

Abstract
Pseudomonas aeruginosa is an opportunistic microorganism with the ability to respond to a wide variety of environmental changes, exhibiting a high intrinsic resistance to a number of antimicrobial agents. This low susceptibility to antimicrobial substances is primarily due to the low permeability of its outer membrane, efflux mechanisms and the synthesis of enzymes that promote the degradation of these drugs. Cephalosporins, particularty ceftazidime and cefepime are effective against P. aeruginosa, however, its increasing resistance has limited the usage of these antibiotics. Encapsulating antimicrobial drugs into unilamellar liposomes is an approach that has been investigated in order to overcome microorganism resistance. In this study, antimicrobial activity of liposomal ceftazidime and cefepime against P. aeruginosa ATCC 27853 and P. aeruginosa SPM-1 was compared to that of the free drugs. Liposomal characterization included diameter, encapsulation efficiency and stability. Minimum Inhibitory Concentration (MIC) was determined for free and liposomal forms of both drugs. Minimum Bactericidal Concentration (MBC) was determined at concentrations 1, 2 and 4 times MIC. Average diameter of liposomes was 131.88 nm and encapsulation efficiency for cefepime and ceftazidime were 2.29% end 5.77%, respectively. Improved stability was obtained when liposome formulations were prepared with a 50% molar ratio for cholesterol in relation to the phospholipid. MIC for liposomal antibiotics for both drugs were 50% lower than that of the free drug, demonstrating that liposomal drug delivery systems may contribute to increase the antibacterial activity of these drugs.
Keywords: Pseudomonas aeruginosa, liposomes, cephalosporins
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen, non-fermenting and, unable to produce energy for cell functioning (14). Non-fermenting bacteria are ubiquitous in nature, particularly in soil and water, and on surfaces in contact with soil or water. In the hospital environment, these microorganisms can be isolated from humidifiers, ventilators, mattresses and other equipment, as well as from the skin of health care professionals (16).
Resistance to antimicrobial drugs exhibited by P. aeruginosa is commonly found, especially in hospitals where resistant microorganisms are frequently detected in intensive care units (ICU). Acquired resistance mechanisms for this pathogen have increased significantly, leading to resistance to multiple antibacterial agents (5).
Low susceptibility of P. aeruginosa to antimicrobial substances is primarily due to the low permeability of its outer membrane, efflux mechanisms and the synthesis of enzymes that promote the degradation of these drugs (11). The outer membrane of P. aeruginosa is almost impermeable to many common antibiotics, such as ampicillin, most cephalosporins and macrolides, when compared to the outer membrane of other Gram-negative bacteria (13).
However, bacterial resistance to β-lactam antibiotics caused by low membrane permeability or efflux pumps is quite frequent for P. aeruginosa, Escherichia coli and Neisseria gonorrhoeae. Efflux mechanisms result in higher minimum inhibitory concentrations for penicillins, broad-spectrum cephalosporins, tetracyclins and fluoroquinolones (12, 30). Studies performed by Sentry (Antimicrobial Surveillance Program) about the activity of broad-spectrum β-lactam antibiotics against P. aeruginosa showed that about 17 to 34% of the bacterial samples were resistant to cefepime and ceftazidime (10, 24).
Since the bacterial outer membrane plays a major role in the permeability of antibiotics, the use of liposomal carriers is an interesting approach to enhance the antimicrobial activity of certain drugs (25). The lipid bilayer of liposomes are able to fuse with the outer membrane of bacteria (8, 27), altering the therapeutic index of a drug (20, 28). Drug encapsulation into liposomes is an effective method to reduce the drug’s toxicity, prolonging its circulation time after intravenous administration and enhancing its accumulation in the target site. These advantages can also improve the efficacy of antibiotics, not only for the treatment of resistant bacterial strains but also contributing to overcome bacterial resistance (4, 9, 15).
In this work, liposomal formulations for cefepime and ceftazidime were prepared and characterized. The antibacterial effect of liposomal cefepime and ceftazidime against P. aeruginosa ATCC 27853 and P. aeruginosa SPM-1 (clinical strain) was investigated by determining the Minimal Inhibitory Concentration (MIC) and Minimal Bactericidal Concentration (MBC) in a time-kill study.
MATERIALS AND METHODS
Chemicals
Cefepime was obtained from Alembic Limited, ceftazidime from Advan Pharma Chem Co. Ltd, soy phosphatidylcholine (PC) was from Lipoid, Gmb; cholesterol from Avanti Polar Lipids, α-tocopherol was purchased from Sigma Co., all other chemicals and reagents were of analytical grade or superior.
Microorganisms
Pseudomonas aeruginosa ATCC 27853 was kindly supplied by INCQS (National Institute for Quality Control in Health, Rio de Janeiro, Brazil), and P. aeruginosa SPM-1 (clinical strain), was donated by IPTSP (Instituto de Patologia Tropical e Saúde Pública, Goiás, Brazil). Microorganisms were kept at 20ºC in trypticase soy broth (Difco), supplemented with 10% (v/v) glycerol. For the experiment, the microorganisms were inoculated into inclined trypticase soy agar and incubated for 24 h at 37°C.
Preparation of Liposomes
Liposomes were prepared by the lipid film hydration method. 40 mM of phosphatidylcholine (PC), 10 or 20 mM of cholesterol (Chol) and 0.04 mM of α-tocopherol were dissolved in 2 mL of chloroform in a round bottom glass tube. Chloroform was removed under a Nitrogen stream and a rotary movement of the glass tube promoted the formation of a thin lipid film on the glass wall. The lipid film was then hydrated with 4mL of a cefepime solution (54 mg·mL-1) in 0.9% NaCl, and 8 mL of a ceftazidime solution (37.66 mg·mL-1). After the hydration, unilamellar liposomes were obtained by sonicating the lipid dispersion for 10 minutes in pulses of 1 minute with a Ti-probe sonicator (Misonix, XL 2020). Liposome diameter was determined by dynamic light scattering (ZetaSizer Nano, Malvern Instruments).
Encapsulation Efficiency
Non-encapsulated drug was removed from liposomes by size exclusion chromatography with a Sephadex G-50 column (10 x 200 mm). Encapsulation efficiency was calculated as the percentage of encapsulated drug (cefepime or ceftazidime) in relation to the total amount of drug added to the formulation. In order to minimize the dilution effect of the column separation process, liposomal formulations were submitted to ultracentrifugation at 50,000 rpm for 90 minutes at 4°C and resuspended in NaCl 0.9% to the desired concentration.
Quantitative determination of cefepime and ceftazidime was performed by high performance liquid chromatography (HPLC) using a ProStar 240 Chromatographer (Varian), equipped with an auto injector and UV detection at 255 nm and 254 nm wavelength, respectively. Separation was made in a C18 column (250 x 4.6 mm) and mobile phase was acetonitrile:water (10:90, v/v) for cefepime and acetonitrile:water (2:98, v/v) for ceftazidime, with an isocratic flow of 1mL·min-1. Results were calculated based on a calibration analytical curve prepared with cefepime reference standard (United States Pharmacopeia) and ceftazidime reference standard (European Pharmacopoeia), in the range of 0.001 to 0.1 mg·mL-1.
Stability of liposomal formulations
Stability of liposomal formulations was determined in order to evaluate the rate of drug leakage from liposomes. After the removal of the non-encapsulated drug, liposome dispersions were maintained at 4°C for 96 hours and at each 24 hours interval, samples were withdrawn for another step of separation in the sephadex column. The remaining amount of entrapped drug was determined by HPLC as described above.
Determination of the Minimum Inhibitory Concentration (MIC)
Antimicrobial activity of cefepime and of ceftazidime against Pseudomonas aeruginosa ATCC 27853 and P. aeruginosa SPM-1, was assayed for the free drugs and liposome entrapped drugs. MIC was determined by the broth dilution technique, as recommended by the Clinical and Laboratory Standards Institute (CLSI) (6). Sequential dilutions of free cefepime and ceftazidime (4096 to 2 µgmL-1) and liposomal cefepime and ceftazidime (4096 to 2 µgmL-1) were prepared in Mueller-Hinton broth. Immediately after the preparation, 1 mL of each drug dilution was inoculated with 1mL of the bacterial suspension, for a final bacterial count of 5 x 105 CFU.mL-1. Tubes were incubated for 18 hours at 35°C. MIC was defined as the lowest concentration at which no visible microbial growth was detected after 18 hours, following the addition of 500 µl of an aqueous solution of triphenyl tetrazolium chloride 0.5%. Positive control tubes were prepared with the culture medium inoculated with either bacterial suspension or bacterial suspension and blank liposomes. Negative (sterility) control tube consisted of culture medium only. Each test was performed in triplicates, in three different days to ensure reproducibility.
Determination of Minimum Bactericidal Concentration (MBC)
MBC was also determined by broth dilution technique. Briefly, overnight cultures of P. aeruginosa ATCC 27853, with a final bacterial count of 5 x 105 CFU.mL-1 were incubated with free cefepime and ceftazidime or their liposomal formulations, at concentrations of 1, 2 and 4 times the previously determined MIC, and P. aeruginosa SPM-1 was incubated with free and liposomal ceftazidime, under the same conditions. Control
tubes did not contain drug. Tubes were incubated at 37°C for 2, 6 and 24 hours. At each time point, serial dilutions were prepared in sterile saline 0.9% and 1 mL of each dilution was inoculated into Trypticase Soy Agar plates, in triplicates. After 18 hours of incubation, the number of CFUs for each dilution was counted. Plates with a number of colonies ranging from 30 to 300 were used for counting. MBC was established as the lowest concentration of either free or liposomal cefepime and ceftazidime, able to promote a 99.9% reduction of the initial bacterial inoculums.
RESULTS
Encapsulation Efficiency
The amount of encapsulated drug was 0.150 mg.mL-1 and 0.106 mg.mL-1 for ceftazidime and cefepime, respectively, following the technique of size exclusion chromatography for the separation of the free drugs.
Stability of liposomal cefepime and ceftazidime formulations
Tables 1 and 2 show respectively, the formulation parameters and drug release data of cefepime and ceftazidime encapsulated in liposomes, indicating that in preparations of cefepime with 10mM of cholesterol there was a loss of 97.19% of the encapsulated drug in the first 24 hours, against 44.91% observed for the preparation containing 20mM cholesterol. For liposomal ceftazidime the loss of the encapsulated drug was 98.25% and 63.25%, respectively, from liposomes with 10 mM and 20 mM of cholesterol, in 24 hours. (Table 1 e 2)
Table 1.
Formulation parameters and stability data for liposomal cefepime.
| Time (h) | CefepE (mg.mL−1) | Diameter (nm) | PdI | Drug release (%) | ||||
|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | A | B | |
| 0 | 0.0748 | 0.1002 | 126 | 151 | 0.278 | 0.313 | – | – |
| 24 | 0.0021 | 0.0552 | 128 | 150 | 0.240 | 0.319 | 97.19 | 44.91 |
| 48 | – | 0.0028 | 130 | 158 | – | 0.330 | nd | 97.20 |
| 72 | – | – | – | – | – | – | – | nd |
CefepE: encapsulated cefepime; PdI: polydispersity Index; nd: not detected; Formulation A: 40mM PC, 10mM Chol, 0.04mM α-tocopherol; Formulation B: 40mM PC, 20mM Chol, 0.04mM α-tocopherol.
Table 2.
Formulation parameters and stability data for liposomal cefepime.
| Time (h) | CefepE (mg.mL−1) | Diameter (nm) | PdI | Drug release (%) | ||||
|---|---|---|---|---|---|---|---|---|
| A | B | A | B | A | B | A | B | |
| 0 | 0.080 | 0.150 | 117 | 136 | 0.329 | 0.291 | – | – |
| 24 | 0.0014 | 0.055 | 118 | 138 | 0.318 | 0.295 | 98.25 | 63.25 |
| 48 | – | 0.0028 | – | 135 | – | 0.301 | nd | 98.10 |
| 72 | – | – | – | – | – | – | – | nd |
CeftzE: encapsulated ceftazidime; PdI: polydispersity Index; nd: not detected; Formulation A: 40mM PC, 10mM Chol, 0.04mM α-tocopherol; Formulation B: 40mM PC, 20mM Chol, 0.04mM α-tocopherol.
Antimicrobial activity of liposomal cefalosporins
Determination of MIC: MIC for free cefepime and ceftazidime against Pseudomonas aeruginosa ATCC 27853 was 8 μgmL-1, which is in accordance to CLSI limits (6), while MIC for liposomal cefepime and ceftazidime was 4 μgmL-1. It was not possible to determine MIC against P. aeruginosa SPM-1 for liposomal cefepime due to the low encapsulation efficiency for this drug, however, the P. aeruginosa SPM-1 had MIC of 1024 μgmL-1 for free ceftazidime and 512 μgmL-1 for liposomal ceftazidime. Blank liposomes did not have any effect on bacterial growth.
Determination of MBC: Figures 1 and 2 show, respectively, the antimicrobial activity of free and liposomal ceftazidime, and free and liposomal cefepime against P. aeruginosa ATCC 27853, and figure 3 for the free and liposomal ceftazidime against P. aeruginosa SPM-1, as a function of time and concentration.
Figure 1.
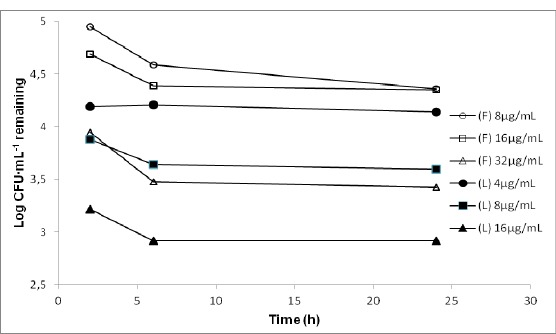
Time-kill curves for P. aeruginosa ATCC 27853 exposed to 1, 2 and 4 times the minimum inhibitory concentration (MIC) of free ceftazidime (F) and liposomal ceftazidime (L).
Figure 2.
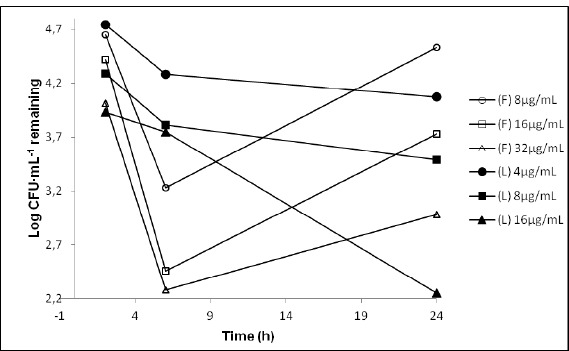
Time-kill curves for P. aeruginosa ATCC 27853 exposed to 1, 2 and 4 times the minimum inhibitory concentration (MIC) of free cefepime (F) and liposomal cefepime (L).
Figure 3.
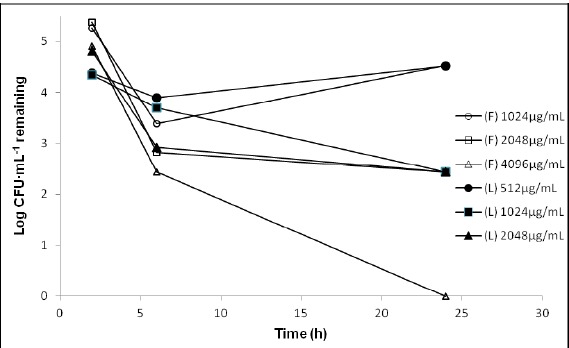
Time-kill curves for P. aeruginosa SPM-1 exposed to 1, 2 and 4 times the minimum inhibitory concentration (MIC) of free ceftazidime (F) and liposomal ceftazidime (L).
None of the concentrations of free or liposomal ceftazidime was able to eliminate 99.9% of the strain of P. aeruginosa ATCC 27853. Nevertheless, the liposomal ceftazidime at 4 x MIC (16 µg ml-1) in 24 hours, succeeded in removing 99.83% of the microorganisms inoculated; whereas this same concentration of free drug, showed a reduction of 96.5% in 24 hours. (Figure 1)
Free cefepime, against P. aeruginosa ATCC 27853, was able to kill 99.9% of the microorganisms inoculated with 2 x MIC (16µgmL-1) in 6 hours. However, at this concentration, the drug was not able to maintain this same antimicrobial performance, with an evident recovery of the bacteria after 24 hours. A reduction of 99.9% of the microorganism after 24 hours was only obtained when the amount of drug was 4 times higher than the MIC (32µgmL-1). (Figure 2)
Cefepime encapsulated into liposomes was able to reduce 99.9% of the inoculums of P. aeruginosa ATCC 27853, with 16µgmL-1 after 24 hours of incubation. In addition, when liposomal cefepime was used no bacterial recovery was observed at any concentration or time of incubation used in this study.
The free ceftazidime was able to kill 99,9% of P. aeruginosa SPM-1 inoculated with 1 x MIC (1024 µgmL-1) in 6 hours. However this concentration failed to maintain this percentage of elimination, with recovery of the microorganisms within 24 hours of contact. The reduction of 99.9% of the resistant strain was maintained in only 24 hours of contact with the free ceftazidime at 2 x MIC (2048 µg mL-1). (Figure 3)
The liposomal ceftazidime proved more effective in reducing 99.9% of the resistant strain with 1024 µg mL-1 in 24 hours. Moreover, when the liposomal ceftazidime was used, no bacterial recovery was observed at any concentrations or time of incubation.
DISCUSSION
Non-fermenting Gram-negative bacteria such as Pseudomonas aeruginosa are major opportunistic pathogens involved in severe infections acquired in hospitals, mainly with patients with nosocomial pneumonia in ICU. The control of these infections is extremely challenging due to the increased levels of resistance presented by this microorganism against most antimicrobial agents (21).
It has been established that encapsulating antibiotic drugs into liposomes can increase antimicrobial activity while reducing toxic effects (4, 9, 15).
This study proved that cefepime and ceftazidime can be successfully encapsulated into liposomes. Natural soy PC was used for the preparation of liposomes due to its low immunogenicity and toxicity when compared to other phospholipids such as cardiolipin, phosphatidylinositol, phosphatidylglycerol and phosphatidic acid (22, 26). Smaller amount of cefepime was encapsulated probably due to high concentrations of L-arginine (725mgg-1), present in the drug as pharmaceutical raw material, which is added to control the pH of the constituted solution at 4,0–6,0.
The encapsulation of hydro soluble drugs like cefepime and ceftazidime has been studied by several researchers. Results found in this work are in agreement with those presented by Antos, Trafny & Grzybowski (3), Omri & Ravaoarinoro (22) and Drulis-Kawa et al (7), who demonstrated encapsulation efficiencies from 3 to 7.2% using several liposome formulations. Conversely, Park et al (23) were able to encapsulate 75.4% of cefoxitine. However, liposomes from that study had a mean diameter higher than 600 nm, which despite the larger aqueous internal volume; these vesicles exhibit a higher content leakage and tendency to agglomerate.
The marked release of cefepime and ceftazidime from liposomes prepared with 10mM of cholesterol (97.19% and 98.25%, respectively) against 44.91% from the preparation of cefepime and of the 63.25% from preparation of ceftazidime, both with 20mM of cholesterol can be a result of a higher membrane fluidity in the 10mM formulation. Kinetics of drug release from liposomes can be controlled by adding different amounts of cholesterol to the membrane bilayers, which increases membrane rigidity, reduces permeability to hydro soluble molecules and promotes better stability of liposome vesicles in a protein rich medium (2, 31).
Higher concentrations of cholesterol (>30 molar%) can completely eliminate membrane phase transition and fluidity (29). In this work, a higher encapsulation efficiency and stability was obtained with liposome formulations using a molar ratio for cholesterol of 50 molar% in relation to PC.
Cefepime and ceftazidime leakage from liposomes was higher than that reported by Drulis-Kawa et al (7) for meropenem encapsulated into liposomes prepared with natural and synthetic lipids, with 24% leakage in 24 hours. When gentamycin was encapsulated into 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) liposomes, approximately 70% of the drug was retained during the first 48 hours (17). However, both meropenem and gentamycin are less hydro soluble than the cephalosporins used in this work. A higher hydrophobicity of the molecule favors its localization in the lipid bilayer or in the lipid-water interface due to its partition coefficient, reducing the leakage of the drug.
Liposomal cefepime and ceftazidime against P. aeruginosa ATCC 27853 and liposomal ceftazidime against Pseudomonas aeruginosa SPM-1 exhibited a higher antibacterial activity when compared to free cefepime and ceftazidime, as indicated by the 4 μg/mL and 512 μg/mL MIC, respectively. This 50% reduction was probably due to the interaction between liposomes and bacterial cells by a fusion mechanism as previously reported (1, 4, 20), which is capable of increasing the intracellular concentration of the drug, reducing the bacteria's ability to develop resistance. Other researchers showed that liposomal encapsulation was able to protect piperacylin from hydrolysis promoted by bacterial β-lactamase, which was probably due to the localization of the drug in the interior of the liposome where β-lactamase cannot penetrate (19).
Results from this work are in agreement with those found by Rukholm et al (25), Mugabe et al (17) and Mugabe et al (18), demonstrating a remarkable reduction of MIC for liposomal antibiotics. Liposomal gentamycin formulations studied by Mugabe et al (17) against resistant P. aeruginosa strains were effective at concentrations 2 to 256 times lower than MIC. Rukholm et al (25) did not find a marked difference between MIC and MBC for liposomal gentamycin against three strains of P. aeruginosa.
MIC reductions have also been found when amicacin, netilmycin and tobramycin were encapsulated into liposomes made of egg yolk PC and cholesterol (7:1 molar ratio). Amicacin MIC was reduced 4 times for S. aureus ATCC 29213 and E. coli ATCC 25 922 and 8 times for S. faecalis ATCC 29212. Conversely, when amicacin liposomes were tested against P. aeruginosa ATCC 27853, antimicrobial activity was lowered 8 times. Netilmycin and tobramycin liposomes were active against P. aeruginosa ATCC 27853 with a MIC of 2 and 8 times lower than that for the free drug, respectively (22).
Drulis-Kawa et al (7) encapsulated meropenem into several liposome formulations and evaluated their activity against eight strains of P. aeruginosa. Cationic liposomes resulted in a MIC 2 to 4 times lower than the MIC for the free drug. Anionic liposomes did not increase the antimicrobial activity, resulting in MICs of equal or higher values than that of the free drug.
MBC for liposomal cefepime and ceftazidime (16 μg·mL-1) was also reduced by 50% when compared to the free drug (32 μg·mL-1). Additionally, when liposomal cefepime was used against P. aeruginosa ATCC 27853, no bacterial recovery was observed during the whole period of incubation, demonstrating a prolonged antibacterial activity of the liposomal formulation when compared to free cefepime. Similar bactericidal effects were obtained by Mugabe et al (18) for liposomal amicacin against P. aeruginosa, where liposomes composed of DPPC/cholesterol in the molar ratio of 2:1 on average eradicated clinical strains of P. aeruginosa with a concentration of 8 mg·mL-1, while the free antibiotic at a concentration of 256 mg.mL-1, was inactive.
From these results, it can be concluded that encapsulating cefepime and ceftazidime into liposomes increases their antibacterial activity against P. aeruginosa ATCC 27853 and P. aeruginosa SPM1, indicating that liposomal formulations can be effective alternative for treating infections caused by these microorganisms and a valid approach against the development of bacterial resistance.
ACKNOWLEDGEMENTS
Authors would like to thank FINEP/MCT and FUNAPE/UFG for partially funding this research.
REFERENCES
- 1.Alipour M., Halwani M., Omri A., Suntres Z.E. Antimicrobial effectiveness of liposomal polymyxin B against resistant Gram-negative bacterial strains. Int J Pharm. 2008;355:293–298. doi: 10.1016/j.ijpharm.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 2.Anderson M., Omri A. The Effect of Different Lipid Componentes on the In Vitro Stability and Release Kinetics of Liposome Formulations. Drug Deliv. 2004;11:33–39. doi: 10.1080/10717540490265243. [DOI] [PubMed] [Google Scholar]
- 3.Antos M., Trafny E.A., Grzybowski J. Antibacterial Activity of Liposomal Amikacin Against Pseudomonas aeruginosa in vitro. Pharmacol Res. 1995;32(n° 1/2):85–87. doi: 10.1016/s1043-6618(95)80013-1. [DOI] [PubMed] [Google Scholar]
- 4.Beaulac C., Sachetelli S., Lagace J. In-vitro bactericidal efficacy of sub-MIC concentrations of liposome-encapsulated antibiotic against Gram-negative and Gram-positive bacteria. J Antimicrob Chemother. 1998;41:35–41. doi: 10.1093/jac/41.1.35. [DOI] [PubMed] [Google Scholar]
- 5.Chastre J., Trouillet J.L. Problem pathogens (Pseudomonas aeruginosa and Acinetobacter) Semin Respir Infect. 2000;15:287–298. doi: 10.1053/srin.2000.20944. [DOI] [PubMed] [Google Scholar]
- 6.Clinical and Laboratory Standards Institute/NCCLS. USA: Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087—1898; 2005. Performance Standards for Antimicrobial Susceptibility Testing; Fifteenth Informational Supplement. CLSI/NCCLS document M100-S15 {ISBN 1 - 56238–556—9} [Google Scholar]
- 7.Drulis-Kawa Z., Gubernator J., Dorotkiewicz-Jach A., Doroszkiewicz W., Kozubek A. In vitro antimicrobial of liposomal meropenem against Pseudomonas aeruginosa strains. Int J Pharm. 2006;315:59–66. doi: 10.1016/j.ijpharm.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Drulis-Kawa Z., Dorotkiewicz-Jach A., Gubernator J., Gula G., Bocer T., Doroszkiewicz W. The interaction between Pseudomonas aeruginosa cells and cationic PC:Chol:DOTAP liposomal vesicles versus outer-membrane structure and envelope properties of bacterial cell. Int. J Pharm. 2009;367:211–219. doi: 10.1016/j.ijpharm.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 9.Ellbogen M.H., Olsen K.M., Gentry-Nielsen M.J., Preheim L.C. Efficacy of liposome encapsulated ciprofloxacin compared with ciprofloxacin and ceftriaxone in rat model of pneumococcal pneumonia. J. Antimicrob Chemother. 2003;52:83–91. doi: 10.1093/jac/dkg024. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara A.M. Potentially multidrug-resistant non-fermentative Gram-negative pathogens causing nosocomial pneumonia. Int J Antimicrob Agents. 2006;27:183–195. doi: 10.1016/j.ijantimicag.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Hancock R.E.W., Speert D.P. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist Updat. 2000;3:247–255. doi: 10.1054/drup.2000.0152. [DOI] [PubMed] [Google Scholar]
- 12.Hardman J.G., Limbird L.E. The Pharmacological Basis of Therapeutics. 10. New York: McGraw-Hill; 2001. Gilmans A.G. Goodman & Gilman’s. [Google Scholar]
- 13.Hauser A.R., Sriram P. Severe Pseudomonas aeruginosa infections. Tackling the comundrum of drug resistance. Postgrad Med. 2005;117(1):41–48. doi: 10.3810/pgm.2005.01.1571. [DOI] [PubMed] [Google Scholar]
- 14.Japoni A., Alborzi A., Kalani M., Nasiri J., Hayati M., Farshad S. Susceptibility patterns and cross-resistance of antibiotics against Pseudomonas aeruginosa isolated from burn patients in the South of Iran. Burns. 2006;32:343–347. doi: 10.1016/j.burns.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Maurer N., Wong K.F., Hope M.J., Cullis P.R. Anomalous solubility behavior of the antibiotic ciprofloxacin encapsulated in liposomes: a 1H-NMR study. Biochim Biophys Acta. 1998;1374:9–20. doi: 10.1016/s0005-2736(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 16.Mcgowan J.E. Resistance in Nonfermenting Gram-Negative Bacteria: Multidrug Resistence to the Maximum. Am J Med. 2006;119(6A):S29–S36. doi: 10.1016/j.amjmed.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 17.Mugabe C., Azghani A.O., Omri A. Liposome-mediated gentamicin delivery: development and activity against resistant strains of Pseudomonas aeruginosa isolated from cystic fibrosis patients. J Antimicrob Chemother. 2005;55:269–271. doi: 10.1093/jac/dkh518. [DOI] [PubMed] [Google Scholar]
- 18.Mugabe C., Halwani M., Azghani R., Lafrenie R.M., Omri A. Mechanism of enhanced Activity of Lipossome-Entrapped Aminoglycosides against Resistant Strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50(n° 6):2016–2022. doi: 10.1128/AAC.01547-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nacucchio M.C., Bellora M.J.G., Sordelli D.O., D’Aquino M. Enhanced Liposome-Mediated Activity of Piperacillin Against Staphylococci. Antimicrob Agents Chemother. 1985;27(n°1):137–139. doi: 10.1128/aac.27.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicolosi D., Scalia M., Nicolosi V.M., Pignatello R. Encapsulation in fusogenic liposomes broadens the spectrum of action of vancomycin against Gram-negative bacteria. Int J Antimicrob Agents. 2010;35:553–558. doi: 10.1016/j.ijantimicag.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Omri A., Suntres Z.E., Shek P.N. Enhanced activity of lipossomal polymyxin B against Pseudomonas aeruginosa in a rat model of lung infection. Biochem Pharmacol. 2002;64:1407–1413. doi: 10.1016/s0006-2952(02)01346-1. [DOI] [PubMed] [Google Scholar]
- 22.Omri A., Ravaoarinoro M. Preparation, properties and the effects of amikacin, netilmicin and tobramycin in free and liposomal formulations on Gram-negative and Gram-positive bacteria. Int J Antimicrob Agents. 1996;7:9–14. doi: 10.1016/0924-8579(96)00003-9. [DOI] [PubMed] [Google Scholar]
- 23.Park J., Suh H., Sung H., Han D., Lee D.H., Park B.J., Park Y.H., Cho B.K. Liposomal Entrapment of Cefoxitin to Improve Cellular Viability and Function in Human Saphenous Veins. Artif Organs. 2003;27(7):623–630. doi: 10.1046/j.1525-1594.2003.07164.x. [DOI] [PubMed] [Google Scholar]
- 24.Pfaller M.A., Sader H.E., Fritsche T.R., Jones R.N. Antimicrobial activity of cefepime tested against ceftazidime-resistant Gram-negative clinical strains from North American Hospitals: report from the SENTRY Antimicrobial Surveillance program (1998–2004) Diagn Microbiol Infect Dis. 2006;56:63–68. doi: 10.1016/j.diagmicrobio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Rukholm G., Mugabe C., Azghani A.O., Omri A. Antibacterial activity of lipossomal gentamicin against Pseudomonas aeruginosa: a time-kill study. Int J Antimicrob Agents. 2006;27:247–252. doi: 10.1016/j.ijantimicag.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 26.Sachetelli S., Beaulac C., Riffon R., Lagacé J. Evaluation of the pulmonary and systemic immunogenicity of Fluidosomes, a fluid liposomal-tobramycin formulation for the treatment of chronic infections in lungs. Biochim Biophys Acta. 1999;1428:334–340. doi: 10.1016/s0304-4165(99)00078-1. [DOI] [PubMed] [Google Scholar]
- 27.Sachetelli S., Khalil H., Chen T., Beaulac C., Senechal S., Lagace J. Demonstration of a fusion mechanism between a fluid bactericidal liposomal formulation and bacterial cells. Biochim Biophys Acta. 2000;1463:2554–2566. doi: 10.1016/s0005-2736(99)00217-5. [DOI] [PubMed] [Google Scholar]
- 28.Schiffelers R., Storm G., Bakker-Woudenberg I. Liposome-encapsulated aminoglycosides in pre-clinical and clinical studies. J Antimicrob Chemother. 2001;48:333–344. doi: 10.1093/jac/48.3.333. [DOI] [PubMed] [Google Scholar]
- 29.Sharma A., Sharma U.S. Liposomes in drug delivery: progress and limitation. Int J Pharm. 1997;154:123–140. [Google Scholar]
- 30.Thomson J.M., Bonomo R.A. The threat of antibiotic resistance in Gram-negative pathogenic bacteria: β-lactams in peril! Curr Opin Microbiol. 2005;8:518–524. doi: 10.1016/j.mib.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Vemuri S., Rhodes C.T. Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm. Acta Helv. 1995;70:95–111. doi: 10.1016/0031-6865(95)00010-7. [DOI] [PubMed] [Google Scholar]