

Summary
Primary somatosensory cortex (S1) contains a complete body map that mirrors subcortical maps developed by peripheral sensory input projecting to sensory hindbrain, thalamus, then S1. Peripheral changes during development alter these maps through ‘bottom-up’ plasticity. Unknown is how S1 size influences map organization and if an altered S1 map feedbacks to affect subcortical maps. We show in mice that S1 is significantly reduced by cortex-specific deletion of Pax6, resulting in a reduced body map and loss of body representations by exclusion of later-differentiating sensory thalamocortical input. An initially normal sensory thalamus was re-patterned to match the aberrant S1 map by apoptotic deletion of thalamic neurons representing body parts with axons excluded from S1. Deleted representations were rescued by altering competition between thalamocortical axons by sensory deprivation or increasing S1. Thus, S1 size determined resolution and completeness of body maps and engaged ‘top-down’ plasticity that re-patterned sensory thalamus to match S1.
Keywords: apoptosis, area patterning, axon elimination, Pax6, cortical specification, systems-matching, topographic maps
In mammals, sensory stimuli originating from peripheral sensory organs or receptors are relayed to the neocortex, where it is processed within modality-specific primary and higher order sensory areas and brought to perception1,2. The sensory periphery is represented within the primary sensory areas in organized maps. For example, tactile receptors distributed over the body are represented in somatotopic maps within the primary somatosensory area (S1)3,4. The size of the representation of a body part within the S1 cortical map directly relates to the relative number and density of peripheral sensory receptors that innervate that body part3,5. This relationship results in the body representation within S1 being organized into a distorted somatotopic map with body parts disproportionally represented based on innervation density rather than size of the body part. Similar body maps are also evident in subcortical sensory nuclei that connect the sensory periphery to S1, first in hindbrain sensory nuclei and ultimately through the projection of thalamocortical axons (TCAs) from the ventro posterior thalamic sensory nucleus (VPN), developing in a ‘peripheral to central’ or ‘bottom-up’ progression6–10 (Supplementary Fig. 1).
The sensory map within a primary cortical area is malleable both in the adult and developing brain and exhibits plasticity in its organization in response to manipulations of the sensory periphery3,4,11–13. Plasticity in sensory maps has been most intensively studied in the somatosensory system of mice because the body map can be revealed using anatomical methods that directly relate to its functional representations throughout the brain, and it can be reproducibly manipulated peripherally with sensory deprivation and centrally with genetic approaches. Development of a normal body map in S1 requires an intact sensory periphery and connectivity from the periphery through to cortex during a critical period of development. Manipulations of the sensory periphery during the critical period that result in sensory deprivation, for example removal of sensory innervation to a body part, produce a defect or absence of the representation of the deprived body part in subcortical and S1 cortical body maps. Similar to map development, this plasticity proceeds in a ‘peripheral to central’ or ‘bottom-up’ progression3,4, beginning with a defective body map in sensory hindbrain nuclei, then VPN thalamus, and finally within S1 cortex14.
In contrast to the vast literature on bottom-up plasticity3,4,8–10,15, little is known about how the size of a primary sensory area influences the cortical representation of the sensory periphery and whether an altered cortical representation might engage a ‘top-down’ plasticity. These issues are of particular significance because the sizes of primary areas in humans reportedly vary as much as 200% across normal individuals16–18, and more sizeable differences are likely in individuals with developmental trauma or congenital defects of cortex, which can result in behavioral deficits and neural dysfunction19,20. Further, tactile and motor behaviors are degraded in adult mice when sensorimotor areas are either reduced or increased in size by embryonic genetic manipulations21 or disconnected from thalamus22. Here we use the mouse somatosensory system as a model to address the influence of cortical area size on sensory representations and the potential for top-down plasticity to modify subcortical sensory nuclei in response to changes in S1.
Results
Cortex-specific deletion of Pax6 reduces S1 size
Transcription factors expressed by cortical progenitors regulate area size and position23. A role for the transcription factor Pax6 in preferentially specifying sensorimotor areas has been suggested by analyses of small eye mutant mice that lack functional Pax6 protein24, but small eye mutants have compromising brain defects and die at birth before the emergence of cortical areas. Therefore, to address whether Pax6 determines S1 size, we crossed mice with floxed alleles of Pax625 to an Emx1-IRES-Cre line26 that generates robust recombination selectively in cortical progenitors (Fig. 1a) at embryonic day 10.526,27, to generate viable Pax6 conditional knockout (cKO) mice28 with a cortex-specific Pax6 deletion (Fig. 1b–c). Our primary analyses were done at postnatal day (PND) 7, when area patterning can be defined and the critical period for morphological plasticity in the patterning of the somatotopic body map in S1 is over11,12,15,23.
Figure 1. Pax6 specifies S1 area identity in cortical progenitors and determines S1 size. a.

Cre mediated recombination by Emx1-IRES-Cre crossed to the R26R reporter line revealed robust βgal staining restricted to dorsal telencephalon including neocortex at embryonic day (E) 12. b: In situ hybridization (ISH) for Pax6 mRNA in Pax6fl/fl Emx1-IRES-Cre− “Wildtype” (WT) E12 embryos reveals graded expression of Pax6 in cortical progenitors (between arrowheads), and strong expression in the rostral migratory stream (RMS) bordering the ganglionic eminence (GE) and in ventral thalamus (vTH). c: In Pax6fl/fl Emx1-IRES-Cre+(cKO) E12 embryos Pax6 mRNA is selectively deleted from cortical progenitors (between arrowheads) but remains at normal levels in the RMS and vTH. d–e: ISH on tangential sections of flattened cortex at PND7 for the sensory area marker RORβ shows that deletion of Pax6 specifically from cortical progenitors using Emx1-IRES-Cre (cKO) results in a significant reduction of S1 size compared to wildtype. Abbreviations: M: medial, P: posterior, dTH: dorsal thalamus, S1: primary somatosensory area, V1: primary visual area, A1: primary auditory area, ALBSF: anterior lateral barrel subfield, PMBSF: posterior medial barrel sub field. Scale bar in a, 0.5mm.
We showed using the S1 marker RORβ on tangential sections of flattened cortex at PND7 that the cortical field with S1 properties was substantially reduced in size in cKO mice compared to wildtype (Fig. 1d,e). This assessment was confirmed using serotonin (5HT) immunostaining, which reveals the somatotopic body map that comprises S1, including the primary body representations, hind paw, forepaw, lower jaw, the posterior medial barrel subfield (PMBSF), and the anterior lateral barrel subfield (ALBSF). PMBSF and ALBSF, which are the most prominent representations in mouse S1(Supplementary Fig. 1), can be accurately delineated and used to exemplify affects of Pax6 deletion on S1 overall. Even following correction for the reduction in overall cortical size in cKO mice compared to wildtype (Fig. 2a), both PMBSF and ALBSF were significantly reduced in size in cKO mice (Fig. 2b,c). These findings demonstrate that Pax6 has a significant role in specifying S1 area identity in cortical progenitors. Further, they show that Pax6 cKO mice provide a model to address the effect of reduced S1 size on cortical sensory representations and whether alterations in the cortical body map result in a top-down plasticity within sensory thalamus and hindbrain.
Figure 2. Quantitation of absolute and relative sizes of S1 representations in cKO mice.

Neocortical surface area and size of S1 barrel fields were quantified at PND7 using 5HT staining on tangential sections of flattened cortex. Overall cKO neocortex (a) was 51.6% +/− 1.89% the size of wildtype (p < 0.0001, t = 18.5, df = 33, n = 6). cKO PMBSF was 72.8% +/− 3.49% the relative size (b, p = 0.0015, t = 6.3, df = 5, n = 6) and 37.1% +/− 1.74% the absolute size (d, p < 0.0001, t = 14.1, df = 10, n = 6)of wildtype PMBSF. ALBSF was only 27.4% +/− 2.88% the relative size (c, p < 0.0001, t = 38.5, df = 5, n = 6) and 14% +/− 1.70% the absolute size (e, p < 0.0001, t = 24.1, df = 10, n = 6) of wildtype ALBSF. Abbreviations: df: degrees of freedom, sem: standard error of the mean, *: statistically significant, ***: highly statistically significant.
Reduced S1 has incomplete body map
Each of the primary body representations that comprise S1 are substantially reduced in cKO mice compared to wildtype (Fig. 3a–d). Quantitation shows that the primary representations are reduced in both absolute and relative size, but vary substantially in the degree of reduction among the representations.. For example, PMBSF was reduced by 62% in absolute size and by 27% in relative size (Fig. 2b,d), whereas ALBSF was reduced by 85% in absolute size and by 72% in relative size (Fig. 2c,e). While the relative measurements demonstrate a role for Pax6 in specifying S1, the measurements of absolute size are more relevant for the issue of plasticity in sensory circuits, and the potential for changes in S1 size and body map to engage a top-down plasticity that may affect VPN and sensory hindbrain nuclei.
Figure 3. S1 size dictates resolution and completeness of body map.

5HT staining on tangential sections of flattened cortex at PND7 reveals that all primary components of the S1 body map and barrels were miniaturized in cKO mice (outlines a–b, cartoons c–d). Most ALBSF barrels and the entire PMBSF barrel row ‘A’ were absent in S1 of cKO mice. Arrows in e and f mark ALBSF (a–b, higher magnification in e–f). g: Comparison of actual mouse body proportions to their representations in the S1 body map illustrates highly magnified representation of the face, including the nose/ALBSF and whiskers/PMBSF (wildtype body map), compared to the remaining body (limbs, trunk). The small reduced and incomplete body representations in S1 of cKO mice produce a distorted body map that illustrates abnormal representations of the sensory periphery (cKO body map) compared to wildtype. Abbreviations: FP: forepaw map, HP: hindpaw map, T: map of the trunk/rest of the body, LJ: lower jaw map. Scale bar in a, 0.5mm, e, 250μm.
Both PMBSF and ALBSF are representations of whiskers on the mouse’s face, and are comprised of anatomically discrete, functional units called ‘barrels’ that are patterned into one-to one representations that mirror the stereotypic distributions of the facial whiskers8,15(Supplementary Fig. 1). PMBSF is the representation of the large facial whiskers (mystacial vibrissae), whereas ALBSF is the representation of the small facial whiskers on the anterior snout8. PMBSF and ALBSF have the highest magnification factor for any body representation within S1, consistent with their behavioral importance to mice and the density of their sensory innervation6,7,15.
The representation of each body part in S1 was not only substantially reduced in size in cKO mice, but also appeared to be incomplete, with the defects in ALBSF possibly being the most substantial (Fig. 3a–d). In addition to the near complete loss of ALBSF, with only 14% of it persisting in cKO mice, virtually all of the small barrels representing the upper jaw whiskers in wildtype ALBSF were absent in cKO ALBSF (Fig. 3e,f). Substantial changes were also evident in the representation of the large facial whiskers in PMBSF of S1. Consistent with the 62% decrease in absolute size of PMBSF in cKO mice, the individual barrels that comprise it were reduced on average by 50% (+/− 4.3%, p< 0.0001, t = 17.04, degrees of freedom = 62, n=8 per genotype) in size compared to wildtype. In contrast to the five barrel rows in wildtype PMBSF, we unexpectedly found only four barrel rows in PMBSF in each of the twenty-eight cKO cases analyzed (Fig. 3a–f). We identified the missing row to be PMBSF row ‘A’ by lesioning the ‘C’ row whisker follicles on P1 in both wildtype and cKO mice to induce bottom-up plasticity that marked the ‘C’ row (Supplementary Fig. 3). Our findings show that cortex-specific deletion of Pax6 resulted in a reduced size of S1, miniaturized representations of body parts within the reduced S1 that varies in degree among the primary presentations, and an incomplete representation of the sensory periphery characterized by the reproducible loss of specific parts of the body map (Fig. 3g).
Thalamus is re-patterned to match S1
The reproducible defects in the body map in S1 of cKO mice provided a model to investigate whether sensory systems exhibit a top-down plasticity that complements in a reverse progression the much-studied bottom-up plasticity3,4,15,29. We addressed whether the reduced size of S1 and importantly its incomplete body map found in cKO mice were retrogradely transferred through a top-down plasticity from S1 cortex to VPN thalamus, and perhaps even further down the sensory neuraxis to the trigeminal hindbrain nuclei. To address this issue, we analyzed the patterning of VPN thalamus and hindbrain trigeminal nuclei at PND7, when cKO mice exhibit an incomplete S1 body map with deletions of specific body representations (Fig. 3).
In PND7 wildtype mice, cytochrome oxidase (COX) histochemical staining of coronal sections through thalamus revealed in ventral VPN the representations of the forepaws and hindpaws laterally and the lower jaw medially, and in dorsal VPN the representations of PMBSF laterally and ALBSF medially (Fig. 4a,c). As in S1 cortex, PMBSF was the dominant body representation in VPN, followed by ALBSF. The PMBSF representation in VPN was patterned into five rows of discrete ‘barreloids’ that mirrored the patterning of cortical barrels in PMBSF of S1 and the large facial whiskers9 (Fig. 4a, c, Supplementary Fig. 1). In PND7 cKO mice, VPN was reduced to half of its wildtype size paralleling the reduction in S1 size (Fig. 4e). COX staining revealed that the somatotopic patterning into the primary representations evident in wildtype VPN was intact in the cKO VPN (Fig 4a–d), but each representation was dramatically reduced in size, closely approximating their reduction within S1 of cKO mice (compare Fig. 2d,e to Fig. 4e–g). For example, the representations of PMBSF and ALBSF in VPN were reduced by 60% and 71%, respectively, closely paralleling the absolute size reductions of these representations in S1 of 62% and 85%, respectively. Thus, in PND7 cKO mice, the overall size of VPN and the primary representations in the VPN body map exhibited significant reductions that matched their S1 counterparts.
Figure 4. VPN thalamus was re-patterned in cKO mice to match reduced size and in complete body map of S1.

Cytochrome oxidase (COX) staining was performed on PND7 coronal sections to measure VPN volume and to reveal barreloid patterning. (a,b) Compared to wildtype, barreloid row ‘A’ and most ALBSF barreloids were absent in VPN of cKO mice. (c,d)Schematics of the VPN body maps as in a and b. Quantification of VPN body map: (e) VPN volume (47.8% +/− 2.11% of wildtype, p< 0.0001, t = 9.9, df = 8 n = 6), (f) PMBSF barreloid volume (40.08% +/− 2.38% of wildtype, p< 0.0001, t = 8.4, df = 8, n = 6) and (g) ALBSF barreloid volume (28.62% +/− 1.83% of wildtype, p< 0.0001, t = 15.6, df = 8, n = 6). Abbreviations: D: dorsal, VPN: ventroposterior nucleus, dLGN: dorsal lateral geniculate nucleus, A–E: barreloids of whisker rows ‘A’–‘E’, ALBSF: barreloids of anterior snout whiskers, FP, HP+T: barreloids of the paws and rest of the trunk/body, LJ: barreloids of the lower jaw map. Scale bar in a,e, 250μm.
In addition to the substantial reduction of VPN ALBSF in cKO mice, most of its discrete patterning evident in wildtype VPN was absent in cKO mice (Fig. 4b,d), paralleling the loss of most of the barrels that normally comprise ALBSF in S1 (Fig. 3b,d). Similar to the defects identified for PMBSF in S1 of cKO mice, COX staining of VPN revealed only four rows of barreloids in PND7 cKO mice (Fig. 4b,d), with a deletion of row ‘A’, corresponding to the absence of row ‘A’ in PMBSF of S1 (Fig. 3e–f). We also used COX staining to investigate whether the defective body maps evident in S1 and VPN of cKO mice were retrogradely transferred to the trigeminal hindbrain nuclei. In contrast to bottom-up plasticity, which induces changes in body maps in a bottom-up progression through the sensory pathway from the hindbrain to the cortex following whisker manipulation3,4,11,12, both the overall size and completeness of the barrelette representations in the trigeminal sensory hindbrain nuclei were indistinguishable between wildtype and cKO mice (Fig. 5a–d).
Figure 5. Trigeminal hindbrain nuclei have normal somatotopic patterning in cKO mice.

COX staining on coronal sections of PND7 hindbrain through the trigeminal nuclei reveals indistinguishable barrelette size, number and patterning between control (a,c) and cKO mice (b,d). Scale bar in a,c, 250μm.
These findings showed that the reduced body map in S1 cortex of PND7 cKO mice and the selective loss of specific representations were retrogradely transferred to VPN thalamus resulting in its re-patterning to mirror the aberrant body map in S1. However, this top-down plasticity did not progress further down the sensory neuraxis to the trigeminal hindbrain sensory nuclei, suggesting that the stability of these nuclei and their axonal projections to VPN were matured beyond the critical period for top-down plasticity and/or afferent input from the trigeminal ganglia was sufficient to maintain them.
Selective apoptosis re-patterns thalamus
To identify mechanisms responsible for top-down plasticity in cKO mice that re-patterned VPN to match its size and body map to S1, we analysed VPN in newborn mice, an age early in the development of VPN TCA input to S1 and during the critical period for bottom-up plasticity11,12. Analyses of thalamic architecture using Nissl stains and serotonin immunolabeling, showed that VPN size and appearance was similar in wildtype and cKO mice at PND0 (Fig. 6a,b,e). Molecular markers that highlight complementary subdivisions of VPN, such as choline acetyltransferase, which marks ventrolateral VPN, and EphA7, which marks dorsomedial VPN, revealed no differences in the molecular patterning of VPN in wildtype and cKO mice at PND0 (Fig. 6c–d). These findings showed that the initial development of VPN in cKO mice was similar to wildtype. Thus, the substantial differences observed at PND7 between VPN of wildtype and cKO mice were due to a postnatal re-patterning of VPN in cKO mice in response to the reduced size and aberrant body map of S1.
Figure 6. Top-down plasticity re-patterns VPN through exaggerated apoptosis selective for VPN neurons representing body parts deleted from S1 in cKO.

At PND0, prior to VPN re-patterning in cKO mice, Nissl-stained coronal sections reveal normal VPN size and architecture in cKO mice compared to wildtype (outlines a,e). Consecutive sections stained for the VPN markers, 5HT (b) and choline acetyltransferase (CHAT) (c) immunostaining, and EphA7 in situ hybridization (d), confirm that VPN in PND0 wildtype and cKO mice have similar size and internal subdivisions revealed by expression of molecular markers in PND0 cKO mice (a–e). In PND0 wildtype mice, adjacent to Nissl-stained sections (e), robust COX histochemical staining was evident throughout VPN (f) and only few apoptotic neurons positive for cleaved Caspase3 were scattered across VPN (g–h). In PND0 cKO mice, VPN resembles wildtype in size and cell density with Nissl staining (e), but has aberrant COX staining with a robust COX-positive central core (arrow in f) and a COX-negative surround that contains dense clusters of Caspase3-positive cells (g–h), that were coincident with representations absent at PND7, including row ‘A’ and much of ALBSF barreloids (arrows in g,h). Abbreviation: dLGN: dorsal lateral geniculate nucleus; vLGN: ventral lateral geniculate nucleus. Scale bar in a,e: 250μm.
COX staining at PND0 revealed the apparent onset of the re-patterning of VPN in cKO mice that was manifested by PND7 (Fig. 4). COX staining in VPN is due to its presence in mitochondria in VPN neurons and in presynaptic terminals on VPN neurons formed by axons ascending from sensory hindbrain nuclei15. In wildtype, robust COX staining was evident throughout VPN (Fig. 6f). However, in VPN of cKO mice, robust COX staining was contracted and did not extend throughout the nucleus (Fig. 6f). This pattern of COX staining in cKO mice correlated with changes in the body map evident later at PND7: robust COX staining centrally within VPN was coincident with the nascent representation of the four barreloid rows preserved at PND7, whereas low intensity COX staining at the VPN periphery was coincident with parts of VPN that contained the representations in the VPN body map most severely reduced or absent at PND7, including ALBSF and barreloid row ‘A’ of PMBSF (compare Fig. 6f to schematics in Fig. 4c–d). These correlations and the role of COX as an important regulator of oxidative stress30–32 suggest that COX staining revealed the viability of VPN neurons that represent different body parts and predicted the likelihood of their survival in cKO mice.
These findings indicated that VPN neurons in cKO mice that would normally form the absent or most severely diminished map representations in S1 were eliminated, resulting in reduced VPN size and re-patterning of the VPN body map observed at PND7. To address this issue, we performed immunostaining for cleaved Caspase3, an early apoptotic marker. In PND0 wildtype mice, we identified only a few Caspase3 labeled cells scattered over VPN (Fig. 6g–h). In contrast, in PND0 cKO mice, large numbers of VPN cells were strongly labeled for Caspase3 (Fig. 6g–h). Rather than being scattered across VPN, they were absent from the domain of robust COX staining in central VPN and instead were clustered in VPN regions with low intensity COX staining coincident with body representations most severely diminished or absent at PND7 in S1 and VPN, including row ‘A’ of PMBSF and most of ALBSF (compare Fig. 6f–h to Fig. 3a–d and Fig. 4a–d). These findings showed that in cKO mice molecularly normal VPN neurons in regions with abnormally low levels of COX were eliminated through apoptosis, resulting in a top-down plasticity that reduced and re-patterned an initially normally sized and patterned VPN to match the reduced S1 in overall size, resolution and completeness of somatotopic representations in the body map.
TCA competition drives map plasticity
Our findings showed that a period of selective and exaggerated apoptosis re-patterned VPN to match the reduced size and incomplete body map of the reduced S1 in cKO mice. We hypothesized that this re-patterning process was driven by competitive interactions between TCAs of VPN neurons played out within the reduced S1 of cKO mice, with the losers eliminated. The exaggerated neuronal death in cKO mice was densely concentrated in selective regions of VPN, leading to the preferential elimination of the body parts they represent (Supplementary Fig. 2). Somatotopic patterning and its relationship to specific body parts is most readily assessed and manipulated for the representation of the large facial whiskers in PMBSF of S1 and VPN3,4,14,15, leading us to study PMBSF as a model for mechanisms of top-down plasticity in this sensory system.
Our findings revealed a mechanism acting in the reduced S1 of cKO mice that favored the selective elimination of specific body map components like the entire barrel row ‘A’ in PMBSF rather than miniaturizing all map components equally. We hypothesized that this mechanism was based upon competitive interactions among VPN TCAs33, with the competitiveness of TCAs depending upon the relative timing of differentiation of their body representations in S1. This competition would be exaggerated in the reduced S1 of cKO mice, which we predicted would result in the preferential elimination of VPN TCAs that formed later-differentiating representations in S1, followed by the death of their parent VPN neurons and the loss of the same body representations in VPN. To test this model, we first examined the timing of the in growth of VPN TCAs into S1 and the differentiation of their S1 body representations.
For this analysis, we used a conditional genetic labeling technique to reproducibly label TCAs from VPN, by crossing a RORα-IRES-Cre line34 to aROSA26-GAP43-eGFP line carrying a conditionally-activated axon reporter35, visualized using GFP immunostaining. During the first postnatal week, when TCA projections were developing36,37, the RORα-IRES-Cre line produces robust recombination throughout VPN but not in neocortex34, and thus within S1 selectively labeled VPN TCAs (Supplementary Fig. 4). Using this labeling approach, we found that barrel row ‘A’ was the last of the five rows in PMBSF to differentiate (Fig. 7a–e), consistent with previous studies on the developmental differentiation of PMBSF in rats38,39 and mice40. Similarly, other representations of the body map severely diminished in the cKO mice, e.g. ALBSF, tended to differentiate later (Fig. 7a–e and References38–40). These findings supported our hypothesis that the timing of differentiation of distinct representations in the S1 body map formed by VPN TCAs dictated the competitive balance among them and the selectivity of the representations most severely affected in the cKO body map in S1 and VPN.
Figure 7. Late differentiation of VPN TCA representations of PMBSF row ‘A’ and ALBSF in S1.
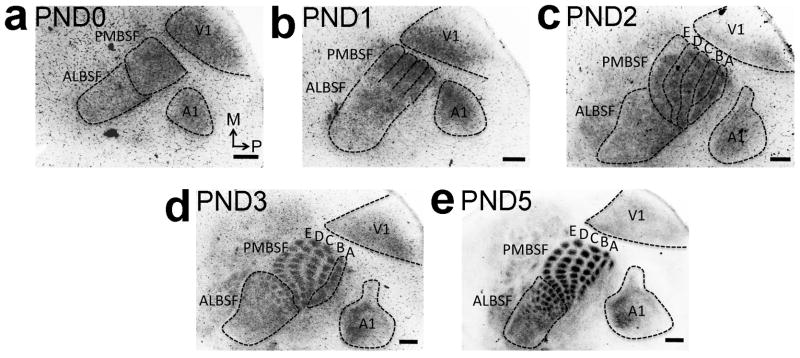
Spatiotemporal progression of the differentiation of body representations formed by TCAs from VPN in the cortical plate of S1 was analyzed by GFP immunohistochemistry on tangential sections of flattened cortex from RORα-IRES-Cre; ROSA26-GAP43-eGFP mice (also see Supplementary Fig. 4). a: At PND0, more GFP-positive TCAs are evident in nascent PMBSF compared to the future ALBSF that is still sparsely innervated by GFP-positive TCAs. b: Although not segregated by septa yet, bands of nascent rostral barrel rows ‘B’, ‘C’, ‘D’ and ‘E’ are evident at PND1 (PMBSF outline b). c: TCAs forming the immature band for nascent caudal row ‘A’ differentiate within the S1 cortical plate significantly later and are first seen at PND2 (outlines of row ‘A’-‘E’ in c). d: By PND3 septa between individual barrels of row ‘B’ to ‘E’ and the later almost ALBSF barrels are formed and fully differentiated. Conversely row ‘A’, still appears as a band (d). e: Individually separated row ‘A’ barrels with differentiated septa are first identifiable later at PND5. Number (n) of brains analysed at each age: PND0: n= 8, PND1: n= 9, PND2: n= 8, PND3: n= 8, PND5: n=7. Scale bar in a–e, 250μm.
Previous studies have shown that competitive interactions between VPN TCAs can be manipulated during a critical period early in the first postnatal week by lesioning whisker follicles on the face, inducing a bottom-up plasticity that ascends the somatosensory pathway to S13,4,11–13. VPN TCAs representing the lesioned follicles are at a competitive disadvantage, resulting in a reduction in the size of their terminations and corresponding barrels in S1 PMBSF and possibly the loss of some VPN neurons representing the lesioned follicles41. Therefore, to test whether enhanced competition among VPN TCAs in the reduced S1 of cKO mice resulted in the selective loss of barrel row ‘A’, we lesioned the follicles of whisker row ‘C’ on the face of wildtype and cKO mice within hours of birth and analysed patterning of S1 PMBSF at PND7 using serotonin immunostaining on tangential sections of flattened cortices. These early PND0 lesions induced a stronger bottom-up plasticity than lesions done on PND1 to identify rows in S1 PMBSF (described in Supplementary Fig. 3).
In unlesioned PND7 cKO mice, four distinct rows of barrels (rows ‘B’-‘D’) were evident, and although the barrels were smaller than in S1 PMBSF of PND7 wildtype, they were clear and present in normal number (Fig. 8a; compare to Fig. 3b,d,f). In contrast, in PND7 cKO mice with the ‘C’ row of whisker follicles lesioned early on PND0, the ‘C’ row in S1 PMBSF was virtually absent with few barrels evident (Fig. 8b). Coincident with this strong bottom-up plasticity, the PND0 lesion also resulted in a clear, albeit partial, rescue of row ‘A’ in PMBSF of S1 evident in PND7 cKO mice (Fig. 8b). We concluded that this deprivation of ‘C’ row input to S1 altered the competitive balance among VPN TCAs and placed those representing row ‘C’ at a competitive disadvantage, leading to their virtual elimination from S1 of cKO mice and allowing TCAs representing the ‘A’ row to compete more effectively for space in the reduced S1 that permits a partial rescue of the ‘A’ row representation.
Figure 8. Altering competitive balance among VPN TCAs rescues representations deleted from S1 body map in cKO.

(a) 5HT staining on tangential sections of flattened cortex at PND7 shows that only 4 barrel rows were evident in PMBSF in S1 in cKO mice, with row ‘A’ being absent. (b) Lesion of the ‘C’ row of large facial whisker follicles in cKO mice within a few hours of birth diminishes ‘C’ barrel row in S1 (blue arrowhead in b) and partially rescues VPN TCA input to row ‘A’ in S1 PMBSF (black arrows b), resulting in five rows in PMBSF as in wildtype compared to four PMBSF rows in un-lesioned cKO mice. (c)Increasing S1 size in Pax6 cKO mice by cortex-specific expression of a Pax6 transgene (cKO; cTG) also partially rescues the ‘A’ row in PMBSF of S1. Abbreviations: A: anterior, TCA: thalamocortical axons. Scale bar in a: 250μm.
Increasing S1 size rescues body map
Our findings suggested that the incomplete body map in the reduced S1 of cKO mice was due to an exaggerated competition among VPN TCAs for limited cortical space inadequate to accommodate the complete body representation. To further test this notion, we complemented the follicle-lesion experiment with a cortex-specific genetic approach designed to increase the size of the reduced S1 in cKO mice, predicting that it would rescue missing body representations. For this approach, we created a novel conditional Pax6 transgenic line (nestin-Pax6cTG), and by crossing it to the Emx1-IRES-Cre line26, ectopic Pax6 was selectively activated in the same cortical progenitors in which endogenous Pax6 was conditionally deleted in cKO mice (Supplementary Fig. 5). We focused our analyses on PMBSF of S1 in cKO mice and in cKO mice crossed to the nestin-Pax6cTG (referred to as cKO; cTG).
Activation of the Pax6 transgene in cKO; cTG mice resulted in a significant increase of ~20% in PMBSF size in S1 compared to cKO mice (Supplementary Fig. 6). At PND7, these cKO; cTG mice exhibited a partial, but clear, rescue of row ‘A’ in PMBSF (Fig. 8c). In contrast to the follicle-lesion experiment (Fig. 8b), the other four PMBSF rows were unaffected and appeared as they do in the cKO S1 (compare Fig. 8c to 8b). These findings supported our conclusion that S1 size has a significant influence on competitive interactions among VPN TCAs in S1 and their ability to survive, therefore S1 size is a critical parameter in determining the completeness of the cortical body map.
Discussion
We exploited features of the mouse somatosensory system that led to its prominence as a model for sensory map development and bottom-up plasticity and used it to show the consequences of reduced cortical area size on the representation of the sensory periphery and to reveal a novel ‘top-down’ plasticity driven by competition-mediated axon elimination and neuronal apoptosis. We found that cortex-specific deletion of Pax6 resulted in a substantially reduced S1, showing a required role for Pax6 in specifying S1 area-identity in cortical progenitors. The reduction in S1 size resulted in a miniaturized body map within the reduced S1, and an unexpected but reproducible loss of specific representations of the sensory periphery from the S1 body map. Thus, genetic mechanisms intrinsic to cortex place strict limitations on the physical dimensions of the cortical field that TCAs from VPN thalamus were able to innervate, thereby determining the overall size, resolution and completeness of the S1 body map. This intrinsic limitation resulted in a defective S1 body map and engaged a top-down plasticity that retrogradely re-patterned VPN thalamus to match the reduced size of S1 and its aberrant body map. Prior to its re-patterning, VPN thalamus had normal size and molecular differentiation, but its re-patterning in cKO mice was predicted by differential levels of COX activity, which correlate with metabolic health and viability of neurons30–32, and molecularly normal VPN neurons with abnormally low COX levels were selectively eliminated through apoptotic cell death. A potentially diminished number of layer 6 corticothalamic axons36,37 may have also contributed to the reduction in VPN size in cKO mice.
Our findings in cKO mice revealed a mechanism that favored the reproducible elimination of selective parts of the body map rather than miniaturizing all map components equally. This mechanism was initiated by the preferential elimination of VPN TCAs representing specific body parts from the reduced S1 and was manifested by an exaggerated apoptosis of VPN neurons densely concentrated in selective regions of VPN rather than being more uniformly distributed across the nucleus. This mechanism appeared to be based on competitive balance among VPN TCAs being determined by the relative timing of differentiation of the body representations that they form within S1, with TCAs forming later-differentiating representations being preferentially excluded from S1 and their parent neurons deleted from VPN. This competitive model for top-down plasticity and its selectivity was further supported by two sets of experiments in cKO mice that showed that altering competitive imbalance among VPN TCAs rescues representations in the S1 body map that were otherwise selectively eliminated, either by inducing classic bottom-up plasticity through peripheral sensory deprivation or by a transgenic approach intrinsic to cortex that partially restored the size of the genetically-reduced S1.
We conclude that S1 size modulates competitive interactions among VPN TCAs, influencing the organization, resolution and completeness of the S1 body map that they form, and in turn initiates a novel top-down plasticity that drives through axon elimination and cell death the re-patterning of VPN thalamus to match the reduced size of S1 and its aberrant body map. It is likely that the reduced size of S1 not only diminished the space available for termination of VPN TCAs but also trophic support for them and their parent neurons. We propose that during normal development, these mechanisms tune sensory maps in cortex and thalamus and account for the precise matching in thalamocortical / corticothalamic circuits required for proper function of sensory systems. The mechanisms demonstrated here can produce relatively minor adjustments in sensory maps as well as the more substantial adjustments required by the prominent disparity in sizes of primary sensory areas even among normal individuals16–18, which is exaggerated in individuals with certain neurogenetic disorders20,42
Potential roles for top-down plasticity complement those proposed for bottom-up plasticity in tuning sensory components in the brain with the sensory periphery to account for normal and abnormal variations in their size and organization. Our findings lead us to speculate that size differences in sensory areas in humans above a critical threshold may have pathological consequences. For example, in autism spectrum disorder (ASD), large-scale changes in the cortical gene expression profile that controls area patterning43 coincide with massive expansion of the frontal cortex42,44 but overall cortical surface area shows little change implying that the more posteriorly positioned sensory cortical areas, including S1 are reduced, which may lead to defects in their sensory maps resembling those that we find in the reduced S1 of cKO mice. In addition, these changes in sensory cortex would likely engage top-down plasticity and lead to similar defects in sensory thalamus, consistent with a reduction in thalamic volume in ASD45,46. Neurodevelopmental mechanisms including intrinsic neocortical area patterning and top-down plasticity and their consequences may contribute to the complex pathologies of ASD and other sensory processing disorders.
Online Methods
Mouse lines
All experiments were approved and conducted following the guidelines of the Institutional Animal Care and Use Committee at the Salk Institute and were in full compliance with the guidelines of the NIH for the care and use of laboratory animals. Analyses were done independent of sex and included both male and female mice according to Mendelian ratio. Background strain was C57/B6. Cortex-specific Pax6 knockout mice (termed: cKO) were generated by crossing Pax6 floxed (Pax6fl/fl) mice25 with Emx1-IRES-Cre expressing mice26 as described28. Pax6 transgenic mice (termed: cTG) were generated by pronuclear injection into fertilized oocytes. To limit activation to neuronal progenitors, the Pax6 transgene includes a CNS-specific enhancer from the second intron of the Nestin gene47 that was subcloned upstream to the Hsp68 promoter driving a ‘stop cassette’ (comprised of an eGFP reporter, a stop codon and three tandem polyA sequences) that was flanked by LoxP sites, followed by the Pax6 cDNA. (see also Supplementary Figure 5). Transgenic founders were backcrossed on the C57Bl/6 background, and screened for germline transmission. To generate dorsal thalamus specific reporter mice and to label TCA’s, RORα-IRES-Cre transgenic mice34 were crossed to GAP43eGFP reporter mice35, termed RORαCRE-GAP43eGFP mice. Mice lacking endogenous Pax6 in cortical progenitors but expressing ectopic Pax6 from the conditional transgene (termed cKO; cTG) were generated by crossing cKO mice to cTG mice. To assess specificity of Cre-recombination in mouse embryos, LacZ reporter mice expressing LacZ under the ROSA26 (R26R) promoter48 were crossed to mice expressing Emx1-IRES-Cre26. Genotyping was done by PCR using the following primers for Pax6 floxed alleles: (FLP6/83: GCG-GTT-GAG-TAG-CTC-AAT-TCT-A, FLP6/84: AGT GGC-TTG-GAC-TCC-TCA-AGA, FLP6/Del: CGT-GTG-CCC-CAG-CTT-CCG-GT), CRE: (creF: ATG-CTT-CTG-TCC-GTT-TGC-CG, creR: CCT-GTT-TTG-CAC-GTT-CAC-CG), GFP: (GFPs: GAC-GTA-AAC-GGC-CAC-AAG-TT, GFPr: GAA-CTC-CAG-CAG-GAC-CAT-GT), Hsp: (HspF: TTC-AGA-ACT-GTG-GGA-GTT-GG, Pax6r: TAG-CCA-GGT-TGC-GAA-GAA-CTC-TGT) and LacZ (β-galactosidase): (lacZF: TTG-GCG-TAA-GTG-AAG-CGA-C, lacZR: AGC-GGC-TGA-TGT-TGA-ACT-G).
In situ hybridization (ISH), Immunostaining, LacZ staining, tangential sections and cytochrome oxidase histochemistry
Mice were perfused with 4% phosphate buffered paraformaldehyde, washed in phosphate buffered saline (PBS) and cryoprotected in 30% sucrose in PBS. Brains were embedded in Tissue-Tek OCT (Sakura Finetek) and sectioned on a Cryostat (Leica). Antisense RNA probes for RORβ, Pax6 and EphA7 were labeled using a DIG-RNA labeling kit (Roche). ISH and immunostaining on 18μm cryostat sections was carried out as previously described49. For tangential sections, cortical hemispheres were dissected, flattened, and post-fixed between slide glasses and then cryoprotected. Tangential sections were cut at 40μm from flattened cortical hemispheres and stained for RORβ or serotonin. For Nissl staining, sections were stained with 0.5% cresyl violet and then dehydrated with graded alcohols. Immunohistochemistry was performed on 18μm coronal, sagittal or 40μm tangential cryosections. Antigens were unmasked by boiling in citrate buffer (Vector). Primary antibodies used were serotonin/5HT (1:20,000, Immunostar, #20080), CHAT (1:1,000, Chemicon, #AB144P), cleaved Caspase3 (1:400, Cell Signaling, #9661) and GFP (1:1,000, Molecular Probes, #A11122). Primary antibodies were incubated overnight at 4°C in blocking solution containing 3% bovine serum albumin (Fisher Scientific) and 0.1% Tween20 (Sigma) in PBS. Biotinylated secondary antibodies (Jackson Immuno Research) were incubated for 45 minutes at room temperature. For immunostaining, sections were developed following the Vecta stain standard DAB (di-amino-benzidine) colorimetric reaction kit (Vector). LacZ staining was performed on E12 whole mount embryos. β-Galactosidase activity was developed in staining solution (PBS, 1mg/ml X-Gal, 2mM MgCl2, 0.01% SDS, 0.02% NP40, 5mM K3Fe(CN)6, 5mM K4Fe(CN)6) for several hours at 30°C. Specimens were then washed in PBS and postfixed in PFA. Cytochrome oxidase (COX) histochemistry was performed on fixed, 18μm cryosections as described previously21. Sections were incubated overnight at 37°C in 0.1M phosphate buffer containing 5% sucrose, 0.03% cytochrome c, 0.02% catalase, and 0.05% DAB.
Whisker lesion
Mice, within hours of birth or at PND1, were anesthetized by cooling. The central ‘C’ whisker row was identified under a surgical microscope and whiskers including follicles plucked using scalpels and tweezers. Mice were revived on a 37°C heating pad and returned to their home cages. At PND7 brains were dissected and processed for serotonin staining on tangential sections.
Measurements and statistical analysis
Data collection and analysis were performed blind to genotype and conditions of the experiments and data were collected and processed randomly and no data points were excluded. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications (for example references38,40). Data met the assumptions of the statistical tests used and data distribution was assumed to be normal, but this was not formally tested. Statistics were calculated with Microsoft Excel. Quantifications show individual data points and mean values of tested groups and are displayed as percent of wildtype. Analyzed numbers of brains (n) for each set of experiments are indicated in the corresponding figures and figure legends or text sections. The examples shown in each figure are representative and reproducible for each set of experiments. Individual experiments were successfully repeated at least five times using different litters. Statistical significance was determined using unpaired two-tailed t-test, p-values < 0.05 (indicated as *) were considered as statistically significant, p-values < 0.001 were considered as highly statistically significant (indicated as **), and p-values < 0.0001 were considered extremely statistically significant (indicated as ***). Variance is indicated in figure graphs and figure legends and was calculated using standard error of the mean (sem). Absolute and relative neocortical surface area and the size of cortical barrel fields stained with serotonin or RORβ was measured using ImageJ50 as shown in schematic drawings in Figure 2, Figure 4 and Supplementary Figure 6. Volume of VPN and barreloid subfields was measured on series of consecutive 18μm COX stained coronal sections using ImageJ50.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 NS31558 and R01 MH086147 and the Vincent J. Coates Chair of Molecular Neurobiology (D.D.M.O’L.). We thank Berta Higgins and Haydee Gutierrez for technical assistance, Jamie Simon (Salk Institute MultiMedia Resources) for help making Adobe Illustrator schematics, Yelena Dayn (Salk Institute Transgenic Core) for help producing the nestin Pax6 conditional transgenic mice, Kevin Jones (U. Colorado) for the Emx1-IRES-Cre mice, Martyn Goulding (Salk Institute) for the ROSA26-GAP43-eGFP mice, and members of the O’Leary lab for discussion.
Footnotes
Supplementary Information is available in the online version of the paper.
Authors’ contributions
All authors have read and approved the final manuscript. Specific contributions are as follows: A.Z. and D.O’L. designed the study, analysed findings, prepared figures and wrote the paper. A.Z. performed the primary experiments. S-J.C. and D.O’L. designed and generated the nestin Pax6 conditional transgenic mice. R.A-P. and A.S. designed and generated the Pax6 conditional knockout mice.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
Contributor Information
Andreas Zembrzycki, Email: azembrzycki@salk.edu.
Shen-Ju Chou, Email: chou@salk.edu.
Ruth Ashery-Padan, Email: ruthash@post.tau.ac.il.
Anastassia Stoykova, Email: astoyko@gwdg.de.
Dennis D.M. O’Leary, Email: doleary@salk.edu.
References
- 1.Kaas JH. The evolution of complex sensory systems in mammals. J Exp Biol. 1989;146:165–176. doi: 10.1242/jeb.146.1.165. [DOI] [PubMed] [Google Scholar]
- 2.Krubitzer L, Kahn DM. Nature versus nurture revisited: an old idea with a new twist. Prog Neurobiol. 2003;70:33–52. doi: 10.1016/s0301-0082(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 3.Erzurumlu RS, Murakami Y, Rijli FM. Mapping the face in the somatosensory brainstem. Nat Rev Neurosci. 2010;11:252–263. doi: 10.1038/nrn2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erzurumlu RS, Gaspar P. Development and critical period plasticity of the barrel cortex. Eur J Neurosci. 2012;35:1540–1553. doi: 10.1111/j.1460-9568.2012.08075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krubitzer LA, Seelke AM. Cortical evolution in mammals: the bane and beauty of phenotypic variability. Proc Natl Acad Sci U S A. 2012;109 (Suppl 1):10647–10654. doi: 10.1073/pnas.1201891109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krubitzer L, Kaas J. The evolution of the neocortex in mammals: how is phenotypic diversity generated? Curr Opin Neurobiol. 2005;15:444–453. doi: 10.1016/j.conb.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Krubitzer L. In search of a unifying theory of complex brain evolution. Ann N Y Acad Sci. 2009;1156:44–67. doi: 10.1111/j.1749-6632.2009.04421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205–242. doi: 10.1016/0006-8993(70)90079-x. [DOI] [PubMed] [Google Scholar]
- 9.Van Der Loos H. Barreloids in mouse somatosensory thalamus. Neurosci Lett. 1976;2:1–6. doi: 10.1016/0304-3940(76)90036-7. [DOI] [PubMed] [Google Scholar]
- 10.Ma PM, Woolsey TA. Cytoarchitectonic correlates of the vibrissae in the medullary trigeminal complex of the mouse. Brain Res. 1984;306:374–379. doi: 10.1016/0006-8993(84)90390-1. [DOI] [PubMed] [Google Scholar]
- 11.Belford GR, Killackey HP. The sensitive period in the development of the trigeminal system of the neonatal rat. J Comp Neurol. 1980;193:335–350. doi: 10.1002/cne.901930203. [DOI] [PubMed] [Google Scholar]
- 12.Durham D, Woolsey TA. Effects of neonatal whisker lesions on mouse central trigeminal pathways. J Comp Neurol. 1984;223:424–447. doi: 10.1002/cne.902230308. [DOI] [PubMed] [Google Scholar]
- 13.Feldman DE, Nicoll RA, Malenka RC. Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. J Neurobiol. 1999;41:92–101. [PubMed] [Google Scholar]
- 14.Fox K, Wong RO. A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron. 2005;48:465–477. doi: 10.1016/j.neuron.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 15.Fox K. Barrel cortex. Cambridge University Press; 2008. [Google Scholar]
- 16.Andrews TJ, Halpern SD, Purves D. Correlated size variations in human visual cortex, lateral geniculate nucleus, and optic tract. J Neurosci. 1997;17:2859–2868. doi: 10.1523/JNEUROSCI.17-08-02859.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White LE, et al. Structure of the human sensorimotor system. II: Lateral symmetry. Cereb Cortex. 1997;7:31–47. doi: 10.1093/cercor/7.1.31. [DOI] [PubMed] [Google Scholar]
- 18.Dougherty RF, et al. Visual field representations and locations of visual areas V1/2/3 in human visual cortex. J Vis. 2003;3:586–598. doi: 10.1167/3.10.1. [DOI] [PubMed] [Google Scholar]
- 19.Higginbotham H, Yokota Y, Anton ES. Strategies for analyzing neuronal progenitor development and neuronal migration in the developing cerebral cortex. Cereb Cortex. 2011;21:1465–1474. doi: 10.1093/cercor/bhq197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubenstein JL. Annual Research Review: Development of the cerebral cortex: implications for neurodevelopmental disorders. J Child Psychol Psychiatry. 2011;52:339–355. doi: 10.1111/j.1469-7610.2010.02307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leingartner A, et al. Cortical area size dictates performance at modality-specific behaviors. Proc Natl Acad Sci U S A. 2007;104:4153–4158. doi: 10.1073/pnas.0611723104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou L, et al. Maturation of “neocortex isole” in vivo in mice. J Neurosci. 2010;30:7928–7939. doi: 10.1523/JNEUROSCI.6005-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Leary DD, Chou SJ, Sahara S. Area patterning of the mammalian cortex. Neuron. 2007;56:252–269. doi: 10.1016/j.neuron.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Bishop KM, Goudreau G, O’Leary DD. Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science. 2000;288:344–349. doi: 10.1126/science.288.5464.344. [DOI] [PubMed] [Google Scholar]
- 25.Ashery-Padan R, Marquardt T, Zhou X, Gruss P. Pax6 activity in the lens primordium is required for lens formation and for correct placement of a single retina in the eye. Genes Dev. 2000;14:2701–2711. doi: 10.1101/gad.184000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorski JA, et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou SJ, Perez-Garcia CG, Kroll TT, O’Leary DD. Lhx2 specifies regional fate in Emx1 lineage of telencephalic progenitors generating cerebral cortex. Nat Neurosci. 2009;12:1381–1389. doi: 10.1038/nn.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinon MC, Tuoc TC, Ashery-Padan R, Molnar Z, Stoykova A. Altered molecular regionalization and normal thalamocortical connections in cortex-specific Pax6 knock-out mice. J Neurosci. 2008;28:8724–8734. doi: 10.1523/JNEUROSCI.2565-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen CC. The functional organization of the barrel cortex. Neuron. 2007;56:339–355. doi: 10.1016/j.neuron.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Diaz F. Cytochrome c oxidase deficiency: patients and animal models. Biochim Biophys Acta. 2010;1802:100–110. doi: 10.1016/j.bbadis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 31.Diaz F, Garcia S, Padgett KR, Moraes CT. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum Mol Genet. 2012;21:5066–5077. doi: 10.1093/hmg/dds350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huttemann M, et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim Biophys Acta. 2012;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallace H, Fox K. Local cortical interactions determine the form of cortical plasticity. J Neurobiol. 1999;41:58–63. [PubMed] [Google Scholar]
- 34.Chou S, et al. Geniculocortical input drives genetic distinctions between primary and higher order visual areas. Science. 2013 doi: 10.1126/science.1232806. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sapir T, et al. Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J Neurosci. 2004;24:1255–1264. doi: 10.1523/JNEUROSCI.3187-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant E, Hoerder-Suabedissen A, Molnar Z. Development of the corticothalamic projections. Front Neurosci. 2012;6:53. doi: 10.3389/fnins.2012.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molnar Z, Garel S, Lopez-Bendito G, Maness P, Price DJ. Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur J Neurosci. 2012;35:1573–1585. doi: 10.1111/j.1460-9568.2012.08119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhoades RW, et al. Development and lesion induced reorganization of the cortical representation of the rat’s body surface as revealed by immunocytochemistry for serotonin. J Comp Neurol. 1990;293:190–207. doi: 10.1002/cne.902930204. [DOI] [PubMed] [Google Scholar]
- 39.Schlaggar BL, O’Leary DD. Early development of the somatotopic map and barrel patterning in rat somatosensory cortex. J Comp Neurol. 1994;346:80–96. doi: 10.1002/cne.903460106. [DOI] [PubMed] [Google Scholar]
- 40.Vitalis T, et al. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: determination of a sensitive developmental period. J Comp Neurol. 1998;393:169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Waite PM, Li L, Ashwell KW. Developmental and lesion induced cell death in the rat ventrobasal complex. Neuroreport. 1992;3:485–488. doi: 10.1097/00001756-199206000-00007. [DOI] [PubMed] [Google Scholar]
- 42.Courchesne E, et al. Mapping early brain development in autism. Neuron. 2007;56:399–413. doi: 10.1016/j.neuron.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 43.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Courchesne E, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- 45.Tamura R, Kitamura H, Endo T, Hasegawa N, Someya T. Reduced thalamic volume observed across different subgroups of autism spectrum disorders. Psychiatry Res. 2010;184:186–188. doi: 10.1016/j.pscychresns.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 46.Cheon KA, et al. Involvement of the anterior thalamic radiation in boys with high functioning autism spectrum disorders: a Diffusion Tensor Imaging study. Brain Res. 2011;1417:77–86. doi: 10.1016/j.brainres.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 47.Yaworsky PJ, Kappen C. Heterogeneity of neural progenitor cells revealed by enhancers in the nestin gene. Dev Biol. 1999;205:309–321. doi: 10.1006/dbio.1998.9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 49.Zembrzycki A, Griesel G, Stoykova A, Mansouri A. Genetic interplay between the transcription factors Sp8 and Emx2 in the patterning of the forebrain. Neural Dev. 2007;2:8. doi: 10.1186/1749-8104-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2013. http://imagej.nih.gov/ij/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.