

Abstract
Aberrant redeployment of the ‘transient’ events responsible for bone development and postnatal longitudinal growth has been reported in some diseases in what is otherwise inherently ‘stable’ cartilage. Lessons may be learnt from the molecular mechanisms underpinning transient chondrocyte differentiation and function, and their application may better identify disease aetiology. Here, we review the current evidence supporting this possibility. We firstly outline endochondral ossification and the cellular and physiological mechanisms by which it is controlled in the postnatal growth plate. We then compare the biology of these transient cartilaginous structures to the inherently stable articular cartilage. Finally, we highlight specific scenarios in which the redeployment of these embryonic processes may contribute to disease development, with the foresight that deciphering those mechanisms regulating pathological changes and loss of cartilage stability will aid future research into effective disease-modifying therapies.
Keywords: bone, cartilage, osteoarthritis, chondrocyte, endochondral ossification
Introduction
The transition of cartilage to bone is the basis by which all long bones form. This transition is tightly regulated to ensure both permissive foetal development through endochondral ossification and postnatal longitudinal growth at the epiphyseal growth plate. Tighter management of these dynamic events may contribute to the inherent stability of some cartilage, such as articular cartilage, and this possibility is supported by emerging evidence reporting the aberrant redeployment of these transient events in the inherently stable cartilage in several pathological states such as osteoarthritis. It is therefore plausible that lessons can be realised from the molecular mechanisms underpinning the physiological transient chondrocyte differentiation and function. This knowledge may help to decipher those mechanisms surrounding joint pathology and ossification, and aid future research into effective disease-modifying therapies.
In this review, we discuss aspects of cartilage and bone physiology, as well as the complex cellular processes involved in the cartilage to bone transition during endochondral ossification and at the postnatal growth plate. Moreover, we will compare this transient cartilage phenotype with the stable phenotype adopted by articular chondrocytes in order to ostensibly safeguard their permanence, and will discuss disease pathology in which the aberrant redeployment of the transient phenotype is observed.
Transient cartilage
During mammalian embryological skeletal development, the appendicular skeleton comprising the pectoral girdle, pelvis and the limbs, arises from the lateral plate mesoderm (Saito et al. 2006). These structures are formed by means of endochondral ossification as does much of the axial skeleton including the vertebrae and ribs, which arise from somites of paraxial mesoderm origin.
Ossification processes are regulated during embryological development to produce the wide range of skeletal forms that we see in different animal species. A major challenge in skeletal morphogenesis is the precision of growth regulation that is required to create functional skeletal proportions, which can vary so widely between species, and to produce paired skeletal elements which are near identical in size. Therefore, there are major questions in embryological skeletal development, including how do condensations get to be the correct size and how is directional growth achieved in limb patterning?
The apical ectodermal ridge and the zone of polarising activity control proximo–distal and anterior–posterior patterning in the growing limb bud. These two centres are regulated by signalling pathways such as Indian hedgehog (IHH) and WNT/β-catenin (reviewed elsewhere; Summerbell et al. (1973), Kronenberg (2003), Tickle (2003), Towers & Tickle (2009) and Wolpert (2010)). Recent emerging evidence has suggested that planar cell polarity, a non-canonical WNT pathway involving the cadherins Fat and Dachsous (Fat/Dchs), is also important in embryonic skeletal development (Goodrich & Strutt 2011). Gradients of Dchs expression appear to regulate cell shape and directional movement during limb morphogenesis and growth (Dabdoub et al. 2003, Vladar et al. 2009, Wada & Okamoto 2009, Romereim & Dudley 2011). There is also substantial evidence, from pharmacological immobilisation of embryonic chicks, that movement-induced mechanical bone loading regulates longitudinal growth of skeletal elements (Osborne et al. 2002, Lamb et al. 2003, Pitsillides 2006). Thus, mechanical stimuli may exert epigenetic control over skeletal development to act as an additional source of phenotypic variation during development.
Endochondral ossification
The majority of the skeleton is first formed during embryonic development as cartilage anlagen; models of future skeletal elements. The transient nature of this cartilage is reflected in its eventual replacement by bone in the fundamental process of endochondral ossification. Endochondral ossification is initiated by embryonic mesenchymal cells migrating to form pre-cartilage condensations, which then undergo differentiation into chondrocytes and secrete an extracellular matrix rich in collagen type II and aggrecan. The chondrocytes of these cartilage condensations undergo an ordered and highly regulated process involving predominant marginal proliferation and central maturation, hypertrophy and cell death. These latter events define the framework of the endochondral bone and are regulated by paracrine signals including parathyroid hormone (PTH)-related peptide and IHH from the perichondrium (Fig. 1; Storm & Kingsley 1996, Tickle & Münsterberg 2001, Wang et al. 2001, Kronenberg 2003, Davey et al. 2006, Villavicencio-Lorini et al. 2010, Mackie et al. 2011).
Figure 1.
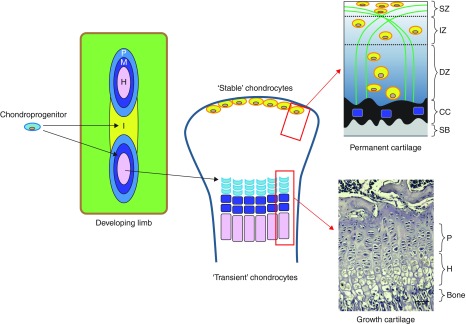
Schematic representation depicting the proposed developmental origins of articular and growth plate cartilage. Mesenchymal aggregation of chondro-progenitors forms the cartilage anlagen and stages of chondrocyte proliferation (P, light blue), maturation (M, purple) and hypertrophy (H, pink) emerge to provide the origins of the future ‘transient’ chondrocytes of the growth plate cartilage. Intervening regions of progressively condensing mesenchyme define the interzone (I, yellow) regions; the position of the future joint and origins of the ‘stable’ articular cartilage chondrocytes. Images (right hand side) depicting the organisation of the mature growth plate (lower) and articular cartilage (upper), consisting of uncalcified cartilage zones (superficial (SZ), intermediate (IZ) and deep (DZ)), and the related chondrocyte and collagen fibril arrangement. The tidemark separates the non-calcified cartilage from the calcified cartilage (CC) which overlies the subchondral bone (SB). Bar=0.1 mm.
Primary ossification originates in the centre of the diaphysis of the developing skeletal element. The growth cartilage is invaded by blood vessels, seemingly attracted by VEGF expression by hypertrophic chondrocyte (Zelzer et al. 2002) and with this, infiltration of bone-resorbing osteoclasts and bone-forming osteoblasts occurs. Furthermore, the perichondrium becomes vascularised around the bone-forming element to create the periosteum. This process of blood vessel invasion facilitated by degradation of the calcified cartilage extracellular matrix around the hypertrophic chondrocytes is critically reliant on the activity of matrix metalloproteinase 13 (MMP13; Stickens et al. 2004). Cartilage matrix resorption is followed by the invasion of osteoblasts, which lay down the newly formed bone. This process spreads longitudinally from the primary ossification centre towards the ends of the bone. Eventually, a secondary ossification centre forms, retaining a cartilaginous growth plate between each epiphysis and the primary ossification centre (Mackie et al. 2008).
Morphology of the postnatal growth plate
The growth plate is a highly specialised developmental region, responsible for postnatal linear bone growth. It consists of chondrocytes, arranged in columns that parallel the axis of the bone, surrounded by their matrix; the precise composition of which is dependent upon the state of chondrocyte differentiation (Fig. 1; Ballock & O'Keefe 2003, Mackie et al. 2011). These chondrocytes undergo a series of tightly regulated differentiation and maturation processes, as is reflected by their changing morphology and matrix production, whilst maintaining their spatially fixed locations (Hunziker et al. 1987).
Interference with the maturational progression of growth plate chondrocytes leads to abnormal cartilage formation and ossification and modifications in the rates of bone formation, for example, in rickets and dwarfism. Details of the many syndromes and diseases and possible therapeutic strategies are out with the scope of this review but have been the subject of discussion in other reviews (Kornak & Mundlos 2003, Krakow & Rimoin 2010).
The primary zone of the growth plate, often known as the ‘resting’ or ‘germinal’ zone, consists of undifferentiated chondrocyte progenitors. Unlike the remainder of the growth plate, the chondrocytes of the resting zone are distributed sporadically and have a low rate of proliferation (Hunziker et al. 1987, Ballock & O'Keefe 2003, Mackie et al. 2011). As chondrocytes progress from the resting zone, they gain a proliferative phenotype and adopt a flattened, oblate shape, arranging themselves into longitudinal columns. It is proposed that the creation of these highly organised columns is directed by the chondrocytes in the resting zone, which have been postulated to produce a growth plate-orientating factor (Hunziker et al. 1987, Abad et al. 2002). The high mitotic activity of these proliferating chondrocytes is controlled by numerous factors, including endocrine/autocrine regulation, circadian rhythm and age. These chondrocytes also undergo a period of high secretory activity as they produce a collagen type-II- and proteoglycan-rich matrix, which with some otherwise marked distinctions closely resembles that seen in the articular cartilage (Farnum et al. 2002).
The chondrocytes undergo major phenotypic changes as they gradually become hypertrophic however, until recently the mechanisms by which this occurs is unclear but may involve membrane transporters in the chondrocyte membrane (Bush et al. 2008, 2010, Loqman et al. 2013). Furthermore, a paper by Cooper et al. has defined three distinct phases through which mammalian chondrocyte volume increases: i) true hypertrophy, indicating a proportionate increase in dry mass production and fluid uptake, responsible for approximately threefold increases in volume, ii) a fourfold enlargement solely by cell swelling and iii) a second distinct phase of true hypertrophy with proportionate dry mass and fluid volume increases. These authors show that this final stage is dependent upon insulin-like growth factor 1 (IGF1), a well-recognised regulator of longitudinal bone growth and chondrocyte hypertrophy (Wang et al. 1999, Ahmed & Farquharson 2010, Cooper et al. 2013, Farquharson & Ahmed 2013). More recent emerging evidence has detailed the regulation of IGF1 signalling in chondrocyte hypertrophy by the WNT signalling pathway, more specifically by WNT-induced secreted protein 3 (Rao et al. 2013). Indeed, it appears that the WNT pathway is critical in both growth plate and articular cartilage function, as we have previously discussed in our recent review (Staines et al. 2012a).
As well as changes in cell morphology, collagen type-II expression decreases during chondrocyte maturation and the hypertrophic chondrocyte initiates the synthesis of collagen type-X (Schmid & Linsenmayer 1985, Kronenberg 2003). The matrix also includes chondrocalcin, osteonectin and osteopontin, as well as having increased membrane activity of alkaline phosphatase, indicating the final maturation phase (Sommer et al. 1996, Shen 2005, Roberts et al. 2007). Together these changes facilitate mineralisation of the matrix, a complex process reliant upon the levels of calcium and phosphate (Castagnola et al. 1988). Mineralisation of the extracellular matrix also enables vascular invasion, through which osteoclasts and osteoblasts gain access via migration to replace the cartilage template with bone (Gerber et al. 1999, Horner et al. 1999, Engsig et al. 2000, Zelzer et al. 2002). Recent evidence has suggested that the terminally differentiated hypertrophic chondrocyte may sense signals that initiate this transformation of the cartilage matrix into bony trabeculae through the expression of receptor activator of nuclear factor-κB ligand, an essential cytokine for osteoclast development, function and survival (Xiong et al. 2011).
The fate of the terminally differentiated chondrocyte at the chondro-osseous junction is still a matter of debate. However, it is well accepted that this chondrocyte does require to be removed so as to maintain the steady-state thickness of the growth plate. There has been significant evidence to suggest that the chondrocytes die by apoptosis and this appears the most accepted mechanism (Magne et al. 2003, Shapiro et al. 2005). Indeed the activation of caspases and the decreased expression of the anti-apoptotic factor, Bcl2, in the hypertrophic chondrocytes certainly support this (Amling et al. 1997, Adams & Shapiro 2002). The distinct lack of typical apoptotic morphological changes in the terminal hypertrophic chondrocytes does, however, challenge this theory (Emons et al. 2009, Carames et al. 2010). It would be expected that the condensation of cellular chromatin and the fragmentation of the cell nucleus would be visible, associated with the eventual break down of the cell into several vesicles which are then phagocytosed. Instead, the presence of autophagic vacuoles and the expression of autophagy-regulating genes by growth plate chondrocytes suggest that these cells undergo processes more similar to autophagy than apoptosis (Roach & Clarke 2000, Shapiro et al. 2005). Furthermore, the transdifferentiation of chondrocytes has also been proposed (Descalzi Cancedda et al. 1995, Roach 1997). This involves the division of the terminal hypertrophic chondrocyte to produce one daughter cell which undergoes apoptosis and one which transdifferentiates into an osteoblast phenotype. However this theory has yet to be fully ratified and would certainly benefit from experiments which use GFP-Cre systems to track the collagen type-X expression of the hypertrophic chondrocyte during its terminal fate.
Longitudinal bone growth occurs at the growth plate until it closes once sexual maturity has been reached. This is initiated through the formation of mineralised tethers between epiphyseal and diaphyseal bone promoting the fusion of the primary and secondary ossification centres (Haines 1975). The chondrocytes of the growth plate reach a state of senescence as they exhaust their proliferative potential, and longitudinal bone growth is ceased. In humans, oestrogen mediates these effects in both males and females and the processes controlling fusion are relevant to understanding the ‘permanent’ loss of this transient chondrocyte phenotype (Nilsson et al. 2005, Mackie et al. 2011).
Stable cartilage
Articular cartilage formation
The earliest emergence of ‘stable’, articular cartilage during skeletal patterning occurs with the formation of interzones; regions of undifferentiated mesenchyme which separate developing skeletal elements in which the joint will later form. There has been some debate as to whether individual skeletal elements are discrete from the outset of their development, or whether these elements form from a single cartilage condensation which is later divided by interzones (Fig. 1; Pitsillides & Ashhurst 2008).
Transient growth cartilage and articular cartilage differ in their collagen composition during their development; in embryonic growth cartilage, expression of collagen type-IIA precedes collagen type-IIB expression, which is not detectable at later stages of development. However in articular cartilage, collagen type-IIA protein does not appear in the interzones, which provides evidence for the belief that the cartilage anlagen that form individual skeletal elements are not continuous (Ng et al. 1993, Pitsillides & Ashhurst 2008). This suggests that the populations of cells which will become the growth and articular cartilage chondrocytes are discrete. Evidence for these discrete origins is also provided by the GDF5-mediated tracking of interzonal cells, and by the unique expression of versican, an extracellular matrix proteoglycan, in the regions of the presumptive articular cartilage in joint morphogenesis (Pacifici et al. 2005, 2006, Snow et al. 2005, Hyde et al. 2007).
Interzones first appear as densely cellular, homogenous regions with GDF5, WNT9A, autotaxin and chordin being known as interzone markers (Pacifici et al. 2005). Non-canonical WNT9A signalling is particularly important early in development as it acts to inhibit chondrocyte differentiation at the presumptive joint site (Hartmann & Tabin 2001). As joint development progresses, the interzone differentiates into three recognisable layers: two chondrogenic layers which cover the articular surfaces of the developing opposed skeletal elements and an intermediate layer which separates them. There is evidence to suggest that the cells derived from this intermediate layer differentiate to become articular chondrocytes, while the outer layer chondrocytes are incorporated into the growing epiphysis (Ito & Kida 2000).
Joint cavity formation is thought to occur due to a combination of extrinsic mechanical factors and intrinsic factors. Cell death is not thought to be primarily responsible for the formation of the joint cleft; rather, changes in extracellular matrix composition including hyaluronan synthesis, mediated by mechanical activation of the MEK–ERK pathway, which result in a loss of tissue cohesion have been implicated (Archer et al. 1994, Ito & Kida 2000, Bastow et al. 2005).
Articular cartilage phenotype
Fully developed uncalcified articular cartilage can be divided into superficial, intermediate and deep zones (Fig. 1). These different zones consist of just one cell type, the chondrocyte. These chondrocytes differ from the transient growth plate chondrocytes as they maintain a stable phenotype characterised by small cell size and expression of tenascin-C, and they do not undergo a sequence of proliferation, maturation, hypertrophy, apoptosis and ossification (Pacifici et al. 2005, 2006). It has been suggested that the expression of the transcription factor ETS-related gene (ERG) is necessary early in joint development to establish the articular chondrocyte phenotype (Pacifici et al. 2006). It is the organisation of cells and their collagen type-II and aggrecan-rich matrix in the differing zones, however, which makes them distinct from one another. Whilst the superficial zone consists of elongated chondrocytes orientated parallel to the surface of the cartilage, chondrocytes in the deep zone are more rounded and aligned along the collagen fibrils. The collagen fibrils in the intermediate zone of the articular cartilage are arranged in arches which allow the transition from the deep and superficial zones (Minns & Steven 1977).
The deep zone of the articular cartilage forms an interface, termed the tidemark, with calcified cartilage which forms through mechanisms that are not quite understood. Within this calcified cartilage, the chondrocytes are hypertrophic and this is accompanied by their expression of collagen type-X and alkaline phosphatase (Heinegard & Oldberg 1989, Gannon et al. 1991, Stephens et al. 1992). Therefore, despite the cellular singularity and stability of the articular cartilage chondrocyte, there is some degree of transiency due to the varying chondrocyte phenotypes which exist within the entirety of the tissue. This notion is supported by the presence of chondrocyte progenitors in the superficial zone both at young stages of development and in the mature cartilage (Dowthwaite et al. 2004). Therefore tighter management of these dynamic events, although much slower than those in the growth plate, may alone underpin the maintenance of the articular cartilage stability.
The calcified cartilage layer is semipermeable and whilst it acts as a physical barrier for vascular invasion of the overlying articular cartilage, it does permit the passage of small molecules from the underlying subchondral bone (Arkill & Winlove 2008). In humans, the calcified cartilage thickness varies widely, from ∼20 to 250 μm however, like the tidemark, this a dynamic structure which can be remodelled during ageing and disease, as highlighted by observed morphological changes in osteoarthritis including tidemark duplication and increased calcified cartilage thickness (Lane & Bullough 1980, Oettmeier et al. 1989, Oegema et al. 1997, Hunziker et al. 2002, Burr 2004). The molecular mechanisms regulating events at this osteochondral interface are yet to be fully defined. Their identification may provide potential targets for therapeutic intervention.
The ectopic initiation of cartilage–bone transitions
The transition of cartilage to bone in the healthy individual is under tight regulation so as to prevent disturbed development and/or longitudinal bone growth. Observations from disorders such as hypothyroidism, rickets and dwarfism may provide insights into the mechanisms underpinning defective hypertrophic differentiation and ossification (Combs et al. 2011, Santos et al. 2013). Regulation is also observed in repair of fractured bone tissue, in which there is a deliberate re-initiation of the endochondral processes (see above) previously discussed in this review. There has been much recent focus upon the WNT signalling pathway in fracture repair and, in particular, the recent discovery of enhanced repair through the administration of neutralizing antibodies against sclerostin, a known WNT inhibitor (Secreto et al. 2009, Ominsky et al. 2011, Virk et al. 2013). In contrast to this desired acceleration in endochondral ossification processes, in certain diseases the ectopic redeployment of these processes is detrimental and considered to be at least contributory to the observed disease pathology. Herein we discuss conditions in which this occurs and touch upon current understanding regarding the role of these processes in their aetiology.
Osteoarthritis
Osteoarthritis is a degenerative joint disease and a massive world-wide healthcare burden. Characterised by articular cartilage loss, subchondral bone thickening and osteophyte formation, the osteoarthritic joint afflicts much pain and disability on its sufferers. Its underpinning molecular mechanisms are, nevertheless, not fully understood; indeed it is even still a matter of debate as to which is the preceding pathology. However, there is increasing evidence implicating the re-initiation of the transient chondrocyte phenotype in osteoarthritic aetiology and pathology (Fosang & Beier 2011, Pitsillides & Beier 2011; Fig. 2). This notion is based upon the previously discussed common embryonic development of cartilage and bone and the current evidence certainly merits thorough examination. However, it has been met with some controversy in the field (Brew et al. 2010). Such controversies may potentially be addressed through a better understanding of the articular cartilage volume to surface area ratios in different species, and thus may provide some explanation as to why the degree of chondrocyte transiency in the articular cartilage differs from species to species.
Figure 2.
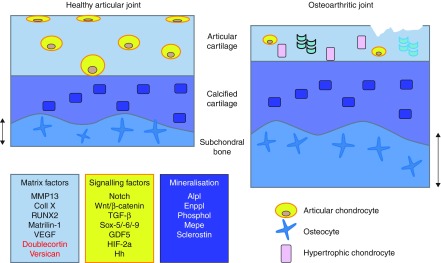
Schematic diagram depicting the healthy articular joint and the osteoarthritic joint in which articular cartilage fibrillation and degradation is observed with concomitant subchondral bone thickening. Contributing to this osteoarthritic pathology, the normally ‘stable’ articular chondrocytes of the articular cartilage adopt a ‘transient’ phenotype with observed chondrocyte hypertrophy and matrix mineralisation similar to that seen in the growth cartilage depicted in Fig. 1. Potential regulation of these processes may include changes in the expression of matrix factors (blue box; factors induced in osteoarthritis development in black, factors lost in red), signalling pathways affecting chondrocyte phenotype and function (yellow box) and the known regulators of mineralisation processes (purple box) (Grover & Roughley 1993, Pacifici et al. 2006, Zhang et al. 2007, Fosang & Beier 2011, Pitsillides & Beier 2011, Staines et al. 2012b). Lessons may be learnt from these and their application may better identify disease aetiology.
In osteoarthritic cartilage, there have been observed decreases in collagen type-II and aggrecan integrity in comparison to normal articular cartilage (Helminen et al. 1993, Garnero et al. 2002, Jalba et al. 2011, Henrotin et al. 2013). Furthermore, markers previously thought to be unique to the hypertrophic chondrocytes of the growth plate have been detected in the uncalcified articular cartilage in both animal models of osteoarthritis, and in patients with the disease. The best recognised of these are MMP13 and collagen type-X, however, also detected are alkaline phosphatase, osteopontin, IHH and osteocalcin (Hoyland et al. 1991, Aigner et al. 1993, Pullig et al. 2000, Appleton et al. 2007, Studer et al. 2012). As in the transient growth plate cartilage, chondrocyte hypertrophy is considered to be a prerequisite for matrix mineralisation and in osteoarthritis, the increased formation of hydroxyapatite has been documented and is consistent with hypertrophic chondrocyte changes (Fuerst et al. 2009).
MMP13, a key marker of chondrocyte hypertrophy, is proving a critical target in osteoarthritis research due to its potent role in the degradation of collagen type-II, proteoglycans, collagen types-IV and IX, osteonectin and perlecan (Wang et al. 2013). Indeed, Mmp13-deficient mice predictably have an endochondral bone growth defect with an expanded hypertrophic chondrocyte zone which was not attributable to increased chondrocyte proliferation, increased proteoglycan turnover and increased trabecular bone (Stickens et al. 2004). Nevertheless, whilst the surgical induction of osteoarthritis in this mouse causes chondrocyte hypertrophy and osteophyte formation, the graded score of cartilage degradation was intriguingly significantly reduced (Little et al. 1999). Deletion of the Mmp13 gene specifically in chondrocytes also produces similar deceleration of osteoarthritis disease progression following meniscal-ligamentous injury in a mouse model (Wang et al. 2013). Consistent with this crucial role for Mmp13, its overexpression was also found to result in pathological osteoarthritis-like changes in the articular cartilage of mice (Neuhold et al. 2001). Recent research has implicated the WNT signalling pathway and its inhibitors as important regulators of MMP13 (van den Bosch et al. 2013, Chan et al. 2013, Staines et al. 2013), which is consistent with critical WNT function in the prevention of joint pathologies (Zhu et al. 2008, 2009, Miclea et al. 2011).
Epigenetics has been strongly implicated in osteoarthritis in recent years with all three currently known mechanisms – DNA methylation, histone modifications and non-coding RNAs – demonstrate a capacity to control the chondrocyte phenotype. This too has highlighted a role for MMP13. More specifically, MMP13 promotor methylation is altered in osteoarthritic cartilage, suggesting that these epigenetic changes can therefore drive the chondrocyte hypertrophy observed in the pathological state (Roach et al. 2005). It should be noted that the precise initiation and control of these events is yet to be established. However, with the surge in epigenetic studies in recent years, it is certainly an exciting and promising time for this field.
This evidence therefore pinpoints the need to decipher the molecular mechanisms underpinning the recapitulation of some of these developmental ‘hallmarks’ by articular cartilage chondrocytes in osteoarthritis. It is envisaged that this will undoubtedly advance understanding into disease pathology and might ultimately define whether the pathogenic pathways in osteoarthritis can be prevented by regulating the stability of chondrocytes by limiting acquisition of their transient, growth-related phenotype.
Certainly the field of tissue engineering would benefit from such understanding. Combining the use of cells and biomaterials, this approach has emerged as a promising target for cartilage repair. The first repair of cartilage defects was described in 1994 and used autologous chondrocytes (Brittberg et al. 1994). Despite the limitations associated with this, including the need to disrupt healthy cartilage and problems with culturing chondrocytes, its emergence has certainly set the platform from which cartilage tissue engineering has greatly progressed in recent years. The use of human mesenchymal stem cells, with their ability to overcome the limitations defined by the use of chondrocytes, has been repeatedly reported in osteoarthritis as a means of providing promise and much excitement in the field (Luyten 2004, Coleman et al. 2010). It has become apparent that human mesenchymal stem cells can also gain expression of the chondrocyte hypertrophy marker, collagen type-X, and it is clear that this still limits their effectiveness as a candidate for tissue engineering. Indeed, a recent study has shown that mesenchymal stem cells express higher levels of the genes associated with osteoarthritis, upon culture, than do chondrocytes derived from the osteoarthritic joints themselves (Mwale et al. 2010, Fernandes et al. 2013).
Intervertebral disc calcification
Intervertebral disc (IVD) degeneration is a major cause of back pain worldwide with complex, expensive surgery which is often prone to failure (Urban & Roberts 1995). Located between the vertebral bodies of the spine, the cartilaginous IVD functions to resist compressive loads (Broberg 1983). Anatomically, it consists of a central nucleus pulposus contained within a fibrocartilage ring, the annulus fibrosus, laterally and the cartilage end plates inferiorly and superiorly (Roberts et al. 2006). The nucleus pulposus shares many similarities with articular cartilage, both in its matrix composition and its cellular metabolism (Raj 2008). Associated with ageing and with abnormal mechanical loading playing a substantial role, the pathogenesis of IVD degeneration is yet to be fully elucidated. There is, however, emerging evidence for the occurrence of hypertrophic differentiation in this process.
The IVDs of patients with degenerative disc disease have increased alkaline phosphatase activity, and also in contrast to healthy IVDs express significant levels of hypertrophic differentiation markers, including collagen type-X, osteoprotegerin (OPG), MMP13 and RUNX2 (Boos et al. 1997, Nerlich et al. 1997, Sato et al. 2008, Rutges et al. 2010, Hristova et al. 2011). Associated with this, microCT analysis clearly shows increased levels of IVD calcification with increasing degeneration (Rutges et al. 2010). A recent in vitro study has sought to examine the mineralisation potential of the cells of the annulus fibrosus, and concluded that under certain conditions these cells can induce mineralisation, as indicated by their increased von Kossa staining, alkaline phosphatase, MMP13 and RUNX2 expression (Nosikova et al. 2013).
Understanding of the mechanisms supporting induction of such hypertrophy in IVD cells is somewhat lacking. However, it was shown recently that PTH can enhance disc repair through its inhibition of collagen type-X and alkaline phosphatase expression, and through the promotion of collagen type-II production. These regenerative properties of PTH are mediated through MAPK signalling (Madiraju et al. 2013). The authors of this study suggest that PTH supplementation may prevent further calcification in degenerated discs and potentially enhance other cell-based therapies. This is certainly promising due to the known critical role for PTH signalling in chondrocyte hypertrophy and matrix mineralisation (Mackie et al. 2011, Yano et al. 2013). The current major problem hindering IVD tissue engineering, like in osteoarthritis, is the undesirable expression of collagen type-X by human mesenchymal stem cells. Recent studies have highlighted that PTH treatment can also inhibit such misexpression and may thus open additional avenues for the treatment of degenerative IVDs with mesenchymal stem cells (Mwale et al. 2010).
Heterotopic ossification
Heterotopic ossification (HO) is a common occurrence in muscle, tendon and ligaments following trauma by injury, disease or surgery. Initiated by cartilage formation, the endochondral ossification and ectopic bone formation in these tissues can produce severe functional impairment (Medici & Olsen 2012).
Besides the trauma-induced HO described, a rare disease called fibrodysplasia ossificans progressiva (FOP) is a hereditary form of HO, presenting itself as painful and highly inflammatory soft tissue swellings which progressively ossify rendering the sufferer immobile (Cohen et al. 1993, Shore et al. 2006, Medici & Olsen 2012). Like in trauma-induced HO, the aberrant bone formation in patients with FOP occurs by means of endochondral ossification processes. Emerging evidence has strongly implicated increased bone morphogenetic protein (BMP) signalling in the pathogenesis of FOP, as well as in trauma-induced HO (Cohen et al. 1993, Kwapisz et al. 2013). The identification of ACVR1/ALK2, one of the four type-I receptors that mediate BMP signalling, as the mutated gene in FOP in 2006 has further highlighted the BMP signalling pathway as an attractive target for future therapy (Shore et al. 2006).
Conclusions
The aberrant redeployment of embryonic processes in diseases such as osteoarthritis and IVD degeneration is now well established. Emerging evidence has further fuelled the hypothesis that lessons for limiting disease progression can be acquired from the regulators of transient cartilage biogenesis and development. This review provides some pointers as to potential targets for future drug therapies or tissue engineering approaches which will further our understanding of the underpinning molecular mechanisms involved in these diseases, and will provide advances towards patient benefit.
Acknowledgements
The authors wish to acknowledge Arthritis Research UK, the Anatomical Society and the BBSRC for funding.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
Funding
This work was supported by Arthritis Research UK (18768), the Anatomical Society and an Institute Strategic Programme Grant from the Biotechnology and Biological Sciences Research Council, UK (C F).
References
- Abad V, Meyers JL, Weise M, Gafni RI, Barnes KM, Nilsson O, Bacher JD, Baron J. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143:1851–1857. doi: 10.1210/en.143.5.1851. [DOI] [PubMed] [Google Scholar]
- Adams CS, Shapiro IM. The fate of the terminally differentiated chondrocyte: evidence for microenvironmental regulation of chondrocyte apoptosis. Critical Reviews in Oral Biology and Medicine. 2002;13:465–473. doi: 10.1177/154411130201300604. [DOI] [PubMed] [Google Scholar]
- Ahmed SF, Farquharson C. The effect of GH and IGF1 on linear growth and skeletal development and their modulation by SOCS proteins. Journal of Endocrinology. 2010;206:249–259. doi: 10.1677/JOE-10-0045. [DOI] [PubMed] [Google Scholar]
- Aigner T, Reichenberger E, Bertling W, Kirsch T, Stoss H, von der Mark K. Type X collagen expression in osteoarthritic and rheumatoid articular cartilage. Virchows Archiv. B, Cell Pathology Including Molecular Pathology. 1993;63:205–211. doi: 10.1007/BF02899263. [DOI] [PubMed] [Google Scholar]
- Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, Philbrick WM, Broadus AE, Baron R. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. Journal of Cell Biology. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis and Rheumatism. 2007;56:1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- Archer CW, Morrison H, Pitsillides AA. Cellular aspects of the development of diarthrodial joints and articular cartilage. Journal of Anatomy. 1994;184:447–456. [PMC free article] [PubMed] [Google Scholar]
- Arkill KP, Winlove CP. Solute transport in the deep and calcified zones of articular cartilage. Osteoarthritis and Cartilage. 2008;16:708–714. doi: 10.1016/j.joca.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Ballock RT, O'Keefe RJ. Physiology and pathophysiology of the growth plate. Birth Defects Research. Part C, Embryo Today: Reviews. 2003;69:123–143. doi: 10.1002/bdrc.10014. [DOI] [PubMed] [Google Scholar]
- Bastow ER, Lamb KJ, Lewthwaite JC, Osborne AC, Kavanagh E, Wheeler-Jones CP, Pitsillides AA. Selective activation of the MEK–ERK pathway is regulated by mechanical stimuli in forming joints and promotes pericellular matrix formation. Journal of Biological Chemistry. 2005;280:11749–11758. doi: 10.1074/jbc.M414495200. [DOI] [PubMed] [Google Scholar]
- Boos N, Dreier D, Hilfiker E, Schade V, Kreis R, Hora J, Aebi M, Boesch C. Tissue characterization of symptomatic and asymptomatic disc herniations by quantitative magnetic resonance imaging. Journal of Orthopaedic Research. 1997;15:141–149. doi: 10.1002/jor.1100150121. [DOI] [PubMed] [Google Scholar]
- van den Bosch M, Blom A, van Erp A, van de Loo F, Davidson EB, van der Kraan P, van Lent P, van den Berg W. Synovial Wnt and Wisp1 expression induces expression of cartilage-degrading metalloproteinases in the synovium. Osteoarthritis and Cartilage. 2013;21:S284–S284. doi: 10.1016/j.joca.2013.02.594. [DOI] [Google Scholar]
- Brew CJ, Clegg PD, Boot-Handford RP, Andrew JG, Hardingham T. Gene expression in human chondrocytes in late osteoarthritis is changed in both fibrillated and intact cartilage without evidence of generalised chondrocyte hypertrophy. Annals of the Rheumatic Diseases. 2010;69:234–240. doi: 10.1136/ard.2008.097139. [DOI] [PubMed] [Google Scholar]
- Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. New England Journal of Medicine. 1994;331:889–895. doi: 10.1056/NEJM199410063311401. [DOI] [PubMed] [Google Scholar]
- Broberg KB. On the mechanical behaviour of intervertebral discs. Spine. 1983;8:151–165. doi: 10.1097/00007632-198303000-00006. [DOI] [PubMed] [Google Scholar]
- Burr DB. Anatomy and physiology of the mineralized tissues: role in the pathogenesis of osteoarthrosis. Osteoarthritis and Cartilage. 2004;12(Suppl A):S20–S30. doi: 10.1016/j.joca.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Bush PG, Parisinos CA, Hall AC. The osmotic sensitivity of rat growth plate chondrocytes in situ; clarifying the mechanisms of hypertrophy. Journal of Cellular Physiology. 2008;214:621–629. doi: 10.1002/jcp.21249. [DOI] [PubMed] [Google Scholar]
- Bush PG, Pritchard M, Loqman MY, Damron TA, Hall AC. A key role for membrane transporter NKCC1 in mediating chondrocyte volume increase in the mammalian growth plate. Journal of Bone and Mineral Research. 2010;25:1594–1603. doi: 10.1002/jbmr.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis and Rheumatism. 2010;62:791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnola P, Dozin B, Moro G, Cancedda R. Changes in the expression of collagen genes show two stages in chondrocyte differentiation in vitro. Journal of Cell Biology. 1988;106:461–467. doi: 10.1083/jcb.106.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BY, Ashton A, Smith MM, Little CB. Sclerostin regulation of Wisp1 and Mmps in chondrocytes is dependent on the Lrp5/6 binding domain but not secretion from the cell. Osteoarthritis and Cartilage. 2013;21:S55–S55. doi: 10.1016/j.joca.2013.02.127. [DOI] [Google Scholar]
- Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. Journal of Bone and Joint Surgery. American Volume. 1993;75:215–219. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- Coleman CM, Curtin C, Barry FP, O'Flatharta C, Murphy JM. Mesenchymal stem cells and osteoarthritis: remedy or accomplice? Human Gene Therapy. 2010;21:1239–1250. doi: 10.1089/hum.2010.138. [DOI] [PubMed] [Google Scholar]
- Combs CE, Nicholls JJ, Duncan Bassett JH, Williams GR. Thyroid hormones and bone development. Minerva Endocrinologica. 2011;36:71–85. [PubMed] [Google Scholar]
- Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, Tabin CJ. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495:375–378. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabdoub A, Donohue MJ, Brennan A, Wolf V, Montcouquiol M, Sassoon DA, Hseih JC, Rubin JS, Salinas PC, Kelley MW. Wnt signaling mediates reorientation of outer hair cell stereociliary bundles in the mammalian cochlea. Development. 2003;130:2375–2384. doi: 10.1242/dev.00448. [DOI] [PubMed] [Google Scholar]
- Davey MG, Paton IR, Yin Y, Schmidt M, Bangs FK, Morrice DR, Smith TG, Buxton P, Stamataki D, Tanaka M, et al. The chicken talpid3 gene encodes a novel protein essential for Hedgehog signaling. Genes and Development. 2006;20:1365–1377. doi: 10.1101/gad.369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi Cancedda F, Melchiori A, Benelli R, Gentili C, Masiello L, Campanile G, Cancedda R, Albini A. Production of angiogenesis inhibitors and stimulators is modulated by cultured growth plate chondrocytes during in vitro differentiation: dependence on extracellular matrix assembly. European Journal of Cell Biology. 1995;66:60–68. [PubMed] [Google Scholar]
- Dowthwaite GP, Bishop JC, Redman SN, Khan IM, Rooney P, Evans DJ, Haughton L, Bayram Z, Boyer S, Thomson B, et al. The surface of articular cartilage contains a progenitor cell population. Journal of Cell Science. 2004;117:889–897. doi: 10.1242/jcs.00912. [DOI] [PubMed] [Google Scholar]
- Emons J, Chagin AS, Hultenby K, Zhivotovsky B, Wit JM, Karperien M, Savendahl L. Epiphyseal fusion in the human growth plate does not involve classical apoptosis. Pediatric Research. 2009;66:654–659. doi: 10.1203/PDR.0b013e3181beaa8c. [DOI] [PubMed] [Google Scholar]
- Engsig MT, Chen QJ, Vu TH, Pedersen AC, Therkidsen B, Lund LR, Henriksen K, Lenhard T, Foged NT, Werb Z, et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. Journal of Cell Biology. 2000;151:879–889. doi: 10.1083/jcb.151.4.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnum CE, Lee R, O'Hara K, Urban JP. Volume increase in growth plate chondrocytes during hypertrophy: the contribution of organic osmolytes. Bone. 2002;30:574–581. doi: 10.1016/S8756-3282(01)00710-4. [DOI] [PubMed] [Google Scholar]
- Farquharson C, Ahmed SF. Inflammation and linear bone growth: the inhibitory role of SOCS2 on GH/IGF-1 signaling. Pediatric Nephrology. 2013;28:547–556. doi: 10.1007/s00467-012-2271-0. [DOI] [PubMed] [Google Scholar]
- Fernandes AM, Herlofsen SR, Karlsen TA, Kuchler AM, Floisand Y, Brinchmann JE. Similar properties of chondrocytes from osteoarthritis joints and mesenchymal stem cells from healthy donors for tissue engineering of articular cartilage. PLoS ONE. 2013;8:e62994. doi: 10.1371/journal.pone.0062994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosang AJ, Beier F. Emerging frontiers in cartilage and chondrocyte biology. Best Practice & Research. Clinical Rheumatology. 2011;25:751–766. doi: 10.1016/j.berh.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Fuerst M, Bertrand J, Lammers L, Dreier R, Echtermeyer F, Nitschke Y, Rutsch F, Schafer FK, Niggemeyer O, Steinhagen J, et al. Calcification of articular cartilage in human osteoarthritis. Arthritis and Rheumatism. 2009;60:2694–2703. doi: 10.1002/art.24774. [DOI] [PubMed] [Google Scholar]
- Gannon JM, Walker G, Fischer M, Carpenter R, Thompson RC, Jr, Oegema TR., Jr Localization of type X collagen in canine growth plate and adult canine articular cartilage. Journal of Orthopaedic Research. 1991;9:485–494. doi: 10.1002/jor.1100090404. [DOI] [PubMed] [Google Scholar]
- Garnero P, Ayral X, Rousseau JC, Christgau S, Sandell LJ, Dougados M, Delmas PD. Uncoupling of type II collagen synthesis and degradation predicts progression of joint damage in patients with knee osteoarthritis. Arthritis and Rheumatism. 2002;46:2613–2624. doi: 10.1002/art.10576. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nature Medicine. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Strutt D. Principles of planar polarity in animal development. Development. 2011;138:1877–1892. doi: 10.1242/dev.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover J, Roughley PJ. Versican gene expression in human articular cartilage and comparison of mRNA splicing variation with aggrecan. Biochemical Journal. 1993;291:361–367. doi: 10.1042/bj2910361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines RW. The histology of epiphyseal union in mammals. Journal of Anatomy. 1975;120:1–25. [PMC free article] [PubMed] [Google Scholar]
- Hartmann C, Tabin CJ. Wnt-14 plays a pivotal role in inducing synovial joint formation in the developing appendicular skeleton. Cell. 2001;104:341–351. doi: 10.1016/S0092-8674(01)00222-7. [DOI] [PubMed] [Google Scholar]
- Heinegard D, Oldberg A. Structure and biology of cartilage and bone matrix noncollagenous macromolecules. FASEB Journal. 1989;3:2042–2051. doi: 10.1096/fasebj.3.9.2663581. [DOI] [PubMed] [Google Scholar]
- Helminen HJ, Kiraly K, Pelttari A, Tammi MI, Vandenberg P, Pereira R, Dhulipala R, Khillan JS, Ala-Kokko L, Hume EL, et al. An inbred line of transgenic mice expressing an internally deleted gene for type II procollagen (COL2A1). Young mice have a variable phenotype of a chondrodysplasia and older mice have osteoarthritic changes in joints. Journal of Clinical Investigation. 1993;92:582–595. doi: 10.1172/JCI116625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrotin Y, Chevalier X, Deberg M, Balblanc JC, Richette P, Mulleman D, Maillet B, Rannou F, Piroth C, Mathieu P, et al. Early decrease of serum biomarkers of type II collagen degradation (Coll2-1) and joint inflammation (Coll2-1 NO2) by hyaluronic acid intra-articular injections in patients with knee osteoarthritis: a research study part of the Biovisco study. Journal of Orthopaedic Research. 2013;31:901–907. doi: 10.1002/jor.22297. [DOI] [PubMed] [Google Scholar]
- Horner A, Bishop NJ, Bord S, Beeton C, Kelsall AW, Coleman N, Compston JE. Immunolocalisation of vascular endothelial growth factor (VEGF) in human neonatal growth plate cartilage. Journal of Anatomy. 1999;194:519–524. doi: 10.1046/j.1469-7580.1999.19440519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyland JA, Thomas JT, Donn R, Marriott A, Ayad S, Boot-Handford RP, Grant ME, Freemont AJ. Distribution of type X collagen mRNA in normal and osteoarthritic human cartilage. Bone and Mineral. 1991;15:151–163. doi: 10.1016/0169-6009(91)90005-K. [DOI] [PubMed] [Google Scholar]
- Hristova GI, Jarzem P, Ouellet JA, Roughley PJ, Epure LM, Antoniou J, Mwale F. Calcification in human intervertebral disc degeneration and scoliosis. Journal of Orthopaedic Research. 2011;29:1888–1895. doi: 10.1002/jor.21456. [DOI] [PubMed] [Google Scholar]
- Hunziker EB, Schenk RK, Cruz-Orive LM. Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone growth. Journal of Bone and Joint Surgery. American Volume. 1987;69:162–173. [PubMed] [Google Scholar]
- Hunziker EB, Quinn TM, Hauselmann HJ. Quantitative structural organization of normal adult human articular cartilage. Osteoarthritis and Cartilage. 2002;10:564–572. doi: 10.1053/joca.2002.0814. [DOI] [PubMed] [Google Scholar]
- Hyde G, Dover S, Aszodi A, Wallis GA, Boot-Handford RP. Lineage tracing using matrilin-1 gene expression reveals that articular chondrocytes exist as the joint interzone forms. Developmental Biology. 2007;304:825–833. doi: 10.1016/j.ydbio.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito MM, Kida MY. Morphological and biochemical re-evaluation of the process of cavitation in the rat knee joint: cellular and cell strata alterations in the interzone. Journal of Anatomy. 2000;197:659–679. doi: 10.1046/j.1469-7580.2000.19740659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalba BA, Jalba CS, Vladoi AD, Gherghina F, Stefan E, Cruce M. Alterations in expression of cartilage-specific genes for aggrecan and collagen type II in osteoarthritis. Romanian Journal of Morphology and Embryology. 2011;52:587–591. [PubMed] [Google Scholar]
- Kornak U, Mundlos S. Genetic disorders of the skeleton: a developmental approach. American Journal of Human Genetics. 2003;73:447–474. doi: 10.1086/377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakow D, Rimoin DL. The skeletal dysplasias. Genetics in Medicine. 2010;12:327–341. doi: 10.1097/GIM.0b013e3181daae9b. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Kwapisz A, Kozlowski P, Szemraj J, Synder M, Sibinski M. Correlations between BMP-4 gene expression, heterotopic ossification and function after uncemented total hip replacement. Ortopedia, Traumatologia, Rehabilitacja. 2013;15:116–124. doi: 10.5604/15093492.1045951. [DOI] [PubMed] [Google Scholar]
- Lamb KJ, Lewthwaite JC, Lin JP, Simon D, Kavanagh E, Wheeler-Jones CP, Pitsillides AA. Diverse range of fixed positional deformities and bone growth restraint provoked by flaccid paralysis in embryonic chicks. International Journal of Experimental Pathology. 2003;84:191–199. doi: 10.1046/j.1365-2613.2003.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane LB, Bullough PG. Age-related changes in the thickness of the calcified zone and the number of tidemarks in adult human articular cartilage. Journal of Bone and Joint Surgery. British Volume. 1980;62:372–375. doi: 10.1302/0301-620X.62B3.7410471. [DOI] [PubMed] [Google Scholar]
- Little CB, Flannery CR, Hughes CE, Mort JS, Roughley PJ, Dent C, Caterson B. Aggrecanase versus matrix metalloproteinases in the catabolism of the interglobular domain of aggrecan in vitro. Biochemical Journal. 1999;344:61–68. doi: 10.1042/0264-6021:3440061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loqman MY, Bush PG, Farquharson C, Hall AC. Suppression of mammalian bone growth by membrane transport inhibitors. Journal of Cellular Biochemistry. 2013;114:658–668. doi: 10.1002/jcb.24408. [DOI] [PubMed] [Google Scholar]
- Luyten FP. Mesenchymal stem cells in osteoarthritis. Current Opinion in Rheumatology. 2004;16:599–603. doi: 10.1097/01.bor.0000130284.64686.63. [DOI] [PubMed] [Google Scholar]
- Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. International Journal of Biochemistry & Cell Biology. 2008;40:46–62. doi: 10.1016/j.biocel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Mackie EJ, Tatarczuch L, Mirams M. The skeleton: a multi-functional complex organ: the growth plate chondrocyte and endochondral ossification. Journal of Endocrinology. 2011;211:109–121. doi: 10.1530/JOE-11-0048. [DOI] [PubMed] [Google Scholar]
- Madiraju P, Gawri R, Wang H, Antoniou J, Mwale F. Mechanism of parathyroid hormone-mediated suppression of calcification markers in human intervertebral disc cells. European Cells & Materials. 2013;25:268–283. doi: 10.22203/ecm.v025a19. [DOI] [PubMed] [Google Scholar]
- Magne D, Bluteau G, Faucheux C, Palmer G, Vignes-Colombeix C, Pilet P, Rouillon T, Caverzasio J, Weiss P, Daculsi G, et al. Phosphate is a specific signal for ATDC5 chondrocyte maturation and apoptosis-associated mineralization: possible implication of apoptosis in the regulation of endochondral ossification. Journal of Bone and Mineral Research. 2003;18:1430–1442. doi: 10.1359/jbmr.2003.18.8.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Olsen BR. The role of endothelial–mesenchymal transition in heterotopic ossification. Journal of Bone and Mineral Research. 2012;27:1619–1622. doi: 10.1002/jbmr.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miclea RL, Siebelt M, Finos L, Goeman JJ, Lowik CWGM, Oostdijk W, Weinans H, Wit JM, Robanus-Maandag EC, Karperien M. Inhibition of Gsk3β in cartilage induces osteoarthritic features through activation of the canonical Wnt signaling pathway. Osteoarthritis and Cartilage. 2011;19:1363–1372. doi: 10.1016/j.joca.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Minns RJ, Steven FS. The collagen fibril organization in human articular cartilage. Journal of Anatomy. 1977;123:437–457. [PMC free article] [PubMed] [Google Scholar]
- Mwale F, Yao G, Ouellet JA, Petit A, Antoniou J. Effect of parathyroid hormone on type X and type II collagen expression in mesenchymal stem cells from osteoarthritic patients. Tissue Engineering. Part A. 2010;16:3449–3455. doi: 10.1089/ten.tea.2010.0091. [DOI] [PubMed] [Google Scholar]
- Nerlich AG, Schleicher ED, Boos N. 1997 Volvo Award winner in basic science studies. Immunohistologic markers for age-related changes of human lumbar intervertebral discs. Spine. 1997;22:2781–2795. doi: 10.1097/00007632-199712150-00001. [DOI] [PubMed] [Google Scholar]
- Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. Journal of Clinical Investigation. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LJ, Tam PP, Cheah KS. Preferential expression of alternatively spliced mRNAs encoding type II procollagen with a cysteine-rich amino-propeptide in differentiating cartilage and nonchondrogenic tissues during early mouse development. Developmental Biology. 1993;159:403–417. doi: 10.1006/dbio.1993.1251. [DOI] [PubMed] [Google Scholar]
- Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Hormone Research. 2005;64:157–165. doi: 10.1159/000088791. [DOI] [PubMed] [Google Scholar]
- Nosikova Y, Santerre JP, Grynpas MD, Kandel RA. Annulus fibrosus cells can induce mineralization: an in vitro study. Spine Journal. 2013;13:443–453. doi: 10.1016/j.spinee.2012.11.050. [DOI] [PubMed] [Google Scholar]
- Oegema TR, Jr, Carpenter RJ, Hofmeister F, Thompson RC., Jr The interaction of the zone of calcified cartilage and subchondral bone in osteoarthritis. Microscopy Research and Technique. 1997;37:324–332. doi: 10.1002/(SICI)1097-0029(19970515)37:4%3c324::AID-JEMT7%3e3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Oettmeier R, Abendroth K, Oettmeier S. Analyses of the tidemark on human femoral heads. II. Tidemark changes in osteoarthrosis – a histological and histomorphometric study in non-decalcified preparations. Acta Morphologica Hungarica. 1989;37:169–180. [PubMed] [Google Scholar]
- Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M, Asuncion FJ, Dwyer D, Han CY, Vlasseros F, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. Journal of Bone and Mineral Research. 2011;26:1012–1021. doi: 10.1002/jbmr.307. [DOI] [PubMed] [Google Scholar]
- Osborne AC, Lamb KJ, Lewthwaite JC, Dowthwaite GP, Pitsillides AA. Short-term rigid and flaccid paralyses diminish growth of embryonic chick limbs and abrogate joint cavity formation but differentially preserve pre-cavitated joints. Journal of Musculoskeletal & Neuronal Interactions. 2002;2:448–456. [PubMed] [Google Scholar]
- Pacifici M, Koyama E, Iwamoto M. Mechanisms of synovial joint and articular cartilage formation: recent advances, but many lingering mysteries. Birth Defects Research. Part C, Embryo Today: Reviews. 2005;75:237–248. doi: 10.1002/bdrc.20050. [DOI] [PubMed] [Google Scholar]
- Pacifici M, Koyama E, Shibukawa Y, Wu C, Tamamura Y, Enomoto-Iwamoto M, Iwamoto M. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Annals of the New York Academy of Sciences. 2006;1068:74–86. doi: 10.1196/annals.1346.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsillides AA. Early effects of embryonic movement: ‘a shot out of the dark’. Journal of Anatomy. 2006;208:417–431. doi: 10.1111/j.1469-7580.2006.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsillides AA, Ashhurst DE. A critical evaluation of specific aspects of joint development. Developmental Dynamics. 2008;237:2284–2294. doi: 10.1002/dvdy.21654. [DOI] [PubMed] [Google Scholar]
- Pitsillides AA, Beier F. Cartilage biology in osteoarthritis – lessons from developmental biology. Nature Reviews. Rheumatology. 2011;7:654–663. doi: 10.1038/nrrheum.2011.129. [DOI] [PubMed] [Google Scholar]
- Pullig O, Weseloh G, Ronneberger D, Kakonen S, Swoboda B. Chondrocyte differentiation in human osteoarthritis: expression of osteocalcin in normal and osteoarthritic cartilage and bone. Calcified Tissue International. 2000;67:230–240. doi: 10.1007/s002230001108. [DOI] [PubMed] [Google Scholar]
- Raj PP. Intervertebral disc: anatomy-physiology-pathophysiology-treatment. Pain Practice. 2008;8:18–44. doi: 10.1111/j.1533-2500.2007.00171.x. [DOI] [PubMed] [Google Scholar]
- Rao RS, Patra M, Sen M. WISP3–IGF1 interaction regulates chondrocyte hypertrophy. Journal of Cell Science. 2013;126:1650–1658. doi: 10.1242/jcs.119859. [DOI] [PubMed] [Google Scholar]
- Roach HI. New aspects of endochondral ossification in the chick: chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. Journal of Bone and Mineral Research. 1997;12:795–805. doi: 10.1359/jbmr.1997.12.5.795. [DOI] [PubMed] [Google Scholar]
- Roach HI, Clarke NM. Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. Journal of Bone and Joint Surgery. British Volume. 2000;82:601–613. doi: 10.1302/0301-620X.82B4.9846. [DOI] [PubMed] [Google Scholar]
- Roach HI, Yamada N, Cheung KS, Tilley S, Clarke NM, Oreffo RO, Kokubun S, Bronner F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis and Rheumatism. 2005;52:3110–3124. doi: 10.1002/art.21300. [DOI] [PubMed] [Google Scholar]
- Roberts S, Evans H, Trivedi J, Menage J. Histology and pathology of the human intervertebral disc. Journal of Bone and Joint Surgery. American Volume. 2006;88(Suppl 2):10–14. doi: 10.2106/JBJS.F.00019. [DOI] [PubMed] [Google Scholar]
- Roberts S, Narisawa S, Harmey D, Millan JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. Journal of Bone and Mineral Research. 2007;22:617–627. doi: 10.1359/jbmr.070108. [DOI] [PubMed] [Google Scholar]
- Romereim SM, Dudley AT. Cell polarity: the missing link in skeletal morphogenesis? Organogenesis. 2011;7:217–228. doi: 10.4161/org.7.3.18583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutges JP, Duit RA, Kummer JA, Oner FC, van Rijen MH, Verbout AJ, Castelein RM, Dhert WJ, Creemers LB. Hypertrophic differentiation and calcification during intervertebral disc degeneration. Osteoarthritis and Cartilage. 2010;18:1487–1495. doi: 10.1016/j.joca.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Saito D, Yonei-Tamura S, Takahashi Y, Tamura K. Level-specific role of paraxial mesoderm in regulation of Tbx5/Tbx4 expression and limb initiation. Developmental Biology. 2006;292:79–89. doi: 10.1016/j.ydbio.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Santos F, Fuente R, Mejia N, Mantecon L, Gil-Pena H, Ordonez FA. Hypophosphatemia and growth. Pediatric Nephrology. 2013;28:595–603. doi: 10.1007/s00467-012-2364-9. [DOI] [PubMed] [Google Scholar]
- Sato S, Kimura A, Ozdemir J, Asou Y, Miyazaki M, Jinno T, Ae K, Liu X, Osaki M, Takeuchi Y, et al. The distinct role of the Runx proteins in chondrocyte differentiation and intervertebral disc degeneration: findings in murine models and in human disease. Arthritis and Rheumatism. 2008;58:2764–2775. doi: 10.1002/art.23805. [DOI] [PubMed] [Google Scholar]
- Schmid TM, Linsenmayer TF. Developmental acquisition of type X collagen in the embryonic chick tibiotarsus. Developmental Biology. 1985;107:373–381. doi: 10.1016/0012-1606(85)90319-7. [DOI] [PubMed] [Google Scholar]
- Secreto FJ, Hoeppner LH, Westendorf JJ. Wnt signaling during fracture repair. Current Osteoporosis Reports. 2009;7:64–69. doi: 10.1007/s11914-009-0012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Adams CS, Freeman T, Srinivas V. Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Research. Part C, Embryo Today: Reviews. 2005;75:330–339. doi: 10.1002/bdrc.20057. [DOI] [PubMed] [Google Scholar]
- Shen G. The role of type X collagen in facilitating and regulating endochondral ossification of articular cartilage. Orthodontics & Craniofacial Research. 2005;8:11–17. doi: 10.1111/j.1601-6343.2004.00308.x. [DOI] [PubMed] [Google Scholar]
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- Snow HE, Riccio LM, Mjaatvedt CH, Hoffman S, Capehart AA. Versican expression during skeletal/joint morphogenesis and patterning of muscle and nerve in the embryonic mouse limb. Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology. 2005;282:95–105. doi: 10.1002/ar.a.20151. [DOI] [PubMed] [Google Scholar]
- Sommer B, Bickel M, Hofstetter W, Wetterwald A. Expression of matrix proteins during the development of mineralized tissues. Bone. 1996;19:371–380. doi: 10.1016/S8756-3282(96)00218-9. [DOI] [PubMed] [Google Scholar]
- Staines KA, Macrae VE, Farquharson C. Cartilage development and degeneration: a Wnt Wnt situation. Cell Biochemistry and Function. 2012a;30:633–642. doi: 10.1002/cbf.2852. [DOI] [PubMed] [Google Scholar]
- Staines KA, Macrae VE, Farquharson C. The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodelling. Journal of Endocrinology. 2012b;214:241–255. doi: 10.1530/JOE-12-0143. [DOI] [PubMed] [Google Scholar]
- Staines KA, Poulet B, Farquharson C, Pitsillides AA. The sclerostin and Mepe axis in the development of osteoarthrttis. Osteoarthritis and Cartilage. 2013;21:S55–S55. doi: 10.1016/j.joca.2013.02.128. [DOI] [Google Scholar]
- Stephens M, Kwan AP, Bayliss MT, Archer CW. Human articular surface chondrocytes initiate alkaline phosphatase and type X collagen synthesis in suspension culture. Journal of Cell Science. 1992;103:1111–1116. doi: 10.1242/jcs.103.4.1111. [DOI] [PubMed] [Google Scholar]
- Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm EE, Kingsley DM. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development. 1996;122:3969–3979. doi: 10.1242/dev.122.12.3969. [DOI] [PubMed] [Google Scholar]
- Studer D, Millan C, Ozturk E, Maniura-Weber K, Zenobi-Wong M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. European Cells & Materials. 2012;24:118–135; discussion 135. doi: 10.22203/ecm.v024a09. [DOI] [PubMed] [Google Scholar]
- Summerbell D, Lewis JH, Wolpert L. Positional information in chick limb morphogenesis. Nature. 1973;244:492–496. doi: 10.1038/244492a0. [DOI] [PubMed] [Google Scholar]
- Tickle C. Patterning systems – from one end of the limb to the other. Developmental Cell. 2003;4:449–458. doi: 10.1016/S1534-5807(03)00095-9. [DOI] [PubMed] [Google Scholar]
- Tickle C, Münsterberg A. Vertebrate limb development – the early stages in chick and mouse. Current Opinion in Genetics & Development. 2001;11:476–481. doi: 10.1016/S0959-437X(00)00220-3. [DOI] [PubMed] [Google Scholar]
- Towers M, Tickle C. Generation of pattern and form in the developing limb. International Journal of Developmental Biology. 2009;53:805–812. doi: 10.1387/ijdb.072499mt. [DOI] [PubMed] [Google Scholar]
- Urban JP, Roberts S. Development and degeneration of the intervertebral discs. Molecular Medicine Today. 1995;1:329–335. doi: 10.1016/S1357-4310(95)80032-8. [DOI] [PubMed] [Google Scholar]
- Villavicencio-Lorini P, Kuss P, Friedrich J, Haupt J, Farooq M, Turkmen S, Duboule D, Hecht J, Mundlos S. Homeobox genes d11–d13 and a13 control mouse autopod cortical bone and joint formation. Journal of Clinical Investigation. 2010;120:1994–2004. doi: 10.1172/JCI41554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Alaee F, Tang H, Ominsky MS, Ke HZ, Lieberman JR. Systemic administration of sclerostin antibody enhances bone repair in a critical-sized femoral defect in a rat model. Journal of Bone and Joint Surgery. American Volume. 2013;95:694–701. doi: 10.2106/JBJS.L.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladar EK, Antic D, Axelrod JD. Planar cell polarity signaling: the developing cell's compass. Cold Spring Harbor Perspectives in Biology. 2009;1:a002964. doi: 10.1101/cshperspect.a002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Okamoto H. Roles of planar cell polarity pathway genes for neural migration and differentiation. Development, Growth & Differentiation. 2009;51:233–240. doi: 10.1111/j.1440-169X.2009.01092.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou J, Bondy CA. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB Journal. 1999;13:1985–1990. doi: 10.1096/fasebj.13.14.1985. [DOI] [PubMed] [Google Scholar]
- Wang Q, Green RP, Zhao G, Ornitz DM. Differential regulation of endochondral bone growth and joint development by FGFR1 and FGFR3 tyrosine kinase domains. Development. 2001;128:3867–3876. doi: 10.1242/dev.128.19.3867. [DOI] [PubMed] [Google Scholar]
- Wang M, Sampson ER, Jin H, Li J, Ke QH, Im HJ, Chen D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Research & Therapy. 2013;15:R5. doi: 10.1186/ar4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpert L. Arms and the man: the problem of symmetric growth. PLoS Biology. 2010;8:e1000477. doi: 10.1371/journal.pbio.1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nature Medicine. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano F, Saito T, Ogata N, Yamazawa T, Iino M, Chung UI, Kawaguchi H. β-Catenin regulates parathyroid hormone/parathyroid hormone-related protein receptor signals and chondrocyte hypertrophy through binding to the intracellular C-terminal region of the receptor. Arthritis and Rheumatism. 2013;65:429–435. doi: 10.1002/art.37779. [DOI] [PubMed] [Google Scholar]
- Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D'Amore PA, Olsen BR. Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ryan JA, Di Cesare PE, Liu J, Walsh CA, You Z. Doublecortin is expressed in articular chondrocytes. Biochemical and Biophysical Research Communications. 2007;363:694–700. doi: 10.1016/j.bbrc.2007.09.030. [DOI] [PubMed] [Google Scholar]
- Zhu M, Chen M, Zuscik M, Wu Q, Wang YJ, Rosier RN, O'Keefe RJ, Chen D. Inhibition of β-catenin signaling in articular chondrocytes results in articular cartilage destruction. Arthritis and Rheumatism. 2008;58:2053–2064. doi: 10.1002/art.23614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Tang DZ, Wu QQ, Hao SY, Chen M, Xie C, Rosier RN, O'Keefe RJ, Zuscik M, Chen D. Activation of β-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult β-catenin conditional activation mice. Journal of Bone and Mineral Research. 2009;24:12–21. doi: 10.1359/jbmr.080901. [DOI] [PMC free article] [PubMed] [Google Scholar]