

Abstract
Plant–bacteria interactions result from reciprocal recognition between both species. These interactions are responsible for essential biological processes in plant development and health status. Here, we present a review of the methodologies applied to investigate shifts in bacterial communities associated with plants. A description of techniques is made from initial isolations to culture-independent approaches focusing on quantitative Polymerase Chain Reaction in real time (qPCR), Denaturing Gradient Gel Electrophoresis (DGGE), clone library construction and analysis, the application of multivariate analyses to microbial ecology data and the upcoming high throughput methodologies such as microarrays and pyrosequencing. This review supplies information about the development of traditional methods and a general overview about the new insights into bacterial communities associated with plants.
Keywords: Plant–bacteria interactions, molecular techniques, multivariate analysis, endophytes, rhizosphere
INTRODUCTION
In nature, bacteria are mainly found in association with different species, composing bacterial communities. These communities occupy all terrestrial niches, colonizing environments such as soil, water, air, plants and animals.
In plants, bacterial communities are associated with different tissues; leaves and roots or as endophytes in inner parts. These bacteria are active in processes of plant development, nutrient supply, plant growth promotion and protection against pathogens. The present review explores aspects of assessing shifts in bacterial communities monitor environmental changes. Different methodologies are to used in such evaluations, based on cultivation or on direct assessment of nucleic acids extracted from environmental samples. Additionally, the recent application of multivariate analysis to data from both techniques has allowed better comprehension of factors determining the composition of bacterial communities, and is highly useful to monitor shifts caused by changes in environmental conditions. Although none of the available techniques completely represents the real environment, the development of these methodologies will provide more accurate information.
Bacterial diversity
Microorganisms are a great source of genetic diversity, still far from completely known and explored (49). Bacteria are an important portion of this diversity, representing one of the three domains in the phylogenetic tree (Archaea, Bacteria and Eucarya) (78).
The bacterial group has a long evolutionary history, conferring the capacity to inhabit most terrestrial niches. Bacteria are the main portion of biomass on Earth, and are responsible for some essential processes for life such as cycling of carbon, nitrogen and sulfur. Hence, additional to bacterial species diversity, there is intra-specific diversity. The bacterial genome is characterized by the total number of genes found in strains, which can be divided into two groups: (i) the core, composed of the group of genes found in at least 95% of strains and are essential for the cell’s life cycle; and (ii) the auxiliary group, found in a maximum of 5% of strains, and responsible for species adaptation in different environments (31). The core is maintained in species by speciation and vertical transmission, while the auxiliary group does not identify the species, since it is different in every strain. This last group of genes is also transmitted from strain to strain and even between species by horizontal gene transfer (6).
This concept clearly demonstrates that bacterial diversity is not static, due to the high reproduction capacity associated with the short life cycle and high cell multiplication rates, which leads to the high adaptation value, and fast responses to environmental change (1, 31).
The dynamic of bacterial communities colonizing plants
A wide diversity of bacteria can interact with plants, composing bacterial communities with important roles in plant development and health status (22). These interactions can vary according to the host plant in a process similar to those widely known for pathogenic microorganisms (65).
Bacterial populations are distributed in the rhizosphere, epiphytic and endophytic communities. The rhizosphere is commonly described as the soil portion directly influenced by root exudates; however, an updated definition of rhizosphere considers it as the soil compartment influenced by the root, including the root itself (25). Epiphytic and endophytic bacteria are characterized by the colonization of surface and inner tissues of plants, respectively. There is an ongoing discussion toward a better definition of these microorganisms; a commonly used definition of endophytes is those whose isolates form on surface-disinfected plant tissues (22). However, in addition to these definitions is the separation of endophytes according to their essentiality in niche occupations. In that case, the endophytic community is divided into “passenger” endophytes, i.e. bacteria that eventually invade internal plant tissues by stochastic events and “true” endophytes, those with adaptive traits enabling them to strictly live in association with the plant (24). Due to the novelty of this separation, and the problems involved in the methodological separation of these endophytic groups, we will consider in this review that the endophytic community is those bacteria that colonize inner tissues of healthy plants.
The cells in the rhizosphere, plant-surface or endophyte communities are variable. A superficial analysis of these communities could lead to the conclusion that there is a strict specificity for niche colonization. However, a more realistic scene is represented by the gradient of population distribution along plants. If a didactic approach is applied to explain bacterial communities associated with plants, it would divide these bacteria into distinct communities, with separation between epiphytic and endophytic communities in accordance with plant organs, such as roots, stems and leaves. However, in nature the gradient of distribution will prevail over separation. It is important to note that bacteria in the rhizosphere are often similar to those in the endophytic community and on leaf surfaces.
This wide distribution is driven by plant development that carries bacteria over the plant tissues (48). Chi et al. (9) demonstrated that similar bacteria were distributed over the rice plant, from roots to leaves. However, the abundance of bacterial types along the different niches can differ, mainly due to differences in these niches in nutrient supply, atmospheric conditions and competitiveness with other components of these communities (52). The behavior of these populations and how they colonize plants is determined by environmental conditions, like formation of biofilms that help bacteria fix to cell walls, avoiding the migration driven by sieve transportation. Similarly, in the parenchymatic region, being single-celled can enable better contact with cells and so better nutritional supply for the bacterium.
Methodologies to assess shifts in bacterial communities associated with plants
A number of methodologies are available in the literature to assess bacterial communities and to compare what is found where (Figure 1); however, all have advantages and drawbacks. Prior establishment of the goals of any work is essential to determine the most suitable technique to answer experimental questions. Below is a description of common techniques used to assess shifts in microbial communities associated with plants.
Figure 1.
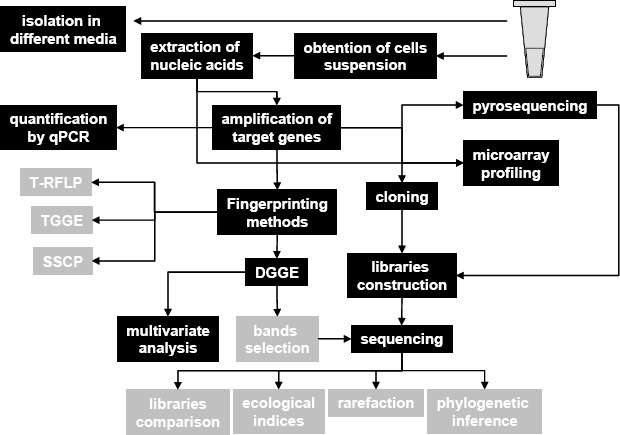
Scheme showing the possibilities for application of molecular microbiology techniques to study bacterial communities associated with plants. In black the techniques presented and discussed in this review.
Isolation and culturability of bacterial communities
Isolation in culture media is the most common methodology to access bacterial communities from different environments, mainly due to its simple application. However, this is a limited analysis of bacterial diversity (Figure 1), influenced by a number of factors, often under or overestimating bacterial diversity (53).
The high diversity of bacteria in most environmental samples is hard to represent on a culture plate, since the vast majority of bacterial species do not grow on standard isolation media (75). It has been demonstrated that isolates obtained by plating do not represent their natural habitat because applied isolation methods only access a small subset of the total microbial community in the environment (15, 16, 75). Tentative improvements to cultivation are based on mimicking the environment that bacteria are from (23). For example, the supplementation of culture media with soil extracts can result in higher diversity of culturable species (23). Using a similar approach, recalcitrant and undescribed species related to Verrucomicrobium and Acidobacteria, which are both rather unexplored groups, were obtained (30). Also, changing incubation conditions of plates can stimulate the culturability of bacteria, e.g. changing atmospheric composition and addition of specific nutrients and signaling molecules (73). More specifically, addition of anti oxidativestress compounds like pyruvate and catalase to culture media can lead to higher recovery of cells from soil and water (4, 5, 76).
Although these tentative studies revealed improvements to culturability of bacterial diversity in distinct environments, it is still essential to use polyphasic approaches for satisfactory evaluation of bacterial communities interacting with plants.
Cultureindependent methodologies to assess bacterial communities associated with plants
The application of techniques based on analysis of nucleic acids (DNA or RNA) directly extracted from environmental samples is essential in microbial diversity studies; they can supply information in a culture-independent way, and exclude the limitations and bias from the low culturable portion of bacteria of these communities (51) (Figure 1). However, it should be noted that culture-independent techniques also have bias, mainly introduced during DNA extraction and amplification of target genes (3, 7).
DNA extraction is the first step to analyze bacterial communities in a culture-independent way, and is crucial for the outcome of any further molecular analysis. The DNA extraction strategy should enable yields representative of the indigenous community as well as enough purity and integrity for polymerase chain reaction (PCR) amplification. There is a wide variety of methodologies which allow retrieving of DNA from samples; most were first developed for soil community analysis, but can also be applied to plant-associated bacteria. These methodologies are divided into two main groups: direct and indirect extraction methods (detailed, described and compared in a book chapter (43). Briefly, indirect methods separate bacterial cells from soil or plant tissues by centrifugation and cells are further chemically lyzed to release DNA. In contrast, the direct methodologies, commonly found in kits, do not separate cells from samples but process the entire sample. In this case the cell lysis is mechanical. Indirect methodologies result in a lower amount but more pure DNA, while direct methodologies result in a high amount of DNA, but possibly have environmental contaminants like humic acids. The final concern about methodologies is the representativeness of extracted DNA; direct extraction techniques have an advantage in this regard as the attachment of specific bacteria groups to soil particles or plant tissues is overcome (43).
Considering the bias during DNA amplification, most techniques applied to microbial communities are based on differences in the ribosomal genes. The 16s rRNA is the most important gene in microbial ecology and bacterial phylogeny (37). However, the named universal primers are designed from sequences that are already described, which could limit the assessment of unknown sequences and consequently, not amplify any new bacterial groups. This primer selectivity was recently demonstrated where selection was driven by the use of different reverse primers (7).
Although these concerns are known and considered, the bacterial diversity is assessed in different environments based on the 16S rRNA gene, and revealing the portion of the community not considered when culture-dependent approaches are applied (27, 49). For example, the bacterial communities in leaves from Atlantic forest trees were described using cloning and sequencing, which allowed the estimation of 400 phylotypes in each tree species (34). It is also important to show the wide applicability of these techniques, from the quantification of target groups and species by real time PCR to the fingerprinting of communities by Denaturing Gradient Gel Electrophoresis (DGGE; Figure 1). A refined analysis can also be considered by combining a culture-independent technique (e.g. DGGE fingerprinting) with multivariate analysis. These applications are discussed below with remarks on the possibilities for use in microbial communities associated with plants.
Quantitative PCR in real time (qPCR)
PCR is a mark on the advent of molecular biology, and has had great impact on techniques in microbial ecology. More recently, the quantitative real time PCR (qPCR) development was incorporated into the toolbox used in studies of bacterial communities associated with plants.
The qPCR is a highly sensitive tool to quantify microbial populations within a sample, since it is based on detecting specific sequences of nucleic acids, and estimating the amount in a sample (33). Briefly, the amplification and detection of the sequences is performed by fluorescent markers present in the reaction. These markers respond to amplification, increasing the fluorescence emission after every amplification cycle, which is detected by the PCR machine. The emitted fluorescence is quantified and a threshold value of fluorescence (Ct) is achieved. The Ct value is used for interpolation on a standard curve of Ct values with known concentrations of target DNA (33).
The most common fluorescent agent used is the SYBR GreenI, which can link to double-strand DNA and emit fluorescence when excited with wavelengths of 494–521 nm. Primer specificity is crucial when relying on SYBR green detection, as lack of specificity may yield non-target (next to target) products whose detection will distort the quantification. Another widely used technique for qPCR analysis uses the so-called Taqman probes, which are based on a third oligonucleotide during annealing. This probe incorporates two molecules, the reporter and the quencher, which when close to each other, repress the fluorescence of the reporter; but after incorporation by DNA amplification, the reporter fluoresces, directly linking the signal intensity with the amount of target DNA amplification. In addition to these two techniques for qPCR, there are other variations in available methodologies, such as Lux and Beacon probes; Zhang and Fang (79) have reviewed these techniques.
Applying the qPCR technique, bacterial species were quantified in association with plants. The pathogen Xylella fastidiosa was quantified in citrus samples (45) while the endophytic bacterium Methylobacterium mesophilicum was targeted by qPCR quantification during its colonization of the model plant Catharanthus roseus (32). In Brassica oleracea, the population of the growth-promoting bacteria Enterobacter radicincitans was monitored by qPCR associated with fluorescent in situ hybridization (61), determining not only the amount of bacteria colonizing plants but also their localization in the host plant. Besides direct quantification of microbial populations, qPCR can quantify microbial gene expression, where it has been used in important studies, as for example, in comparing the ammonium-oxidizer activities present in Bacteria and Archaea domains in soils (35).
Fingerprinting techniques of bacterial communities
Fingerprinting is the most common culture-independent approach for assessing the structure of bacterial communities. It gives an overview of the most abundant members in each sample, evidenced by patterns of bands or peaks. Amongst the techniques for fingerprinting bacterial communities the most widely used are restriction fragment length polymorphism (T-RFLP), single-strand conformation polymorphism (SSCP), automated rRNA intergenic spacer analysis (ARISA), temperature gradient gel electrophoresis (TGGE) and DGGE. These techniques were first developed and applied to soil microbiology and reviewed by Ranjard et al. (51) and Oros-Sichler et al. (46). The present review will focus on DGGE; however, some other fingerprinting methods will be briefly described.
Analysis by T-RFLP is based on a differential display of restriction sites in bacteria of different taxonomical affiliations. T-RFLP uses a combination of fluorescent labeling of the forward primer after electrophoresis, thus a bacterial community is converted into a diagram of peaks, where each signal is related to a distinct bacterial taxon (36).
SSCP is a fingerprinting methodology based on differential migration of secondary structures formed by single-strand DNA fragments (67). After denaturation of double-strand DNA from PCR amplification on environmental samples, samples that form secondary structures are electrophoresed in a non-denaturant gel and a band pattern produced, indicating the constitution of bacterial communities. In a recent study, Smalla et al. (70) compared the resolving power of DGGE, T-RFLP and SSCP; the three methodologies gave similar distinction of samples. Thus the choice of technique is based on the equipment availability in each laboratory.
Denaturing Gradient Gel Electrophoresis (DGGE)
DGGE is used in descriptions and comparisons of bacterial communities in different environments (51). A DGGE gel separates amplicons with similar numbers of nucleotides, based on differences in composition (the GC percentage) (41). This technique has a high resolution and permits rapid processing of many samples (42).
In the beginning, the comparisons among communities were based on amplicons obtained with so-called universal primers, e.g. for the domain Bacteria (29, 41). Later, specific primers targeting microbial groups were developed, resulting in better assessment of some populations and better band-pattern definition for complex communities, such as soil and rhizosphere communities. Some of these specific primers target large bacterial groups like classes α and β-proteobacteria (21) or orders like Actinobacteria (29). More specifically, primers were also developed to access restricted populations, e.g. genera Pseudomonas (19, 40, 77), Paenibacillus (12) and Burkholderia (63). A remark should be made about using DGGE to access bacterial communities in plants, especially in endophytic communities. Due to the prokaryotic origin of chloroplasts, they harbor copies of the 16S rRNA gene, which compete for amplification with bacterial DNA. It was originally a problem when fingerprinting endophytic communities due to the high amount of chloroplast DNA, which limit the detection of bacteria in these samples. However, a first PCR that uses primers 799F (8) and 1492R, prior to GC clamp amplification, increases the bacterial DNA concentration. All these primers, universal and specific, were based on the sequence of the 16S rRNA gene (Table 1).
Table 1.
Available primers to assess specific bacterial communities based on phylogenetic or functional genes in environmental samples.
| Target | Primer | Sequence (5’→3’) | Reference |
|---|---|---|---|
| 16S rRNA | 968F* | AACGCGAAGAACCTTAC | (29) |
| R1387 | CGGTGTGTACAAGGCCCGGGAACG | (29) | |
| Bacterial 16S rRNA | 1492R | TACGGYTACCTTGTTACGACT | (54) |
| 799F | AACMGGATTAGATACCCKG | (8) | |
| Alphaproteobacteria | Alpha-U | CCGCATACGCCCTACGGGGGAAAGATTTAT | (21) |
| Betaproteobacteria | Beta-2 | CGCACAAGCGGTGGATGA | (21) |
| Actinobacteria | F243 | GGATGAGCCCGCGGCCTA | (29) |
| Pseudomonas spp. | PsF | GGTCTGAGAGGATGATCAGT | (19) |
| Pseudomonas spp. | PsR | TTAGCTCCACCTCGCGGC | |
| Pseudomonas spp. | F311PS | CTGGTCTGAGAGAGGATGATCAGT | (40) |
| Pseudomonas spp. | R1459PS | AATCACTCCGTGGTAACC | |
| Paenibacillus spp. | PAEN515F | GCTCGGAGAGTGACGGTACCTGAGA | (12) |
| Burkholderia spp. | BurkR | TGCCATACTCTAGCYYGC | (63) |
| Burk3* | CTGCGAAAGCCGGAT | ||
| rpoB | 1698F* | AACATCGGTTTGATCAAC | (13, 47) |
| 2041R | CGTTGCATGTTGGTACCCAT | ||
| nifH | NHA1* | TCCACTCGTCTGATCCTG | (58) |
| NHA2 | CTCGCGGATTGGCATTGCG | (58) | |
| mxaF | mxaF1001* | GCGGCACCAACTGGGGCTGGT | (17, 39) |
| mxaR1557 | GGGCAGCATGAAGGGCTCCC | ||
| amoA | AmoA1F* | GGGGTTTCTACTGGTGGT | (59) |
| AmoA2R | CCCCTCKGSAAAGCCTTCTTC | ||
| gacA Pseudomonas spp. | gacA1-F* | ATTAGGGTGCTAGTGGTCGA | (10) |
| gacA2-R | GGTTTTCGGTGACAGGCA |
forward primers, where the GC clamp was added at the extremity 5´
Primers based on other genes have also been developed. The first usage of optional genes to be targeted in a DGGE analysis was based on the rpoB gene, which codifies for the RNA polymerase enzyme (47). Other functional genes exploited for DGGE analysis are the nifH, present in nitrogen-fixing bacteria (58) and the mxaF gene, present in Methylobacterium spp. and other methylotrophic bacteria (17, 28). More recently DGGE was used to screen ammonium-oxidizing bacteria and archaea (18, 59, 62) in association with clone sequencing. The advantage of analysis based on functional genes is the possibility of answering not only who is there, but how diverse the active community is in the process in that environment.
Considering the many primers available (Table 1) and the capacity for fast processing of many samples, DGGE is an important approach to study bacterial communities, supplying information about shifts on composition.
Ordination methods of multivariate analysis applied to DGGE fingerprints
The increasing application of DGGE in determining the composition of bacterial communities and the shifts caused by environmental changes revealed that simple visual analysis of DGGE fingerprints is not sufficient to explore the data generated. The improvement of information extraction from DGGE patterns was first made by clustering fingerprints, based on correlation of densitometric curves. Furthermore, areas of bands along fingerprintings were used to associate numbers with DGGE patterns, such as using the Shannon diversity index. More recently, multivariate analyses were applied to DGGE data, presenting results in ordination plots (11, 64). A detailed review of applications of multivariate analysis in microbial ecology was recently published (50).
Multivariate analysis can be defined as data processing that combines different measurements from the same sample, and inferring correlations and interactions of factors. Although a simple clustering analysis based on different parameters is a multivariate analysis, the most common presentations of these techniques are ordination methods. Ordination is the collective term for multivariate techniques that arrange sites along axes on the basis of data on species composition, resulting in a diagram in which sites are represented by points in a two-dimensional plot (20, 74).
Focusing on microbial ecology related to plants, the main advantage of multivariate analysis is the possibility of including in the one analysis all factors which may influence composition of bacterial communities. This approach was first applied in bacterial communities from soils (72) and further in the analysis of historical usage of soil and plant species cultivation in the structure of the Burkholderia spp. community (64). Similarly, using multivariate analysis, it was demonstrated that the rhizosphere environment determines the selection of Pseudomonas spp. to interact with plants, in detriment to the local of plant cultivation (11).
A series of steps to apply multivariate analysis is presented in Figure 2. Briefly, gels with fingerprints generated from DGGE analysis are converted into matrices where presence and absence of each band is considered in each sample. Alternatively, the normalized intensity of the band can be used as an inference of the frequency of species in samples. These matrices can be first used for species-based analyses like Principal Components Analysis (PCA) or Correlation Analysis (CA). These analyses are more exploratory and trace correlations between samples, not considering environmental factors. In a comparison, these plots are similar to the commonly presented trees generated by clustering analysis. The decision of whether CA or PCA should be used is made on the basis of the gradient size of the species distribution. The gradient size is commonly estimated by Detrended Correspondence Analysis (DCA), where the gradient size of the first axis is considered. It is assumed that values > 4.0 indicate normal distribution of data, and values < 3.0 indicate that a mathematical model based on linear distribution would be better. For intermediate values both methods can be applied, and normally the one which better represents factors is presented in articles. Species-based techniques often use PCA for linear data and CA for normal distributed data (Figure 2).
Figure 2.
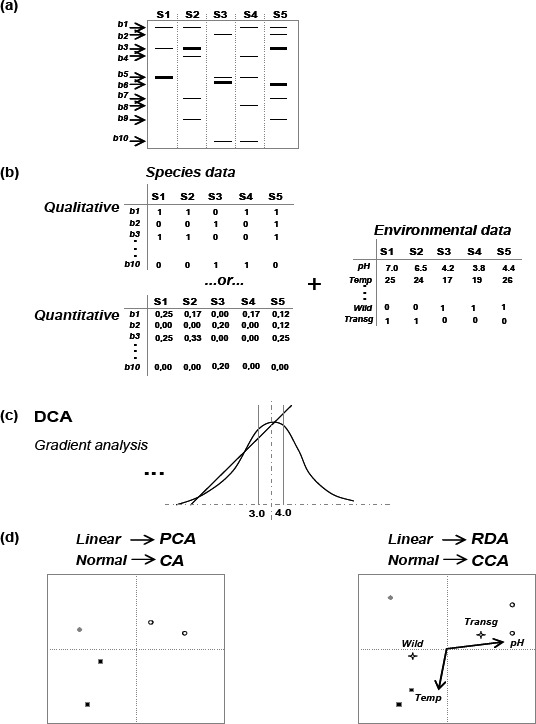
Steps for multivariate analysis using data from molecular microbiological methods. The sequence is the obtaining of the fingerprint (a), the conversion in matrices with qualitative or quantitative data and the combination with data for samples classification (b), the gradient analysis (c) and the application of the best fitted mathematical model (d).
In addition, the correlation between species occurrence and environmental data from samples can be analyzed. These environmental data can be nominal, characterized by classification of samples with qualitative information; or quantitative, when parameters are measured in samples or in their place of collection. Examples of nominal variables are: whether the sample is obtained from transgenic plants or the location where samples were collected. Quantitative variables are numerically determined values, such as temperature or pH.
Multivariate analysis is performed by combination of species and environmental data in a species–environmental data analysis. The distribution of the species data, determined by DCA, indicates the best model: linear data indicates Redundancy Analysis (RDA), and normal data indicates Canonical Correspondence Analysis (CCA). Hence, to infer the significance of each environmental factor in the composition of species in samples, a Monte Carlo permutation test is used, generating P values for each considered environmental factor.
Another concern about multivariate analysis is interpretation of graphics and values for either RDA or CCA analyses. A brief description is that samples and environmental variables are distributed over the axes revealing separation on different quadrants. The bi-dimensional distribution makes the first separation on the x-axis, revealing main differences in horizontal separation, while the second factors of variation are plotted on the y-axis. Considering that, as far from the center of the plot are samples, as stronger are vectors, and higher is the separation of samples or the importance of such factor in the composition of bacterial communities. Also, vectors pointing in the same direction are variables which respond similarly to variations, and samples where vectors point, are those where the factor is more intense. However, nominal variables, represented by qualitative data cannot be represented by vectors, and become centroids in plots. Samples related to these variables are commonly distributed around the centroid named after the variable.
For better understanding, we present an example where a DGGE gel (Figure 2a) is converted into a matrix of relative band surface, further subjected to multivariate analysis, of correlating bands with two quantitative (pH and temperature) and two qualitative (transgenic or wild type) environmental variables (Figure 2b). After measurement of gradient distribution of data (Figure 2c), multivariate analysis produced a plot (Figure 2d). If environmental variables are not considered, PCA or CA only shows the correlation between samples; if RDA or CCA analyses are made, information about environmental factors is achieved. In this case, pH and plant transgenic status were revealed to be determinant of the composition of species in such communities (vector and centroids related to the x-axis), while there were secondary effects due to variation in temperature (y-axis related vector) (Figure 2d).
Such observations are consistent with all ordination plots obtained by multivariate analysis. In the beginning the graphical representation seems complicated, but after initial dealing it can very clearly be seen to show the relationships between samples and variables.
Construction and analysis of clone libraries
The construction and analysis of ribosomal gene libraries is a highly sensitive tool to study environmental microbial ecology, allowing comparison of sequences from different samples, with resolution at different taxonomic levels. This technique is also a culture-independent approach that overcomes problems found when the culturable fraction of microbial communities is sampled (66).
This technique was used to describe fungal community diversity in forest soils, by sequencing 863 clones of fungal Internal Transcribed Spacer, determining the presence of 412 phylotypes composing this community (44). In a similar study, the bacterial community colonizing the leaf surfaces in Atlantic forest was assessed, and showed that every tree species harbored a different bacterial community, indicating that bacterial diversity in the forest had been underestimated (34). The application of the pyrosequencing revealed > 10,000 bacterial phylotypes in an ocean community (71). Clone libraries were used to describe bacteria on the roots of maize plants (8), determining qualitatively and quantitatively which species were present in this niche.
The main advantage of this technique is the variety of analyses possible with the sequence groups. The libraries can be used for comparisons, ecological index estimates, rarefaction analyses and phylogenetic inferences on members of the determined community (Figure 2; (66, 69). Recently, the application of this technique has expanded to other genes beside the 16S rRNA. Diversity of functional genes has been investigated by this technique, for example, the nitrate redutace gene (nirK) in water and plant-related environments (2, 60).
High throughput approaches to assess the diversity of bacterial communities
Another important scope in every review of microbial ecology is the upcoming high throughput techniques, such as microarray-based profiling of bacterial communities (68, 80) and pyrosequencing (14). These are very promising to aid understanding of communities and interactions of bacteria associated with plants.
Microarrays are a technique widely used for high-scale gene detections and gene expression quantification in different organisms. Recently, this technique was adapted to profiling of environmental communities. The main advantage of the microarray technique over other methodologies is quantitative assessment of bacterial diversity. For a review of application of this approach see Zhou (80). The most recent version of microarray profiling is the Geochip (26), consisting of 24,243 oligonucleotide probes (with 50 bases) which covers > 10,000 genes spread over > 150 functional bacterial groups, involved in metal reduction and resistance, organic contaminant degradation, and nitrogen, carbon, sulfur and phosphorus cycling (26).
Pyrosequencing has broken the barrier of sequence limitations in study of bacterial diversity. With the ability to generate megabases of sequences in a few hours, it allows deep exploration of species in any environmental sample (14). This technique differs from Sanger methodology, which is based on incorporation and further detection of fluorescently labeled ddNTP (dideoxynucleotide triphosphate). During pyrosequencing, only one dNTP (deoxynucleotide triphosphate)is available at a time, and incorporation of this nucleotide generates the signal detected by the equipment (38, 56, 57). The signal is emitted once complementarity is achieved and the base is incorporated; then a reaction is catalyzed and pyrophosphate is released, activating ATP sulfurilase, producing energy to the luciferase to convert luciferin into oxyluciferin, releasing the light signal. Using these reaction steps, an extremely low amount of reaction is required, allowing up-scaling of the process. In this way, the latest version of instruments can produce around 300,000 reads with approximately 200–400 bp in about 5 h.
This technique has been used to describe bacterial communities in different environments. The deep ocean biosphere was described by pyrosequencing of samples collected at different depths (71), and soil bacterial communities (55) were similarly investigated. Both studies showed that diversity of these organisms was extremely high, and although 30,000 sequences were obtained, the complete description of species and sequences in both environments were not completed. The authors showed that the great majority of species were described, but there remained the ‘rare biosphere tail’ that was not completely explored, even with the high amount of sequences. Although the microbial diversity associated with plants seems to be less diverse, particularly for the endophytes, the future application of the technique in this field is promising.
Final considerations
Characteristics of bacterial communities associated with plants show it to be an interesting area for research, promising to supply information on microbial ecology, environmental changes and also acting as a reservoir of species with potential usage in agriculture and industry. The application of molecular microbiology techniques allows culture-independent approaches in the investigations of bacterial communities. Although development of new techniques and insights allows better assessment and description of bacterial communities, the dynamics of these organisms are still assessed similarly to photographs, taken at the moment of sampling, but not guaranteed to be the same five minutes later.
RESUMO
AVALIAÇÃO DA DIVERSIDADE DE COMUNIDADES BACTERIANAS ASSOCIADAS ÀS PLANTAS
As interações planta-bactéria resultam de um reconhecimento recíproco de ambas espécies. Estas interações são responsáveis por processos biológicos essenciais para o desenvolvimento e a proteção das plantas. Este trabalho revisa as metodologias aplicadas na investigação de alterações nas comunidades bacterianas associadas Às plantas. Uma descrição das técnicas é feita, desde o isolamento até a aplicação de técnicas independentes de cultivo, destacando as técnicas de qPCR, Gel de Eletroforese em Gradiente Desnaturante (DGGE), construção e análise de bibliotecas de clones, a aplicação de análise multivariada em dados de ecologia microbiana, e as novas metodologias de alto processamento de amostras como microarranjos e pirosequenciamento. Em resumo, esta revisão fornece informações sobre o desenvolvimento das técnicas tradicionais e uma visão geral sobre as novas tendências dos estudos de comunidades bacterianas associadas às plantas.
Palavraschave: interação plantabactéria, técnicas moleculares, análise multivariada, endofíticos, rizosfera
REFERENCES
- 1.Abby S., Daubin V. Comparative genomics and the evolution of prokaryotes. Trends Microbiol. 2007;15(3):135–141. doi: 10.1016/j.tim.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Adhitya A., Thomas F.I.M., Ward B.B. Diversity of assimilatory nitrate reductase genes from plankton and epiphytes associated with a seagrass bed. Microbial Ecol. 2007;54:587–597. doi: 10.1007/s00248-006-9175-0. [DOI] [PubMed] [Google Scholar]
- 3.Ahn J.H., Kim M.C., Shin H.C., Choi M.K., Yoon S.S., Kim T., Song H.G., Lee G.H., Ka J.O. Improvement of PCR amplification bias for community structure analysis of soil bacteria by denaturing gradient gel electrophoresis. J. Microbiol. Biotechn. 2006;16(10):1561–1569. [Google Scholar]
- 4.Barer M.R., Harwood C.R. Bacterial viability and culturability. Adv. Microb. Physiol. 1999;41:93–137. doi: 10.1016/s0065-2911(08)60166-6. [DOI] [PubMed] [Google Scholar]
- 5.Bogosian G., Bourneuf E.V. A matter of bacterial life and death. EMBO Rep. 2001;2(9):770–774. doi: 10.1093/embo-reports/kve182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucher Y., Nesbo C.L., Doolittle W.F. Microbial genomes: dealing with diversity. Curr. Opin. Microbiol. 2001;4(3):285–289. doi: 10.1016/s1369-5274(00)00204-6. [DOI] [PubMed] [Google Scholar]
- 7.Brons J.K., van Elsas J.D. Analysis of bacterial communities in soil by use of denaturing gradient gel electrophoresis and clone libraries, as influenced by different reverse primers. Appl. Environ. Microb. 2008;74:2717–2727. doi: 10.1128/AEM.02195-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chelius M.K., Triplett E.W. The diversity of archaea and bacteria in association with the roots of Zea mays L. Microbial Ecol. 2001;41(3):252–263. doi: 10.1007/s002480000087. [DOI] [PubMed] [Google Scholar]
- 9.Chi F., Shen S.H., Cheng H.P., Jing Y.X., Yanni Y.G., Dazzo F.B. Ascending migration of endophytic rhizobia, from roots to leaves, inside rice plants and assessment of benefits to rice growth physiology. Appl. Environ. Microb. 2005;71(11):7271–7278. doi: 10.1128/AEM.71.11.7271-7278.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa R., Gomes N.C.M., Krogerrecklenfort E., Opelt K., Berg G., Smalla K. Pseudomonas community structure and antagonistic potential in the rhizosphere: insights gained by combining phylogenetic and functional gene-based analyses. Environ. Microbiol. 2007;9(9):2260–2273. doi: 10.1111/j.1462-2920.2007.01340.x. [DOI] [PubMed] [Google Scholar]
- 11.Costa R., Salles J.F., Berg G., Smalla K. Cultivation-independent analysis of Pseudomonas species in soil and in the rhizosphere of field-grown Verticillium dahliae host plants. Environ. Microbiol. 2006;8(12):2136–2149. doi: 10.1111/j.1462-2920.2006.01096.x. [DOI] [PubMed] [Google Scholar]
- 12.Da Silva K.R.A., Salles J.F., Seldin L., van Elsas J.D. Application of a novel Paenibacillus-specific PCR-DGGE method and sequence analysis to assess the diversity of Paenibacillus spp. in the maize rhizosphere. J. Microbiol. Meth. 2003;54(2):213–231. doi: 10.1016/s0167-7012(03)00039-3. [DOI] [PubMed] [Google Scholar]
- 13.Dahllof I., Baillie H., Kjelleberg S. rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl. Environ. Microb. 2000;66(8):3376–3380. doi: 10.1128/aem.66.8.3376-3380.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards R.A., Rodriguez-Brito B., Wegley L., Haynes M., Breitbart M., Peterson D.M., Saar M.O., Alexander S., Alexander Jr. E.C., Rohwer F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eilers H., Pernthaler J., Glockner F.O., Amann R. Culturability and in situ abundance of pelagic bacteria from the North Sea. Appl. Environ. Microb. 2000;66(7):3044–3051. doi: 10.1128/aem.66.7.3044-3051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felske A., Wolterink A., van Lis R., de Vos W.M., Akkermans A.D.L. Searching for predominant soil bacteria: 16S rDNA cloning versus strain cultivation. FEMS Microbiol. Ecol. 1999;30(2):137–145. doi: 10.1111/j.1574-6941.1999.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 17.Fjellbirkeland A., Torsvik V., Ovreas L. Methanotrophic diversity in an agricultural soil as evaluated by denaturing gradient gel electrophoresis profiles of pmoA, mxaF and 16S rDNA sequences. Anton. Leeuw. Int. J. G. 2001;79(2):209–217. doi: 10.1023/a:1010221409815. [DOI] [PubMed] [Google Scholar]
- 18.Francis C.A., Roberts K.J., Beman J.M., Santoro A.E., Oakley B.B. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. P. Natl. Acad. Sci. USA. 2005;102(41):14683–14688. doi: 10.1073/pnas.0506625102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garbeva P., van Veen J.A., van Elsas J.D. Assessment of the diversity, and antagonism towards Rhizoctonia solani AG3, of Pseudomonas species in soil from different agricultural regimes. FEMS Microbiol. Ecol. 2004;47(1):51–64. doi: 10.1016/S0168-6496(03)00234-4. [DOI] [PubMed] [Google Scholar]
- 20.Gauch Jr. H.G. Cambridge University Press: Cambridge; 1999. Multivariate Analysis in Community Ecology. p. 168. [Google Scholar]
- 21.Gomes N.C.M., Heuer H., Schonfeld J., Costa R., Mendonca-Hagler L., Smalla K. Bacterial diversity of the rhizosphere of maize (Zea mays) grown in tropical soil studied by temperature gradient gel electrophoresis. Plant Soil. 2001;232(1-2):167–180. [Google Scholar]
- 22.Hallmann J., QuadtHallmann A., Mahaffee W.F., Kloepper J.W. Bacterial endophytes in agricultural crops. Can. J. Microbiol. 1997;43(10):895–914. [Google Scholar]
- 23.Hamaki T., Suzuki M., Fudou R., Jojima Y., Kajiura T., Tabuchi A., Sen K., Shibai H. Isolation of novel bacteria and actinomycetes using soil-extract agar medium. J. Biosci. Bioeng. 2005;99(5):485–492. doi: 10.1263/jbb.99.485. [DOI] [PubMed] [Google Scholar]
- 24.Hardoim P.R., van Overbeek L.S., van Elsas J.D. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 2008;16(10):463–471. doi: 10.1016/j.tim.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann A., Lemanceau P., Prosser J.I. Multitrophic interactions in the rhizosphere. Rhizosphere microbiology: at the interface of many disciplines and expertises. FEMS Microbiol. Ecol. 2008;65:179. doi: 10.1111/j.1574-6941.2008.00558.x. [DOI] [PubMed] [Google Scholar]
- 26.He Z., Gentry T.J., Schadt C.W., Wu L., Liebich J., Chong S.C., Huang Z., Wu W., Gu B., Jardine P., Criddle C., Zhou J. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J. 2007;1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- 27.Head I.M., Saunders J.R., Pickup R.W. Microbial evolution, diversity, and ecology: A decade of ribosomal RNA analysis of uncultivated microorganisms. Microbial Ecol. 1998;35(1):1–21. doi: 10.1007/s002489900056. [DOI] [PubMed] [Google Scholar]
- 28.Henckel T., Friedrich M., Conrad R. Molecular analyses of the methane-oxidizing microbial community in rice field soil by targeting the genes of the 16S rRNA, particulate methane monooxygenase, and methanol dehydrogenase. App. Environ. Microb. 1999;65(5):1980–1990. doi: 10.1128/aem.65.5.1980-1990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heuer H., Krsek M., Baker P., Smalla K., Wellington E.M.H. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microb. 1997;63(8):3233–3241. doi: 10.1128/aem.63.8.3233-3241.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janssen P.H., Yates P.S., Grinton B.E., Taylor P.M., Sait M. Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl. Environ. Microb. 2002;68(5):2391–2396. doi: 10.1128/AEM.68.5.2391-2396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konstantinidis K.T., Ramette A., Tiedje J.M. Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl. Environ. Microb. 2006;72(11):7286–7293. doi: 10.1128/AEM.01398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lacava P.T., Li W.B., Araujo W.L., Azevedo J.L., Hartung J.S. Rapid, specific and quantitative assays for the detection of the endophytic bacterium Methylobacterium mesophilicum in plants. J. Microbiol. Meth. 2006;65(3):535–541. doi: 10.1016/j.mimet.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Lacava P.T., Azevedo J.L. Técnicas moleculares aplicadas ao estudo de ecologia microbiana. In: Melo I.S., Azevedo J.L., editors. Microbiologia Ambiental. Brasil: Embrapa Meio Ambiente, Jaguariùna; 2008. pp. 107–124. [Google Scholar]
- 34.Lambais M.R., Crowley D.E., Cury J.C., Bull R.C., Rodrigues R.R. Bacterial diversity in tree canopies of the Atlantic forest. Science. 2006;312(5782):1917–1917. doi: 10.1126/science.1124696. [DOI] [PubMed] [Google Scholar]
- 35.Leininger S., Urich T., Schloter M., Schwark L., Qi J., Nicol G.W., Prosser J.I., Schuster S.C., Schleper C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442(7104):806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 36.Liu W.T., Marsh T.L., Cheng H., Forney L.J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl. Environ. Microb. 1997;63(11):4516–4522. doi: 10.1128/aem.63.11.4516-4522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louws F.J., Rademaker J.L.W., de Bruijn F.J. The three Ds of PCR-based genomic analysis of phytobacteria: Diversity, detection, and disease diagnosis. Annu. Rev. Phytopathol. 1999;37:81–125. doi: 10.1146/annurev.phyto.37.1.81. [DOI] [PubMed] [Google Scholar]
- 38.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., Dewell S.B., Du L., Fierro J.M., Gomes X.V., Godwin B.C., He W., Helgesen S., Ho C.H., Irzyk G.P., Jando S.C., Alenquer M.L., Jarvie T.P., Jirage K.B., Kim J.B., Knight J.R., Lanza J.R., Leamon J.H., Lefkowitz S.M., Lei M., Li J., Lohman K.L., Lu H., Makhijani V.B., McDade K.E., McKenna M.P., Myers E.W., Nickerson E., Nobile J.R., Plant R., Puc B.P., Ronan M.T., Roth G.T., Sarkis G.J., Simons J.F., Simpson J.W., Srinivasan M., Tartaro K.R., Tomasz A., Vogt K.A., Volkmer G.A., Wang S.H., Wang Y., Weiner M.P., Yu P., Begley R.F., Rothberg J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald I.R., Kenna E.M., Murrell J.C. Detection of methanotrophic bacteria in environmental samples with the PCR. Appl. Environ. Microb. 1995;61(1):116–121. doi: 10.1128/aem.61.1.116-121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milling A., Smalla K., Maidl F.X., Schloter M., Munch J.C. Effects of transgenic potatoes with an altered starch composition on the diversity of soil and rhizosphere bacteria and fungi. Plant Soil. 2004;266(12):23–39. [Google Scholar]
- 41.Muyzer G., Dewaal E.C., Uitterlinden A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S ribosomal RNA. Appl. Environ. Microb. 1993;59(3):695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muyzer G., Smalla K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Anton. Leeuw. Int. J. G. 1998;73(1):127–141. doi: 10.1023/a:1000669317571. [DOI] [PubMed] [Google Scholar]
- 43.Nybroe O., Brandt K.K., Nicolaisen M.H., Sorensen J. Methods to detect and quantify bacteria in soil. In: van Elsas J.D., Jansson J.K., Trevors J.T., editors. Modern Soil Microbiology - second edition. FL, USA: CRC Press, Boca Raton; 2007. pp. 283–316. [Google Scholar]
- 44.O’Brien H.E., Parrent J.L., Jackson J.A., Moncalvo J.M., Vilgalys R. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microb. 2005;71(9):5544–5550. doi: 10.1128/AEM.71.9.5544-5550.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oliveira A.C., Vallim M.A., Semighini C.P., Araujo W.L., Goldman G.H., Machado M.A. Quantification ofXylella fastidiosa from citrus trees by real-time polymerase chain reaction assay. Phytopathology. 2002;92(10):1048–1054. doi: 10.1094/PHYTO.2002.92.10.1048. [DOI] [PubMed] [Google Scholar]
- 46.Oros-Sichler M., Costa R., Heuer H., Small K. Molecular fingerprinting techniques to analyze soil microbial communities. In: van Elsas J.D., Jansson J.K., Trevors J.T., editors. Modern Soil Microbiology - second edition. FL, USA: CRC Press, Boca Raton; 2007. pp. 355–386. [Google Scholar]
- 47.Peixoto R.S., Coutinho H.L.D., Rumjanek N.G., Macrae A., Rosado A.S. Use of rpoB and 16S rRNA genes to analyse bacterial diversity of a tropical soil using PCR and DGGE. Lett. Appl. Microbiol. 2002;35(4):316–320. doi: 10.1046/j.1472-765x.2002.01183.x. [DOI] [PubMed] [Google Scholar]
- 48.Prieto P., Mercado-Blanco J. Endophytic colonization of olive roots by the biocontrol strain Pseudomonas fluorescens PICF7. FEMS Microbiol. Ecol. 2008;64(2):297–306. doi: 10.1111/j.1574-6941.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- 49.Prosser J.I., Bohannan B.J.M., Curtis T.P., Ellis R.J., Firestone M.K., Freckleton R.P., Green J.L., Green L.E., Killham K., Lennon J.J., Osborn A.M., Solan M., van der Gast C.J., Young J.P.W. Essay - The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 2007;5(5):384–392. doi: 10.1038/nrmicro1643. [DOI] [PubMed] [Google Scholar]
- 50.Ramete A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007;62(2):142–160. doi: 10.1111/j.1574-6941.2007.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ranjard L., Poly F., Nazaret S. Monitoring complex bacterial communities using culture-independent molecular techniques: application to soil environment. Res. Microbiol. 2000;151(3):167–177. doi: 10.1016/s0923-2508(00)00136-4. [DOI] [PubMed] [Google Scholar]
- 52.Rao D., Webb J.S., Kjelleberg S. Microbial colonization and competition on the marine alga Ulva australis. Appl. Environ. Microb. 2006;72(8):5547–5555. doi: 10.1128/AEM.00449-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rasche F., Hodl V., Poll C., Kandeler E., Gerzabek M.H., van Elsas J.D., Sessitsch A. Rhizosphere bacteria affected by transgenic potatoes with antibacterial activities compared with the effects of soil, wild-type potatoes, vegetation stage and pathogen exposure. FEMS Microbiol. Ecol. 2006;56(2):219–235. doi: 10.1111/j.1574-6941.2005.00027.x. [DOI] [PubMed] [Google Scholar]
- 54.Rochelle P.A., Fry J.C., Parkes R.J., Weightman A.J. DNA extraction for 16S ribosomal RNA gene analysis to determine genetic diversity in deep sediment communities. FEMS Microbiol. Lett. 1992;100(1-3):59–65. doi: 10.1111/j.1574-6968.1992.tb14019.x. [DOI] [PubMed] [Google Scholar]
- 55.Roesch L.F.W., Fulthorpe R.R., Riva A., Casella G., Hadwin A.K.M., Kent A.D., Daroub S.H., Camargo F.A.O., Farmerie W.G., Triplett E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007;1:283–290. doi: 10.1038/ismej.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ronaghi M., Karamohamed S., Pettersson B., Uhlen M., Nyren P. Realtime DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 1996;242(1):84–89. doi: 10.1006/abio.1996.0432. [DOI] [PubMed] [Google Scholar]
- 57.Ronaghi M., Uhlen M., Nyren P. A sequencing method based on real-time pyrophosphate. Science. 1996;281(5375):363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 58.Rosado A.S., Duarte G.F., Seldin L., Van Elsas J.D. Genetic diversity of nifH gene sequences in Paenibacillus azotofixans strains and soil samples analyzed by denaturing gradient gel electrophoresis of PCR-amplified gene fragments. Appl. Environ. Microb. 1998;64(8):2770–2779. doi: 10.1128/aem.64.8.2770-2779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rotthauwe J.H., Witzel K.P., Liesack W. The ammonia monooxygenase structural gene amoA as a functional marker: Molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microb. 1997;63(12):4704–4712. doi: 10.1128/aem.63.12.4704-4712.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruiz-Rueda O., Trias R., Garcia-Gil T.J., Baneras L. Diversity of the nitrite reductase gene nirS in the sediment of a free-water surface constructed wetland. Int. Microbiol. 2007;10:253–260. [PubMed] [Google Scholar]
- 61.Ruppel S., Ruhlmann J., Merbach W. Quantification and localization of bacteria in plant tissues using quantitative real-time PCR and online emission fingerprinting. Plant Soil. 2006;286(1-2):21–35. [Google Scholar]
- 62.Sahan E., Muyzer G. Diversity and spatio-temporal distribution of ammonia-oxidizing Archaea and Bacteria in sediments of the Westerschelde estuary. FEMS Microbiol. Ecol. 2008;64(2):175–186. doi: 10.1111/j.1574-6941.2008.00462.x. [DOI] [PubMed] [Google Scholar]
- 63.Salles J.F., De Souza F.A., van Elsas J.D. Molecular method to assess the diversity of Burkholderia species in environmental samples. Appl. Environ. Microb. 2002;68(4):1595–1603. doi: 10.1128/AEM.68.4.1595-1603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salles J.F., van Veen J.A., van Elsas J.D. Multivariate analyses of Burkholderia species in soil: effect of crop and land use history. Appl. Environ. Microb. 2004;70(7):4012–4020. doi: 10.1128/AEM.70.7.4012-4020.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salvaudon L.G.T., Shykoff J.A. Genetic diversity in natural populations: a fundamental component of plant-microbe interactions. Curr. Opin. Plant Biol. 2008;11:135–143. doi: 10.1016/j.pbi.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 66.Schloss P.D., Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microb. 2005;71(3):1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwieger F., Tebbe C.C. A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl. Environ. Microb. 1998;64(12):4870–4876. doi: 10.1128/aem.64.12.4870-4876.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sessitsch A., Hackl E., Wenzl P., Kilian A., Kostic T., Stralis-Pavese N., Sandjong B.T., Bodrossy L. Diagnostic microbial microarrays in soil ecology. New Phytol. 2006;171:719–736. doi: 10.1111/j.1469-8137.2006.01824.x. [DOI] [PubMed] [Google Scholar]
- 69.Singleton D.R.F., Rathbun S.L., Whitman W.B. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microb. 2001;67(9):4374–4376. doi: 10.1128/AEM.67.9.4374-4376.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smalla K., Oros-Sichler M., Milling H.H., Baumgarte S., Becker N.G., Kropf S., Ulrich A., Tebbe C.C. Bacterial diversity of soils assessed by DGGE, T-RFLP and SSCP fingerprints of PCR-amplified 16S rRNA gene fragments: do the different methods provide similar results? J. Microbiol. Meth. 2007;69:470–479. doi: 10.1016/j.mimet.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 71.Sogin M.L., Morrison H.G., Huber J.A., Welch D.M., Huse S.M., Neal P.R., Arrieta J.M., Herndl G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. P. Natl. Acad. Sci. USA. 2006;103(32):12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steenwerth K.L., Jackson L.E., Calderon F.J., Stromberg M.R., Scow K.M. Soil microbial community composition and land use history in cultivated and grassland ecosystems of coastal California. Soil Biol. Biochem. 2002;34(11):1599–1611. [Google Scholar]
- 73.Stevenson B.S., Eichorst S.A., Wertz J.T., Schmidt T.M., Breznak J.A. New strategies for cultivation and detection of previously uncultured microbes. Appl. Environ. Microb. 2004;70(8):4748–4755. doi: 10.1128/AEM.70.8.4748-4755.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ter Braak C.J.F., Šmilauer P. CANOCO Reference Manual and CanoDraw for Windows User’s Guide: Software for Canonical Community Ordination (version 4.5). 2002:500. [Google Scholar]
- 75.Tyson G.W., Banfield J.F. Cultivating the uncultivated: a community genomics perspective. Trends Microbiol. 2005;13(9):411–415. doi: 10.1016/j.tim.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 76.van Overbeek L.S., Bergervoet J.H.H., Jacobs F.H.H., van Elsas J.D. The low-temperature-induced viable-but-nonculturable state affects the virulence of Ralstonia solanacearum biovar 2. Phytopathology. 2004;94(5):463–469. doi: 10.1094/PHYTO.2004.94.5.463. [DOI] [PubMed] [Google Scholar]
- 77.Widmer F., Seidler R.J., Gillevet P.M., Watrud L.S., Di Giovanni G.D. A highly selective PCR protocol for detecting 16S rRNA genes of the genus Pseudomonas (sensu stricto) in environmental samples. Appl. Environ. Microb. 1998;64(7):2545–2553. doi: 10.1128/aem.64.7.2545-2553.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woese C.R. There must be a prokaryote somewhere - microbiologists search for itself. Microbiol. Rev. 1994;58(1):1–9. doi: 10.1128/mr.58.1.1-9.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang T., Fang H.H. Applications of real-time polymerase chain reaction for quantification of microorganisms in environmental samples. Appl. Microbiol. Biot. 2006;70(3):281–289. doi: 10.1007/s00253-006-0333-6. [DOI] [PubMed] [Google Scholar]
- 80.Zhou J. Microarrays for bacterial detection and microbial community analysis. Curr. Opin. Microbiol. 2003;6:288–294. doi: 10.1016/s1369-5274(03)00052-3. [DOI] [PubMed] [Google Scholar]