutic vaccine procedures against BPV are being developed to control, prevent or treat the related diseases (5). “Virus like particles” (VLPs), equivalent to viral capsids that are devoid of a genome, are prime candidates for the production of prophylactic vaccines (5) since they can produce high levels of humoral neutralizing antibodies. These antibodies afford protection in challenges with homologous virus types, since L1 epitopes preserve their original conformation (5). Papillomavirus VLPs can be produced by L1 protein over-expression in compatible expression systems. However, stable in vitro expression of the virus major late sequence is still hard to accomplish (5). Irregular shaped or smaller sized L1 proteins can occur when produced without codon modification in insect cells when baculovirus is used as a vector or in Saccharomyces cerevisiae (7). These irregular proteins are unable to produce an immunological response. Aiming at the production of large amounts of papillomavirus L1 protein VLPs with proper antigenic profiles, we examined the possibility of expressing large amounts of papillomavirus antigens in Drosophila S2 cells (6). In this study, we describe the processes of producing and transfecting constructions, including BPV4 L1 sequences in Drosophila melanogaster S2 cells.
The pMT-V5-His-TOPO vector (Invitrogen, DES® TOPO® TA Expression Kit K4125-01) was used for inducible expression of BPV4 L1 fused to carboxy-terminal V5- and His6 epitopes in Drosophila S2 cells (Invitrogen, Drosophila Expression System K4120-01). This vector contains a metallothionein (MT) promoter for high-level, metal-inducible expression of the inserted gene, a pUC origin and an ampicillin resistance gene for the selection of the E. coli transformants and a TOPO® cloning site for rapid cloning of Taq-amplified PCR products. The coding sequence of the BPV-4 L1 gene with its translational stop codon removed, was amplified by PCR using the viral DNA present in the vector pAT153-BPV-4 as a template and was then fused with the expression vector. Specific primers were designed for the amplification of the BPV4 L1 gene, based on sequences obtained from GeneBank (accession number X05817): 5’-GTT GGA TCC GAG ATG TCT TTC TGG GTT CCA AAT TCT-3’, positions 5590-5662 and 5’-GAA AGA TCT TCA TAT TGC TCA TAC TGC TGC AGG-3’, positions 7129-7155 of the BPV4 genome containing the L1 protein gene, produced an amplification product of 1565 bp. Aiming the improvement of the exogen L1 gene expression, all upstream and downstream nonconding sequences were removed and the forward primer included a specific sequence for transcription initiation close to the Kozak sequence. The reverse primer used did not include a stop codon, allowing for the production of a V5 C terminal tail and a histidine epitope encoded from the vector expression sequence. Oligonucleotide primers were designed with BglII and BamHI restriction sites for insertion direction analysis. PCR reactions for the BPV-4 L1 gene amplification were carried out using the “TripleMaster® PCR System” using its high-fidelity buffer (Eppendorf) in a T-personal thermal cycler (Biometra, Germany). The conditions used for the reaction were as follows: an initial denaturation at 94ºC for 2 min, followed by 26 cycles of denaturation at 94ºC for 20 s, primer annealing at 57ºC for 20 s and extension at 72ºC for 1min, and a final extension at 72ºC for 5 min. The PCR product was purified using a “GFX PCR DNA and Gel Purification Kit” (Amersham Biosciences K27-9602-01) and was introduced into a pMT-V5-His-TOPO vector (Invitrogen). Taq polymerase has a non-template-dependent terminal transferase activity that adds a single deoxyadenosine (A) to the 3' ends of PCR products allowing for its efficient insertion into the vector with a single, overhanging 3' deoxythymidine (T) residue. The mixture was used to transform DH5a competent cells and transformed colonies were selected on Luria-Bertani agar plates containing ampicillin at 75 µg/ml. The clones were expanded by overnight incubation at 37ºC in Luria-Bertani medium containing 75 µg/ml of ampicillin and plasmidial DNA was isolated using the “S.N.A.P. Miniprep Kit” (Invitrogen, K1900-01). The plasmids were screened for the presence of the L1 ORF and direction by enzymatic digestion with enzymes BamHI (Gibco BRL), on the L1 gene site, and XhoI (Gibco BRL), on the site present in the vector sequence, and were subsequently sequenced in order to verify the direction of the reading frame in an ALFexpress DNA Sequencer (Pharmacia Biotech.), using a “Cyä 5 Thermo Sequenase Dye Terminator Kit” (Amersham Pharmacia Biotech, K 27-2682-01). The forward primer used (5'-CAA GTG AAT CAT CTC AGT GCA ACT AAA-3') is complementary to the vector sequence, and the reverse primer used (5'-AAG CTT GTG CCT TAT CCC AA-3') is complementary to the L1 gene at positions 6096-6115 bp, amplifying a 525 bp fragment. The sequences were aligned with the aid of the Esee software (The Eyeball Sequence Editor, Version 1.09) and analyzed by Blast (30). Drosophila melanogaster Schneider line-2 (S2) cells (Invitrogen) were grown in Schneider medium (Gibco) and kept at 27ºC, supplemented with 10% fetal calf serum (Gibco). Cell viability was assessed to verify if it was at the appropriate level for transfection and cytogenetic analyses were performed in 50 cells. The cells were exposed to colchicin (16 µg/ml) (Cultilab) for 1h, at 27ºC and after hypotonization with KCl 0.075M, 27ºC, for 15 minutes, they were fixed in methanol:acetic acid (3:1) twice and stained in Giemsa 3% for 5 minutes (41). The transfection of S2 cells was performed using a “Calcium Phosphate Transfection Kit” (Invitrogen -Drosophila Expression System K4120-01) and the expression was induced by adding copper sulphate (500 mM), 24h before L1 detection analysis. SDS-PAGE was carried out as described by manufacturer’s protocol (Invitrogen - Drosophila Expression System K4120-01). Briefly, after transfection, S2 samples from a 35 mm plate (~3 × 106 cells) were lysed, mixed to the SDS-PAGE sample buffer (Bio-Rad)), bromophenol blue and 0.8% SDS, and submitted to electrophoresis (12.5% polyacrylamide gel and a 5% stacking gel, 43). Samples of 5 and 10 µl of each S2 cell strain and of 10 µl of the molecular weight marker (Bio-Rad) were loaded into sample wells. The gel was stained with 0.25% Coomassie blue. The samples obtained were diluted in buffer (EDTA 25mM, Tris-HCL 10mM pH 8.0), receiving or not additional treatment with β-mercaptoethanol for additional reduction, and transferred to a nitrocellulose membrane using a slot blot apparatus (Bio-Rad) and air-dried. The membrane was exposed to Anti-V5 horseradish peroxidase (HRP)-labeled antibody (Invitrogen Anti-V5-HRP Antibody KR961-25) for 2h and developed in a Choronaphtol solution. All procedures were carried out according to the instructions suggested by the manufacturers of the vector and antibody (Invitrogen). ELISA was carried out following the recommendations of the manufacturer of the Anti-V5-HRP antibody (Invitrogen) and was incubated overnight at 4ºC (nontransfected S2 cells were used as a negative control). The samples were exposed to different antibody dilutions, namely, 1:100, 1:500, 1:1000, 1:2000 and incubated for 2h at room temperature, washed and developed in OPD (orthophenylenediamine) solution (Sigma) in the dark for 30 min at room temperature according to the manufacturer’s guidelines. Eleven clones which included the insert were submitted to enzymatic digestion.
Three clones presented inserts that had been included in the correct direction (Fig. 1A). The reading frame was confirmed by DNA sequencing. The first 525 nucleotides of the L1 gene did not present mutations when compared to the viral genome when analysed using Blast software. Drosophila S2 cell samples, selected as a heterologous system for expression, were evaluated for cell viability and shown to be able to reach the appropriate transfection rate at 5×106 cells. Notwithstanding the correct transfection and verification of appropriate conditions for all the procedures including cell growth, cell viability and insert position in the correct frame, no evidence for the expression of the 55-60 kDa L1 protein could be detected, as observed by SDS-PAGE (Fig. 2), Dot Blot analyses (Fig. 2) or ELISA.
Figure 1.
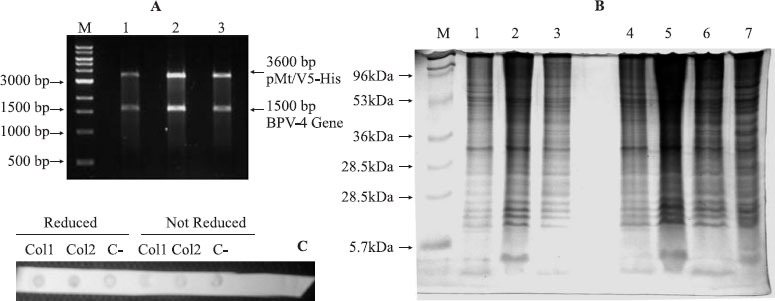
A. Enzymatic digests of the 3 clones (lanes1-3) of the BPV4 L1 capsid protein ORF DNA containing pMT/V5-His expression vector developed in an ethydium bromide-stained agarose gel. Molecular size is referenced to a GeneRulerTM 1kb DNA ladder (M); B. Total protein expression in the three BPV L1 ORF-expression vector transfected Drosophila S2 cells before (lanes 1-3) and after (lanes 4-6) mercaptoethanol reduction S2 non-transfected cells (lane 7) and molecular size references (M) in silver-stained polyacrylamide gel. C. Dot Blot analysis for BPV4 L1 capsid protein transcripts of transfected 1 and 2 (Co11 and Col2) and non-transfected (C-) S2 clones before and after β-mercaptoethanol reduction
Figure 2.
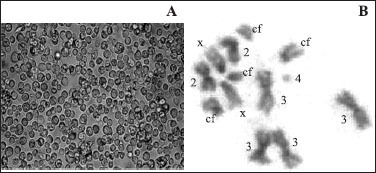
D. melanogaster S2 cell culture (A) and S2 cell metaphase (B) with four copies of chromosome 3 (3), centric fragments (cf) and breakages of X and 2 chromosomes (arrows).
Drosophila melanogaster S2 cells were evaluated as an expression system for BPV-4 L1 capside protein. These cells have been used as a heterologous expression system for many other VLPs and are considered to have advantages in the production of these proteins (6). In this study, we used all the products recommended, analyzed S2 cell karyotype and growth rate. We could verify that the S2 cells presented suitable growth rates, expected confluence patterns, were successfully transfected, and the inserts were present in the correct reading frame direction and apparently lacked mutations in the protein sequence. However, we did not obtain any significant L1 expression as evaluated by SDS page, Dot Blot and ELISA.
As previously stated, viral and host cell defences may hinder transcription in heterologous systems. Also, papillomaviruses encode different proteins using a single gene by means of alternate splicing to express their proteins in a cell differentiation-specific and temporal manner since posttranscriptional regulation of papillomavirus gene expression is linked to cell differentiation (2). In papillomavirus, one ORF can span two separate exons and each transcript from the BPV genome could be bicistronic, tricistronic, or even polycistronic with two or more ORFs. Furthermore, a papillomavirus ORF could exist in multiple mRNAs transcribed from different promoters as shown by mapping of BPV-1 transformed C127 cells as well as productively infected bovine fibropapillomas. Four hypotheses may explain the limited efficiency of viral capsid protein production in the heterologous S2 cell expression system: (i) premature poly-adenylation; (ii) alternative processing and mRNA instability; (iii) impediment of nuclear mRNA exportation, and (iv) critical mismatch between viral codon usage and specific tRNA available in host cells ().
Successful L1 gene expression in yeast expression systems could, hypothetically, be a consequence of (i) cytoplasmic mRNA production circumventing down regulated nuclear export; (ii) down regulation of host cell protein synthesis, (iii) matching of codons in the viral genome and host cell genome or (iv) absence of effective host cell defences. The in vitro expression in bacterial cells, transgenic plants, and mammalian cells with strong promoter plasmid vectors may yet be a distant goal (1).
The changes detected in the S2 expression system genome could also explain the failure of L1 protein expression. Cytogenetic analysis carried out to verify the cell line karyotype stability showed aneuploid cells with different centric fragments (Figure 1B) differing significantly from their original description (6). The most common metaphasic karyotype (74% of the cells examined) was composed of X, 2 and 4 chromosome trisomies, chromosome 3 tetrasomies and additional four centric fragments: cells with 1, 2 or 3 copies of chromosome 4 were also frequent as well as breakage of chromosomes X and 2 (Figure2). This configures a probable polyploidy event and subsequent chromosome loss, rearrangements and fragmentation in the cell lineage. Thus, the S2 expression system used here, derived from a tetraploid female lineage, a common feature of the S2 gene expression host cell line (6), with partial chromosome X to where the D. melanogaster rDNA genes have been mapped (6) and chromosome losses, may harbour unpredictable gene inactivation mechanisms impairing the heterologous BPV4 L1 protein expression. Further studies of the expression system and of the transcript products of the L1 papillomavirus gene will certainly yield interesting results on the ensuing topic of virus gene expression.
RESUMO
Transfecção em células de Drosophila S2 usando o gene L1 do Papilomavírus Bovino tipo 4: ausência de expressão da proteína L1
O desenvolvimento de uma vacina para papilomavirus bovino (BPV) consiste em grande desafio. A transfecção do gene codificante da proteína L1 de BPV em sistema de células S2 de Drosophila melanogaster não logrou sucesso, apesar da correta inserção da seqüência gênica em vetor apropriado. Graves alterações genéticas na linhagem celular S2, que incluem aberrações cromossômicas, provavelmente estão relacionadas à ausência da expressão da proteína desejada.
Palavras chave: papilomavírus bovino, BPV4, células S2.
REFERENCES
- 1.Baker C.C., Howley P.M. Differential promoter utilization by the bovine papillomavirus in transformed cells and productively infected wart tissues. EMBO J. 1987;6(4):1027–1035. doi: 10.1002/j.1460-2075.1987.tb04855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barksdale S., Baker C.C. Differentiation-specific alternative splicing of bovine papillomavirus late mRNAs. J. Virol. 1995;69(10):6553–6. doi: 10.1128/jvi.69.10.6553-6556.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campo M.S. Vaccination against Papillomavirus in Cattle. Clin. Dermatol. 1997;15:275–283. doi: 10.1016/s0738-081x(96)00165-4. [DOI] [PubMed] [Google Scholar]
- 4.Campo M.S. Papillomavirus and disease in humans and animals. Vet. Comp. Oncology. 2003;1(1):3–14. doi: 10.1046/j.1476-5829.2003.00001.x. [DOI] [PubMed] [Google Scholar]
- 5.Kirnbauer R., Chandrachud L.M., O’neil B.W., Wagner E.R., Grindlay G.J., Armstrong A., Mcgarvie G.M., Schiller J.T., Lowy D.R., Campo M.S. Virus-like Particles of Bovine Papillomavirus Type 4 in Prophylactic and Therapeutic Immunization. Virology. 1996;219:37–44. doi: 10.1006/viro.1996.0220. [DOI] [PubMed] [Google Scholar]
- 6.Schneider I. Cell Lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 1972;27:353–365. [PubMed] [Google Scholar]
- 7.Zhou J., Liu W.J., Peng S.W., Sun X.Y., Frazer I. Papillomavirus Capsid Protein Expression Level Depends on the Match between Codon Usage and tRNA Availability. J. Virol. 1999;73(6):4972–4982. doi: 10.1128/jvi.73.6.4972-4982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]