

Abstract
The primary objective of radiation oncology is to exploit the biological interaction of radiation within tissue to promote tumor death while minimizing damage to surrounding normal tissue. The clinical delivery of radiation relies on principles of radiation physics that define how radiation energy is deposited in the body, as well as technology that facilitates accurate tumor targeting. This review will summarize the current landscape of recent biological and technological advances in radiation oncology, describe the challenges that exist, and offer potential avenues for improvement.
Keywords: Radiation oncology, radiobiology, radiation physics, radiosensitizers, novel treatments, prognostic factors
I. Introduction
The field of radiation oncology—where ionizing radiation is used to treat a variety of cancers and as well as benign conditions—was born shortly after the discovery of X-rays and their effects on tissue in 1895 (1). At first, treatments were typically delivered in single doses using low-energy cathode ray tubes or radium-filled glass tubes positioned close to tumors. Technological developments between 1920 and 1945 focused on improving beam output and energy. The low energy (200–500 kV) of x-rays used in that period was associated with skin toxicity due to poor penetration, thereby limiting the use of radiotherapy for deep tumors. The development of Cobalt-60 units and linear accelerators in the 1950’s to deliver “supervoltage” radiation energies (≥1 MeV) was a critical advance, as these high energy x-rays could penetrate further to reach deeper seated tumors. Fractionated therapy, which dates back to the 1920’s, dividestreatment into multiple small doses rather than one large radiation dose, and has allowed for further improvement in tolerance of normal tissues to treatment. Advances in technology, imaging, and cancer biology over subsequent decades have pushed the field of radiation oncology closer towards the idealized goal of non-invasively achieving maximal local cancer control with minimal normal tissue toxicity.
II. Basic principles and current uses of radiotherapy
Cancer may grow locally or spread systemically, via lymphatic or hematogenous routes. Successful treatment requires therapy targeted towards all sites of involvement. The three major modalities of cancer therapy – surgery, radiation therapy, and chemotherapy – can be used alone or in combination to address all sites at risk for harboring disease. In a curative-intent approach, surgery and/or radiation therapy are generally used to address local-regional areas of risk. Surgery remains the most commonly used modality to treat local disease. By its nature, surgery can be both therapeutic and diagnostic, since tumor excision expeditiously provides tissue for histologic examination and staging. Radiation therapy as a sole modality can sometimes offer a non-invasive alternative to the therapeutic role of surgery, with the possibility for organ preservation, such as with bladder and laryngeal cancer. As an adjuvant therapy, radiation therapy can facilitate resection when given prior to surgery, or treat microscopic residual disease when given after surgery, such as treatment after breast-conserving lumpectomy. On the other hand, chemotherapy is given to treat known metastatic disease or as an adjuvant to reduce the risk of potential micrometastasis. Chemotherapy is often also combined with radiation therapy to act as a radiosensitizer for the purpose of increasing local control. The optimal use of each modality of cancer therapy is tailored according to the cancer cell histology, anatomic location, stage of cancer, and other patient factors. Some common diseases that serve as examples of the role of radiotherapy in integrated multimodality cancer treatment are summarized in Table 1. It is crucial for treatment approaches to consider quality of life in survivorship, as each modality carries a different set of risks that need to be balanced against one another to provide the optimal risk-benefit ratio for each individual patient.
Table 1.
Multidisciplinary management of several common cancers treated with curative intent
| Role of Surgery, and Type of Surgery | Role of Chemotherapy or Other Systemic Therapy, and Example Agents | Role of Radiation Therapy, and Type of Therapy* | |
|---|---|---|---|
| Prostate cancer |
Primary local therapy Radical prostatectomy Pelvic lymph node dissection |
Concurrent therapy with radiation Hormonal therapy (bicalutamide, leuprolide, zoladex) |
Primary local therapy Adjuvant therapy after surgery External beam RT to the prostate or prostate bed, with or without pelvic lymph nodes Brachytherapy to the prostate |
| Breast cancer |
Primary local therapy Lumpectomy Modified radical mastectomy Axillary lymph node dissection or sentinel lymph node biopsy |
Neoadjuvant therapy before surgery Adjuvant therapy after surgery Cyclophosphamide, docetaxel, doxorubicin, paclitaxel, trastuzumab, Hormonal therapy (anastrazole, letrozole, tamoxifen) |
Adjuvant therapy after surgery External beam RT to the breast or chest wall, with or without axillary and internal mammary lymph nodes |
| Lung cancer |
Primary local therapy Lobectomy Pneumonectomy Hilar and mediastinal lymph node dissection |
Adjuvant therapy after surgery Concurrent therapy with radiation cisplatin, carboplatin, etoposide, paclitaxel |
Primary local therapy Adjuvant therapy after surgery External beam RT to the lung with or without hilar or mediastinal lymph nodes External beam RT to the mediastinum |
| Colorectal cancer |
Primary local therapy Colonic resection Total mesorectal excision Peri-colonic lymph node dissection |
Adjuvant therapy after surgery Concurrent therapy with radiation Capecitabine, fluorouracil, irinotecan, oxaliplatin |
Neoadjuvant therapy before surgery External beam RT to the mesorectum and pelvic lymph nodes |
| Melanoma |
Primary local therapy Wide local excision Lymph node dissection or sentinel lymph node biopsy |
Adjuvant therapy after surgery Immunotherapy (interferon alpha-2b) |
Adjuvant therapy after surgery External beam RT to the tumor bed and lymph nodes |
| Bladder cancer |
Primary local therapy Radical cystectomy Transurethral resection of tumor Pelvic lymph node dissection |
Neoadjuvant therapy before surgery Adjuvant therapy after surgery Concurrent therapy with radiation Cisplatin, gemcitabine, methotrexate, vinblastine |
Primary local therapy External beam RT to the bladder and pelvic lymph nodes |
| Non-Hodgkin lymphoma |
Local and systemic control (primary therapy) Cyclophosphamide, doxorubicin, etoposide, fludarabine, ifosfamide, methotrexate, rituximab, vincristine |
Primary local therapy Adjuvant therapy after chemotherapy External beam RT to the tumor and adjacent lymph nodes Radioimmunotherapy (ibritumomab, tositumomab) |
|
| Head and neck cancer (oral cavity, oropharynx, larynx) |
Primary local therapy Total or partial glossectomy Supraglottic, hemi-, or total laryngectomy Cervical lymph node dissection |
Adjuvant therapy after surgery Neoadjuvant therapy before radiation Concurrent therapy with radiation Cetuximab, cisplatin, docetaxel, fluorouracil, methotrexate |
Primary local therapy Adjuvant therapy after surgery External beam RT to the head and neck tumor and lymph nodes Brachytherapy to the head and neck tumor |
| Glioblastoma |
Primary local therapy Maximal tumor removal |
Concurrent therapy with radiation Temolozomide |
Primary local therapy Adjuvant therapy after surgery External beam RT to the tumor or resection cavity |
| Cervical cancer |
Primary local therapy Extrafascial or radical hysterectomy Pelvic lymph node dissection |
Concurrent therapy with radiation Cisplatin, fluorouracil |
Primary local therapy Adjuvant therapy after surgery External beam RT to the cervix or tumor bed, and pelvic lymph nodes Brachytherapy to the cervix |
| Anal cancer |
Primary local therapy Abdominoperineal resection |
Concurrent therapy with radiation Cisplatin, fluorouracil, mitomycin |
Primary local therapy External beam RT to the anus, mesorectum, and pelvic and inguinal lymph nodes |
Where indicated as primary local therapy, radiation as a primary therapy can be given as an organ sparing alternative to surgery.
Empiric clinical observations in the early 20th century demonstrated that daily radiation exposures can induce death in cancerous cells while allowing for normal tissue recovery, provided that the daily doses (expressed in units of Gray, or Gy) are relatively small. This observation formed the basis for fractionated radiation therapy. Many mechanistic explanations for this effect have been proposed by radiobiologists. The ability of normal tissue to repopulate itself with healthy cells clearly represents one important component of normal tissue tolerance of fractionated radiotherapy. A common course of radiation therapy can average 6–8 weeks of treatment, with 5–6 daily treatments per week.
At the tissue level, the impact of radiation therapy on tissue function with increasing radiation dose can be graphically represented by a sigmoid-shaped curve (Figure 1). The sigmoidal curve that describes tumor control probability is situated to the left of the sigmoidal curve that describes normal tissue complication probability. The degree of separation between these curves defines a therapeutic window, in which the dose of radiation is predicted to eradicate tumor while maintaining normal tissue tolerance. The sigmoidal relationship between dose and response implies that for any given tissue, there is a dose threshold above or below which incremental changes in dose yield little additional impact. However, within a critical range on the steep portion of the curve (such as cumulative doses of 40–100 Gy when given with “conventional” fractionation of 1.8–2.0 Gy/fraction), small increases in dose may result in large increases in clinical impact. Dose prescriptions in radiation therapy are determined with consideration to the unique relationship between the dose response curves for the specific tumor and surrounding normal tissues, which vary widely for each clinical circumstance. The figure is an oversimplification because families of curves may exist for the tumor clones comprising a tumor and for the complex components of normal tissues intertwined with the tumor.
Figure 1. An idealized graphical representation of tissue effects vs. radiation dose.

The solid sigmoid-shaped curves describe tumor control probability and normal tissue complication probability. The interrupted sigmoid-shaped curves describe the predicted effects in response to drugs that have tumor-specific radiosensitization or radioprotection that is specific to normal tissues.
III. Advances in Radiotherapy: biological aspects
Novel targets for radiosensitization
A common challenge in clinical radiotherapy is that tumors are frequently located immediately adjacent to radiosensitive normal tissues. Therefore drugs that can preferentially sensitize tumors to radiotherapy are of great interest in oncology. There are several clinical examples where radiosensitizers could play a critical role by boosting the anti-tumor effects of radiotherapy. One example is in malignant gliomas where lethality occurs as direct consequence of local tumor recurrence after treatment. By contrast, some tumors such as prostate cancer are already highly curable with radiotherapy but require very high doses of radiotherapy and/or hormonal ablation that carry significant toxic risks. In such situations, a radiosensitizer capable of exerting tumor-specific effects might allow for dose de-escalation, thereby reducing complication risks to normal tissues. Even in other situations where distant metastases are relatively common, local tumor recurrences can still represent a significant component of treatment failures. For example, local recurrence rates can approach 50–70% in locally advanced non-small cell lung cancers after aggressive chemoradiotherapy. Therefore, attempts at improving local control with radiosensitizers may be clinically valuable in these situations as well.
Standard chemotherapeutic agents are the most common agents utilized for increasing the local efficacy of radiotherapy, however, this review will focus on more recent strategies of identifying targeted radiosensitizers, particularly those that have been tested in the clinic. It is important to remember that inhibition of specific proteins can frequently generate sensitization of cells in culture, but ultimately a drug must improve the therapeutic index of radiation to be clinically useful (see illustration of this concept in Figure 1).
The molecular pathophysiology of radiotherapy: sensors, transducers, and effectors of DNA damage
One Gy generates approximately 105 ionization events per cell, producing about 1000–2000 single strand DNA breaks (SSBs) and 40 double strand DNA breaks (DSBs) per nucleus. A large and growing list of non-DNA repair /checkpoint related mechanisms contribute to cellular responses to radiation. Although ionizing radiation is known to generate DNA base damage, SSBs, and DSBs, the DSBs are generally thought to represent the principal lethal events and the most critical lesions to radiotherapy. DSBs initiate a complex set of cellular responses including DNA damage recognitionand transduction of the signal, resulting in many downstream effects including cell cycle checkpoint activation, induction and coordination of stress response genes, DNA repair, and/or activation of the apoptotic cascade (Figure 2).
Figure 2.

An overly simplified representation of various cellular targets and responses that occur after radiation exposure.
Targeting of DSB response and repair
The MRN protein complex (Mre11, Rad 50, Nbs1), a principle sensor of DNA damage, accumulates at DSBs very rapidly after radiation and participates in activating ataxia telangiectasia mutated (ATM) protein. Although ATM and ATR (ataxia telangiectasia and RAD3-related) perform partially overlapping functions, ATM preferentially recognizes DSBs while ATR preferentially senses replication-blocking DNA lesions. Both ATM and ATR phosphorylate downstream targets that regulate cell cycle checkpoints and apoptosis, as well as other forms of cellular responses like senescence, autophagy, and DNA repair. One centrally important phosphorylation target is the chromatin protein histone H2AX, since chromatin structure subsequently becomes less condensed and allows for the recruitment of repair proteins. Since H2AX phosphorylation is easily detectable with fluorescent microscopic methods, it has become a common marker for DSB induction and resolution (repair) in both experimental and clinical settings. Following DNA damage recognition, phosphorylated H2AX promotes the recruitment of other sensor/effector proteins including 53BP1, MDC1, and BRCA1, which regulate the processing of damaged DNA ends in preparation for repair. This process is regulated by several ubiquitin and SUMO ligases (PIAS1, PIAS4, RNF4, and RNF8) and the PSMD4 proteasome.
Homologous recombination (HR) and non-homologous end-joining (NHEJ) repair pathways are the two pathways that contribute to the repair of DSBs. Both repair pathways occur after the recruitment of the sensor/effector proteins discussed above. HR involves the identification of a stretch of homologous DNA and replication of the missing genetic information from this homologous DNA template(2). Alternatively, the NHEJ pathway processes the broken DNA ends and re-ligates them, frequently by making use of a region of micro-homology(3). These pathways appear to have somewhat overlapping and complementary roles.
The NHEJ pathway is the dominant pathway for repairing radiation-induced DSBs, particularly during G0/G1 portions of the cell cycle. The generally accepted model for this repair process begins with the binding and processing of DNA ends by Ku70/80, Artemis, and DNA-PKcs complexes. Ultimately the DSB is re-ligated by the Ligase IV/XRCC4 complex(4). Because NHEJ often requires processing of the DSB ends before re-ligation, repair by this pathway is error-prone. HR, by contrast, guides repair using an undamaged sister chromatid, and is thus an error-free mode of DSB repair. Also noteworthy is that HR-proteins allow cells to tolerate and repair replication-blocking lesions (like damage by inter-strand DNA cross-linkers) and collapsed replication forks. The initial steps of the HR pathway require 5′ to 3′ nuclease activity that generates a 3′ single-stranded DNA (ssDNA) tail at the site of damaged DNA, which is promoted by several proteins including CtIP, BRCA1, and the MRN complex. This processing of DSB ends occurs primarily in S/G2, and this cell-cycle selectivity is accomplished by phosphorylation of CtIP and Mre11 by cyclin dependent kinase 2 (CDK2). The next step in HR involves the 3′ ssDNA tail being coated with RAD51, which requires several mediator proteins including BRCA2, RAD52, the RAD51 paralogs (XRCC2, XRCC3, RAD51B, RAD51C, RAD51D), and several other proteins. This RAD51-coated 3′ tail then invades a homologous template DNA to form a joint molecule, the template sequence is essentially copied by polymerase activity and branch migration, and this Holliday junction is ultimately resolved to yield a repaired chromatid(2). The ability of some common chemotherapeutic agents to act as radiosensitizers may be, in part, accounted for by interference with these pathways.
Many components of the DSB repair pathways have been investigated as therapeutic targets, and some chemical inhibitors might be considered as possible lead compounds in oncology drug development (Figure 3). For example, mirin is a chemical inhibitor of the MRN complex(5). Consistent with the upstream role of MRN in sensing and signaling DNA damage, mirin generates a broad range of cellular effects, including inhibition of ATM activation, loss of G2/M cell cycle checkpoint, down-regulation of HR, and down-regulation of NHEJ repair efficiency(5, 6). This lack of cancer cell specificity may limit the utility of mirin in the clinic. Similar upstream signaling functions have been targeted with inhibitors of the family of phosphatidylinositol 3-kinase-related kinases (PIKK), which include ATM, ATR, mammalian target of rapamycin (mTOR), human suppressor of morphogenesis in genitalia-1 (hSMG-1), the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), and transformation/transcription domain-associated protein (TRRAP)(7). Several broad-spectrum inhibitors of PIKKs have been developed, including wortmanin or LY294002, however at least some have toxicities that limit their utility in the clinical setting. More specific inhibitors of ATM and/or ATR include KU-55933, CGK733, NU6027, and CP466722. KU-55933 has been the focus of particular attention, as it is a specific ATP-competitive inhibitor of ATM that is capable of sensitizing cells to radiation and several chemotherapeutic drugs, including etoposide, doxorubicin, and camptothecin(8).
Figure 3. Agents that may be potentially useful in modulating radiation effects.

Agents that have radioprotective effects are shown in red, and agents that have radiosensitizing effects are shown in green.
Several inhibitory compounds have also been developed to modulate specific DSB repair pathways. HR has been inhibited by specifically targeting RAD51 protein or RAD51 paralogs (see Budke et. al. and references therein (9)). Other drugs have been also been shown to lower HR efficiency by non-specifically reducing RAD51 protein levels. These strategies are promising since RAD51 protein is highly expressed in many human cancers (10, 11) and since HR inhibition has been shown to promote preferential sensitization of tumor cells relative to normal cells(12, 13). These observations suggest that human tumors may develop ‘addictions’ to abnormally high RAD51 levels that can be exploited pharmacologically. Likewise, compounds have been developed to target components of the NHEJ repair. Again, this can be accomplished by blocking PIKKs. However, more targeted NHEJ inhibitors have also been developed, including several different DNA-PK inhibitors that include NU7441, Vanillin, SU11752, IC87102, IC87361, NU7026, CC-115, and Salvicine.
Relatively few of these inhibitors of DSB repair have transitioned into clinical trials, perhaps due to lack of commercial interest and the recognition of radiotherapy as fertile ground for pharmaceutical development. One interesting new strategy that is being developed clinically by DNA Therapeutics involves small DNA molecules that act as DNA bait. One such agent, termed Dbait or DT01, mimics DNA double-strand breaks and acts to disorganize damage signaling and DNA repair(14). DT01 is currently being studied in a phase I trial for patients with metastatic melanoma.
PARP inhibitors
Poly ADP-ribose polymerase inhibitors (PARPi) are currently being studied in numerous clinical trials with chemotherapy and a few trials with radiotherapy. The PARP family of proteins is defined by their capacity to modify target proteins by the covalent addition of poly ADP-ribose polymers. PARP1 is the most abundant of this protein family, and it accounts for approximately 80% of PARP activity in cells. PARP1 and PARP2 possess DNA binding domains, and their catalytic function is activated when they bind sites of DNA damage. When PARP1 becomes activated, it generates long and branching poly ADP-ribose chains on histones and other proteins located near DNA breaks. These polymer scaffolds are important in recruiting other DNA repair proteins (for example the base excision repair protein XRCC1) to the break site. PARPi compounds have generated intense interest by the oncology community since 2005, following the demonstration of synthetic lethality in BRCA-defective cells(15, 16). Synthetic lethality is a concept whereby a tumor is defective in one survival pathway, and inhibition of an escape pathway is an effective cytotoxic strategy. A common feature of these drugs is the exquisite hypersensitivity observed with HR-defective tumor cells, including triple-negative breast cancers which exhibit epigenetic deregulation of HR.
Several studies have demonstrated that PARPi compounds can radiosensitize tumors in preclinical models(17–19). Ionizing radiation, as discussed earlier, induces both SSBs and DSBs. Although the PARPi-mediated mechanism of radiosensitization remains somewhat unclear, PARP inhibitors are known to sensitize cells to agents that generate SSBs(20). Bristow and colleagues have pointed out two key aspects of PARPi effects pertinent to this issue: 1) radiosensitization occurs primarily in replicating cells and, 2) PARPi compounds delay rather than abolish SSB repair(18). These factors suggest that components of replication machinery may collide with unrepaired PARP-bound SSB lesions, thereby generating more toxic lesions than the starting SSBs. Other possible mechanisms of PARPi-mediated radiosensitization may include re-oxygenation of hypoxic tumors; PARPi compounds have demonstrated vasoactive properties and could potentially counteract the radioresistance associated with hypoxia. Finally, recent studies have shown that chronic hypoxia in tumors can generate a reduction in HR protein expression and function. As such, hypoxia within tumors may generate specific anatomic compartments that behave like BRCA-defective tumors, wherein a contextual synthetic lethality occurs for PARPi(21).
Several clinical trials are presently evaluating combinations of PARPi drugs plus radiotherapy, either with or without chemotherapeutic drugs for diseases including rectal, brain, and breast cancers (http://clinicaltrials.gov). Again, it is unknown whether or not these agents will improve the therapeutic index of radiotherapy, and specific disease site trials will be necessary to determine potential efficacy.
Histone deacetylase inhibitors
Histone deacetylases (HDACs) represent a family of at least 18 enzymes that remove acetyl groups from lysine residues in core histone proteins. De-acetylation exposes the positive charges on histones, thereby increasing their interaction with the negatively charged phosphate backbone of DNA. This results in more compacted chromatin that is relatively inaccessible to modulation by transcription factors. This pathway has relevance to radiation biology, given that HDAC inhibitors stimulate radiation-induced cell-cycle arrest, apoptosis, and DSB formation(22, 23). The mechanism is unclear, but studies suggest that HDAC inhibitors suppress DNA repair efficiency.
Traditional HDAC-inhibitory drugs, like valproic acid, have a long history as mood stabilizers and anti-epileptics. Several second generation inhibitors have more recently been developed, including vorinostat, belinostat, and panobinostat. On-going clinical trials are evaluating these drugs plus radiotherapy in many tumor types and in a wide range of clinical settings, including fractionated radiotherapy, stereotactic radiosurgery, and together with an infused 131-I MIBG radiopharmaceutical. One recently completed phase I study showed that vorinostat (at 300 mg once daily) was tolerable in combination with a palliative radiotherapy course to the pelvis, consisting of 30 Gy over two weeks(24). Unfortunately, efficacy results from phase 2 trials are not available.
Cell cycle arrest following radiation
It has been known for decades that mammalian cells arrest in G1 and G2 following radiation exposure. The pathways that govern cell cycle checkpoints were first discovered in yeast by Hartwell, Nurse, and Hunt, for which they received the 2001 Nobel Prize. Briefly, radiation-induced activation of ATM and ATR leads to phosphorylation of the downstream effector kinases Chk1 and Chk2. These in turn phosphorylate the phosphatase CDC25A, which in turn becomes degraded and thus unable to dephosphorylate and activate CDK2; the net result is cell-cycle arrest. Additionally, Chk2 phosphorylation of p53 activates expression of p21, which also induces cell-cycle arrest.
Targeted inhibition of the cell cycle checkpoint machinery has been explored as a method to sensitize cells to DNA damage. Some of these strategies have been based on the tendency of tumor cells to have abnormal G1 checkpoint functions but intact G2 checkpoint mechanisms. Targeted inhibition of G2 checkpoints can promote progression of damaged cells to mitotic catastrophe, and this effect may be tumor-specific since normal tissues are preferentially protected by intact G1 checkpoints. This may also potentially counteract the radiation resistance that has been observed in some cancer stem cells, such as CD133-positive glioma stem cells(25).
A number of chemical inhibitors have been developed to target Chk1 and/or Chk2, including UCN-01, AZD7762 (AstraZeneca), XL844 (EXEL-9844, Exelixis), LY2606368 (Eli Lilly), and PF-00477736 (Pfizer). All of these compounds have been tested in early-phase clinical trials against solid tumors. Many of these are being tested in combination with various chemotherapeutic drugs. However, it remains an open question as to whether these drugs can be safely combined with radiotherapy, and whether they can improve the therapeutic index.
Modifiers of cellular death following radiation
Cells subsequently face a critical period, particularly if radiation damage is not completely repaired(26). These critically injured cells may progress unrepaired to mitotic catastrophe or simply lose proliferative capacity by undergoing senescence. Radiation-induced senescence is commonly associated with activation of the p16/RB and p53/p21 tumor suppressor pathways. Alternatively, cells may progress to apoptotic death, which classically involves p53-dependent activation of the caspase cascade. Apoptotic death can also occur following activation of cell surface ‘death receptors’ (receptors of TNF, Fas, or TRAIL), which themselves can be up-regulated by radiation. Death receptors can also generate cyto-protective effects by activating transcription factor nuclear factor kappa B (NFκB), which increases expression of genes that promote proliferation or oppose apoptosis.
Pathways of intracellular signaling following radiation
The majority of radiation-induced radical oxygen species (ROS) interact with cellular contents other than DNA. ROS such as superoxide and hydroxyl radicals are also known to deplete cellular stores of antioxidants like glutathione(27). The resulting cellular stresses stimulate a complex set of signaling cascades (reviewed in detail by Schmidt-Ullrich and colleagues(28)).
Radiation can also activate signaling pathways that cells normally use to respond to mitogens, which in turn promote survival, anti-apoptotic responses, and transcriptional changes. The net effect can be variable and cell-specific; however a common theme includes activation of cell surface receptors, like the ErbB family that includes epidermal growth factor receptor (EGFR). Receptor activation subsequently signals downstream pathways, including the mitogen-activated protein kinase (MAPK) superfamily of cascades (ERK, JNK, p38) and the phosphatidyl inositol 3 kinase (PI3K) pathways. These pathways deliver anti-apoptotic signals via Akt and Erk signaling. Radiation can also activate these pathways via autocrine mechanisms, like through the production of transforming growth factor alpha (TGFα) which binds and activates EGFR(28). Radiation also activates the pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α) and Interleukin-6(29), which in part may account for bystander effect (discussed below).
Finally, radiation-induced damage of the plasma membrane induces the breakdown of sphingomyelin to ceramide, which is pro-apoptotic independent of DNA damage(30). Radiation also activates cytosolic phospholipase A2 (cPLA2), an enzyme that recognizes phospholipids on the cell membrane and degrades them into inflammatory products like arachidonic acid and eventually eicosanoids. Lysophosphatidylcholine (LPC) is one such product formed by cPLA2, and its production leads to activation of Akt and enhanced cell death(31).
One interesting strategy for radiosensitization has been the blockade of pro-growth signaling from receptor tyrosine kinases, i.e. insulin-like growth factor 1 receptor (IGF-1R) and EGFR. Numerous antibodies and chemicals have been developed to target different levels of this signaling cascade. One successful pharmacologic effort was the inhibition of EGFR during radiotherapy for head and neck cancers, using the chimeric (mouse/human) monoclonal antibody cetuximab. A randomized trial demonstrated benefit of weekly cetuximab in addition to 6–7 weeks of radiotherapy. The updated data show a 5-year overall survival of 45% with cetuximab/radiotherapy group and 36% with radiotherapy alone(32). Many related strategies are being evaluated in this and other anatomic tumor sites, using inhibitory antibodies or small molecule-based tyrosine kinase inhibitors.
Bystander effects and importance of tumor microenvironment
Non-irradiated cells often exhibit stress responses, following even low dose (<0.1 Gy) radiation exposures to neighboring cells. This ‘bystander effect’ has been well described in detail previously (33, 34). Bystander responses appear to be cell type specific; in general they consist of a broad range of effects including gene induction, genomic instability, differentiation, and changes in apoptotic potential. These processes are mediated, at least in part, by diffusible substances, given that the effects occur when bystander cells are physically separated from the irradiated cells. Some authors have additionally implicated cell-cell contacts (gap junctions) as contributing to this effect.
This illustrates an important concept- that one must consider radiation effects in the context of the entire tumor micro-environment, rather than simply the sensitivities of individual cancer cells. This notion is further demonstrated by the contribution of host stromal components within tumors to radiation responsiveness. Studies in mice, for example, show that host-derived blood vessels are a key determinant of tumor control with radiotherapy(35).
Impact of cancer stem cells (tumor initiating cells) within irradiated tumors and normal tissues
Although many researchers disagree over various aspects of cancer stem cell biology and nomenclature, there clearly exists a population of tumor cells that exhibit an exclusive ability for self-renewal and differentiation into the heterogeneous lineages that promote tumor maintenance. These concepts have been supported by three recent important papers demonstrating the existence of cancer stem cells in mouse models of brain, skin, and intestinal tumors(36–38). Results of these studies indicate that targeting cancer stem cells may improve therapeutic outcomes. In response to fractionated radiation, cell populations become enriched for cells expressing putative markers of ‘stemness.’ Specific phenotypes vary based on tumor type and methodology, but common features of these cells after radiation often include increased survival, reduced apoptosis, and rapid resolution of H2AX foci (i.e. fast repair of DSBs). For example, one study demonstrated an enrichment of glial cancer stem cells after radiation, and that these cells exhibited preferential activation of the DNA damage checkpoint responses and increases in DNA repair capacity(25).
The basic mechanisms of radiation resistance remain unclear in stem cells, however aldehyde dehydrogenase 1 (ALDH1) may represent a partial explanation. High expression of ALDH1 protein in tumors is known to be an adverse prognostic factor, perhaps because its aldehyde-catabolizing activity confers a survival advantage. Also, aldehyde-catabolizing enzymes are known to cooperate with Fanconi anemia genes, suggesting that ALDH activity might play a role in repairing adducts on DNA(39). Hence, since ALDH1 appears to be preferentially expressed in cancer stem cells, this may represent a therapeutic target to reverse radioresistance in stem cell clones. Another interesting feature of these cells is their propensity to repair DSBs using HR repair. Breast cancer-derived stem cells (CD24−, ESA+) were “effectively sterilized” by inhibition of the HR pathway, while non-sorted cells from the same cell line (MDA-MB231) were unaffected(40). This finding suggests that HR inhibitors represent a promising therapeutic strategy for depleting cancer stem cell reservoirs in tumors. Also, screening of small molecule libraries led to the identification of salinomycin and thioridazine, both of which appear to target cancer stem cells(41, 42) and may be used to improve radiotherapy.
Normal tissues also contain stem cell niches that likely influence their ability to tolerate radiotherapy. Thomas Helleday’s group recently reported a study, in which punch biopsies were taken from normal skin during the first and last weeks of a 5-week clinical radiotherapy course(43). The week-5 skin biopsies showed a significant enrichment for proliferating cells that were undergoing HR (Ki67+ and RAD51 focus+), as well as epidermal stem cells (β1 integrin+ and Ki67−). These results suggest that tumor tissue and normal tissues share some features in terms of stem cell responses, which may pose challenges to targeting cancer stem cells. In some situations, the anatomic locations of normal stem cell niches are known and can perhaps be spared from radiation exposure. One example is the hippocampus, in which neural stem cells are known to reside. An ongoing trial by the Radiation Therapy Oncology Group (RTOG) is prospectively testing whether reduced exposure to the hippocampus can decrease neurocognitive toxicity associated with whole brain radiotherapy. Another example comes from stem cell niches located within the walls of larger ducts in salivary glands(44), which could potentially be excluded from radiotherapy treatment target volumes.
Targeting tumor hypoxia and redox conditions
The investigation of tumor hypoxia as an effector and biomarker of tumor resistance has exploded in the past decade, and modalities that exploit tumor hypoxia have been widely tested. Tumors are known to outgrow their blood supply, thereby generating regions of necrosis that are surrounded by areas of hypoxia. Hypoxia promotes activation of the hypoxia-inducible transcription factor (HIF) family of proteins that regulate a variety of downstream genes that promote angiogenesis, cell survival, anaerobic energy metabolism, and treatment resistance(45). Hypoxia can also select for highly aggressive tumor cell clones. This topic is particularly relevant to radiotherapy, because the presence of molecular oxygen during delivery of ionizing radiation enhances radiation-induced cell kill by 2.5 to 3.5 fold. The commonly accepted mechanism for this observation is that oxygen ‘fixes’ free radical-induced DNA damage into a permanent state. Hence, hypoxic regions of tumor are generally considered radiation resistant, and reversal of hypoxia has long been a goal for radiosensitization.
Early approaches to improve tumor oxygen status, which included the administration of hyperbaric oxygen during radiotherapy, generated mixed results(46). Inhaled carbogen (98% oxygen and 2% carbon dioxide) combined with oral nicotinamide (a vasoactive agent) is a less cumbersome alterative, since a hyperbaric chamber is not necessary. A phase III randomized trial evaluated this strategy in T2-4 laryngeal cancers(47). Compared to radiotherapy alone, the addition of carbogen/nicotinamide was non-toxic and yielded some gains in regional control (sterilization of lymph node metastasis) but no benefit in terms of overall local tumor control. A similar trial in bladder cancer showed benefit of borderline significance in both local control and overall survival associated with carbogen/nicotinamide(48). Related efforts have attempted to increase oxygen delivery to tumors using other modalities. Erythropoietin was combined with radiotherapy in a randomized trial, however this actually appeared to radioprotect head and neck cancers(49). Efaproxiral (RSR-13) is an allosteric modifier of hemoglobin that increases oxygen delivery by reducing hemoglobin-oxygen binding capacity. This compound was proven tolerable when combined with radiotherapy for brain metastases or lung cancers(50, 51), but it did not demonstrate sufficient efficacy for approval for routine clinical use. Additional drugs that target hypoxia include oxygen mimetics, such as the nitroimidazoles. Unfortunately, few have shown an improvement in the therapeutic index for radiotherapy. One such hypoxia radiosensitizing drug, nimorazole, did reduce the risk of head and neck cancer recurrences after radiotherapy, but the drug was effective only in patients with high serum levels of osteopontin, a biomarker that predicts clinically relevant tumor hypoxia(52). However, this agent has not been widely adopted.
A newer class of radiation sensitizers has been developed to target hypoxia, based on reduction/oxidation conditions that predominate in anaerobic environments. Tirapazamine and porfiromycin are hypoxic cell cytotoxins/sensitizers, which act as bio-reductive alkylating agents(53). Tirapazamine differs from oxygen mimetics in that it does not ‘fix’ radiation damage, but instead it is metabolized into a highly reactive radical species in anaerobic conditions. Preclinical data regarding tirapazamine was extremely promising, however several phase III trials failed to demonstrate significant clinical benefit in combination with radiotherapy (54, 55). This disappointing outcome may reflect poor penetration of tirapazamine into hypoxic tumor regions. In an interesting related development, RAD51 and other HR-related DNA repair proteins have been shown to be transcriptionally down-regulated in response to chronic hypoxia(56). Contrary to traditional concepts of hypoxia, this down-regulation of DNA repair may actually render cells more sensitive to radiation.
Related efforts have used porphyrin-like macrocycles that form complexes with large metal cations and participate with the cellular redox cycle. Motexafin gadolinium (also known as MGd,) is one such drug that inhibits antioxidant proteins such as thioredoxin reductase, and it exhibits preferential localization to tumor over normal tissue(45). In combination with whole brain radiotherapy, MGd provided modest improvements for patients with brain metastases from lung cancer(57), however these effects have not been impressive enough for Xcytrin to gain FDA approval. MGd is also being evaluated in ongoing or completed clinical trials involving radiotherapy for other CNS tumors (glioblastoma multiforme and pediatric brainstem gliomas), as well as various carcinomas including pancreaticobiliary, non-small cell lung, and head and neck cancers.
Pharmacologic approaches are being developed to target cellular signaling that occurs in response to hypoxia and redox status in tumors. Many of these have focused on the transcription factor HIF-1, using agents that modulate its transcription, stability, association with binding partners, or signal transduction(45). EZN-2968 (Enzon Pharmaceuticals), an antisense oligonucleotide to HIF-1α, demonstrated safety and potential activity in phase I testing, however data from subsequent testing are still awaited.
A somewhat related strategy arose from an siRNA screen, looking for tumor-selective radiosensitizing targets in head and neck cancers. Surprisingly, knockdown of uroporphyrinogne decarboxylase (UROD), a regulator of heme synthesis, sensitized head and neck cancer cells to radiation and some cytotoxic agents such as cisplatin, paclitaxel, and 5-fluorouracil. This may occur due to alterations in iron homeostasis, which promotes an increased production of reactive oxygen species(58). The role of hypoxia in tumor killing by radiotherapy remains to be resolved. Perhaps biomarkers or non-invasive physical measures of hypoxia will determine which patients might benefit from modification of hypoxia.
Anti-angiogenic drugs and radiotherapy
New blood vessel development is required for tumors to grow beyond 1–2 mm. Vascular endothelial growth factor (VEGF) is a key pro-angiogenic growth factor that is secreted by solid tumors and acts through one of three VEGF receptors (VEGFRs). The most widely studied VEGF inhibitor is a humanized monoclonal antibody (bevacizumab) that binds to and inhibits the activity of human VEGF. Preclinical data have demonstrated that blockade of VEGF signaling increases the anti-tumor effects of radiation. Additional studies have suggested that VEGF protein levels can become up-regulated in tumors in response to ionizing radiation, suggesting that VEGF might mediate the development of tumor endothelial cell radioresistance (59, 60). The therapeutic combination of radiotherapy plus VEGF inhibition could be considered counterintuitive, since antagonism of tumor vasculature might be expected to increase tumor hypoxia. However, tumor angiogenesis has proven to be a dysregulated process that generates networks of tortuous and hyper-permeable vessels, resulting in spatial heterogeneity in tumor oxygenation and elevated interstitial fluid pressure. Current research in pre-clinical models has demonstrated that VEGF blockade can “normalize” the tumor vasculature, thereby reducing tumor hypoxia and interstitial pressure and improving the metabolic profile of the tumor microenvironment(61).
The broad interest generated from these findings led to clinical trials that have combined radiotherapy and/or chemotherapy with bevacizumab in a wide range of cancer types. Encouraging results have emerged from multiple phase II trials in a variety of disease sites, though few of these regimens have proceeded to phase III testing. For example, bevacizumab has been safely combined with oxaliplatin, capecitabine, and radiation in the pre-operative setting for rectal cancer(62). Importantly, a large international phase III trial has recently completed accrual to compare radiation and temozolomide with or without bevacizumab in newly diagnosed glioblastomas, and test the impact of bevacizumab on overall survival. Bevacizumab and potentially other anti-angiogenic therapies may increase the risks of radiation-induced complications in normal tissues. For example, clinical trials combining thoracic radiotherapy plus bevacizumab have reported unexpectedly high rates of radiation pneumonitis and tracheoesophageal fistula (63, 64). Likewise, the combination of bevacizumab, capecitabine, and radiation (RTOG 0411) for pancreatic cancer resulted in grade 3–4 gastrointestinal toxicity in 35% of patients (65). The risk of such serious complications probably depends on the tumor type and the radiation sensitivity of the adjacent anatomic structures.
Gene therapy and radiotherapy
There has been extensive preclinical work over the past 15 years on combining gene therapy with radiotherapy. Replication-defective viruses have been used to deliver radiosensitizing prodrugs, cytokines, tumor suppressor genes, and immune-activating compounds. Other related strategies have employed replication-competent viruses to generate oncolytic anti-tumor effects. The following section will focus on gene therapy modalities that have been tested with radiotherapy in clinical trials.
TNFerade (Ad.Egr-TNF11D) is a replication-defective adenoviral vector that contains radio- and chemo-inducible elements from the Egr-1 promoter upstream to cDNA encoding TNF-α. This strategy takes advantage of TNF’s function as a direct radiosensitizer and immune activator. Irradiation promotes spatial and temporal control of TNF-α transcription from Egr-1 promoter elements, such that TNF-α is specifically secreted within the radiotherapy target volume(66). In phase 1–2 trials with radiotherapy or chemoradiotherapy, TNFerade generated impressive tumor response rates in patients with soft tissue sarcomas, as well as carcinomas of the esophagus, head and neck region, and rectum(67). An additional phase 1 trial is ongoing in prostate cancer. A phase 3 randomized trial of 5-FU and radiotherapy +/− TNFerade was recently completed in patients with unresectable pancreatic cancers. TNFerade appears to have prolonged survival in patients with relatively small T1-T3 pancreatic tumors on subset analysis (not larger T3 and or T4 tumors), but it has not been approved for this use (personal communication, Kenneth Chang).
Other trials have employed enzyme/prodrug strategies. For example, virus-directed expression of herpes simplex virus thymidine kinase (HSV-tk) can phosphorylate the prodrug gancyclovir into a toxic metabolite that interferes with DNA replication, leading to chain terminations and single-strand DNA breaks. A phase 3 randomized trial was unable to show a survival benefit with this strategy, when it was combined with surgery and radiation for glioblastoma multiforme(68), although the agent was delivered after therapy. Related strategies involve the use of an additional ‘suicide gene’ to viral vectors. Freytag and colleagues have tested replication-competent adenovirus that carries both HSV-tk and cytosine deaminase, which converts the prodrug 5-fluorocytosine into 5-FU(69). This gene therapy strategy with prostate radiotherapy appears to be tolerable, but efficacy remains to be demonstrated in phase 2–3 trials.
Oncolytic viruses have also been combined with radiotherapy, based on interesting pre-clinical evidence that some viruses can cooperate with radiotherapy by preferentially infecting and lysing cancer cells. Replication-competent herpes simplex virus I (HSV-1) has been genetically modified to allow safe use with radiotherapy. For example, a modified HSV-1, called G207, has been safely administered via intra-tumoral injection immediately before radiotherapy in recurrent or progressive malignant gliomas. More recently, a genetically modified HSV-1 encoding granulocyte macrophage colony stimulating factor was combined with chemoradiotherapy in a phase 1/2 trial for locally advanced head and neck cancer(70). Also, intra-tumoral injections of a reovirus were recently combined with palliative radiotherapy in a phase I trial(71).
Immune modulation and radiotherapy
Pre-clinical evidence shows that radiation can stimulate anti-tumor responses by the immune system, which has important potential implications clinically. Radiation generates an inflammatory microenvironment in tumors, whereby damaged cells increase antigen presentation and immune recognition. More specifically, radiation-induced immune effects include an elevation of major histocompatibility complex class I (MHC-I) expression, changes in antigenic peptide repertoire, and decreases in regulatory T cells. Radiation also up-regulates cytokines and adhesion molecules that recruit and activate CD8+ cytotoxic T lymphocytes and dendritic cells. The evolving understanding of these processes may allow for them to be exploited therapeutically(72–74).
NCI researchers reported a randomized phase 2 trial that treated prostate cancer with standard radiotherapy, with or without gene therapy-based vaccine that encodes PSA(75). The therapy consisted of a “priming” vaccination with recombinant vaccinia (rV) PSA plus rV containing a T-cell co-stimulatory molecule, followed by monthly booster vaccines with recombinant fowlpox PSA. Most of the vaccinated patients did successfully develop increases in PSA-targeted T cells. Other clinical trials have explored related strategies of radiotherapy in combination with different vaccines, intra-tumoral injection of immature dendritic cells, or adoptive immunotherapy via infusion with expanded tumor-infiltrating lymphocytes (reviewed by Kamrava and colleagues (72)). Many of these strategies have demonstrated an impressive induction of immune responses; however, it remains unclear whether these effects will translate to improved clinical outcomes. A recent phase 1 trial reported that patients with metastatic melanoma or kidney cancer treated with high dose IL-2 demonstrated impressive tumor responses when 1–3 tumors were treated with stereotactic body radiotherapy, compared with historical controls treated with high dose IL-2 alone(76).
A more recent strategy that is likely to be tested in the near future is radiotherapy in combination with antagonists of cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) or programmed death 1 (PD-1). Antibodies (ipilimumab, tremelimumab, anti-PD-1, and anti-PD-L1) against these targets are known to block the mechanisms used by tumor cells to evade the immune system(77). An interesting outcome of an anti CTLA-4 treatment was recently reported in a patient with metastatic melanoma who required radiotherapy for symptomatic disease - unexpected, significant responses occurred outside of the radiation field (78). In this phenomenon known as the “abscopal effect,” local radiation can sometimes produce distant anti-tumor effects. Since its first report in the literature by Mole in 1953 (79), there have been few studies that have elucidated potential mechanisms for this effect. Preclinical studies demonstrate the critical role of a functional immune system (with abscopal effects mediated in part by IL-12 (80) and dendritic cells (81). Clinical data demonstrate that radiation can modulate the immune cell subsets, which likely underlie these abscopal effect (78). This has great importance for radiotherapy since interaction of radiation with the immune system may have both local and distant effects. Another interesting new strategy utilizes radiation to generate cell-based cancer vaccines. The PARPi drug veliparib plus radiation was shown to induce unrepaired DNA injury in cells, leading to an accelerated senescence phenotype. These senescent tumor cells express immune-stimulatory cytokines capable of activating cytotoxic T lymphocytes. Injection of the resulting senescent tumor cells into mice successfully potentiated the anti-tumor effect of radiotherapy in a syngeneic mouse tumor model(82). This approach has yet to be tested clinically.
Protection of normal tissues from radiation injury
Acute side effects of radiotherapy are principally caused by disruption of rapidly proliferating cell renewal systems, whereas the late side effects are often due to vascular damage, fibrosis, and poor repopulation of normal tissue components by healthy cells. Radiation can also generate mutations that ultimately induce secondary cancers, which are especially problematic for the pediatric patient population. As oncologic treatments continue to improve clinical outcomes, a growing population will be surviving with treatment-induced complications that can negatively impact quality of life. For this reason, efforts are underway to generate treatment strategies that minimize damage of radiotherapy on normal tissues. Moreover, a successful radioprotective drug might also allow clinicians to escalate radiotherapy doses higher, which could lead to better tumor control rates. Radioprotectors can be thought of as drugs given prophylactically before radiation exposure, whereas radiation mitigators can be administered during or shortly after radiotherapy. A third category consists of therapeutic agents, which are drugs that modulate normal tissue response or healing. The major challenge in all of these pursuits has been generating drugs that do not simultaneously protect tumor cells (see illustration of this concept in Figure 1). Ongoing efforts are summarized below.
Free radical scavengers
Sulfhydryl-containing compounds act as free-radical scavenger agents, thereby absorbing radicals that are generated by radiation-induced ionization(83). As a result, these compounds reduce chromosome abnormalities and increase cell survival following radiation, suggesting a potential use in reducing normal tissue complication and radiation-induced malignancies. The thiol-containing drug amifostine (originally called WR-2721) is the only FDA approved drug for clinical use as a protector of radiation-induced cell death, and it remains the best studied drug of this class. In one randomized trial, Brizel and coworkers demonstrated that amifostine reduces radiation-induced salivary gland injury(84), however other trials have observed considerably less impressive normal tissue protection(85). Some pre-clinical studies have also observed protection of tumor cells by amifostine, however there is little evidence suggesting that this occurs in clinically relevant situations(86). Pyridoxamine (which is the active ingredient in the drug Pyridorin) is a newer and more active free radical scavenger than amifostine. It has shown some success in phase I/II trials, although these trials included non-radiation indications such as diabetic renal injury.
Non-thiol agents for radioprotection and mitigation
Modern development programs have placed an emphasis of generating radioprotector drugs that are preferentially active in normal cells. One such strategy is based on the relative radiation resistance of G1 phase cells, compared to cells in G2/M or the G1/S transition. The cell cycle is regulated by cyclins and cyclin-dependent kinases (CDKs), which are commonly dysregulated in tumor cells. Two CDK4/6 inhibitors (PD0332991 and 2BrIC) have been shown to induce a reversible G1 arrest preferentially in normal cells. Furthermore, PD0332991 was shown to mitigate hematopoietic toxicity in mice following total body radiotherapy (TBI) without affecting growth of melanoma tumors (87). The p53 inhibitor pifithrin was shown to protect mice from lethality following total body radiation; this strategy could potentially be normal-tissue specific since many cancers carry p53 mutations(88). Another strategy of radioprotection involves inhibitors (lithium, SB216763, and SB415286) of glycogen synthase kinase 3β (GSK-3β), which is involved in radiation-induced apoptosis. The proposed mechanism of normal tissue specificity is based on distinct GSK-3β-dependent signaling patterns that are observed in normal vs. cancer cells. These GSK-3β inhibitors were shown to block radiation-induced hippocampal neuronal apoptosis, leading to improved cognitive function in irradiated mice(89). Several early stage clinical trials are currently evaluating lithium in combination with brain radiotherapy. The anti-ceramide monoclonal antibody 2A2 represents yet another potentially radioprotective agent, in this case protecting against endothelial apoptosis in the small intestine, thereby preventing death of mice from radiation-induced GI toxicity(90). Finally, anti-apoptotic compounds (TPP-IOA and TPP-ISA) that target the cytochrome c/cardiolipin complex have been used to protect mice against lethal doses of irradiation(91).
Therapeutic agents
Vascular damage is a major underlying cause of late radiation tissue injury. In small vessels, endothelial cell damage initiates inflammatory and coagulation cascades which cause micro-thrombus formation and tissue ischemia. Additionally, overproduction of cytokines such as transforming growth factor beta (TGF-β) can drive smooth muscle proliferation. Interventions that reduce TGF-β have proven to reduce lung and intestinal fibrosis in animal models. Likewise, anticoagulants and the blood flow-promoting drug pentoxifylline have been shown to modulate vascular responses and/or reduce microvascular damage after radiotherapy, but neither has generated large enough effects to gain widespread use(92). The renin-angiotensin system has a key role in the regulation of hemodynamics in multiple organ systems. Angiostatin II up-regulates TGF-β and thus may also contribute to fibrosis. The angiotensin-converting-enzyme (ACE) inhibitor captopril was tested in a randomized trial for patients undergoing total body radiation for bone marrow transplantation. Patients who received captopril experienced significantly less pulmonary mortality and modestly reduced renal failure(93). Some (though not all) retrospective trials of thoracic radiotherapy for lung cancer also suggested that ACE inhibitors may reduce radiation pneumonitis. Statin drugs have known anti-inflammatory and anti-thrombotic effects. Multiple retrospective studies suggest they may enhance radiation effects in several tumor types, while possibly reducing toxic risks. Various types of growth factor support have been used to promote normal cell proliferation. One notable example is keratinocyte growth factor (palifermin), for which a randomized trial showed less mucositis during chemoradiotherapy for locally advanced head and neck cancer(94). Finally, bevacizumab was recently shown to be an effective treatment of symptomatic radiation necrosis of the brain(95).
Molecular predictors of outcome in radiotherapy
In the past decade we have witnessed an explosion in new diagnostic technologies capable of sub-classifying malignancies and predicting clinical outcomes, based on genetic and epigenetic tumor characteristics. Such studies have fueled a vision in the oncology community of ‘personalized medicine,’ a system by which individualized treatment courses can be tailored to specific patient and tumor characteristics. These studies represent an expansive work that cannot be adequately encompassed by this review, and most of these studies have focused on predictors of overall prognosis.
Many studies have reported gene expression signatures that predict overall tumor behavior for different cancer types, and some of these signatures have been validated with clinical data. However few have attempted to use expression array technology to predict radiocurability per se. One such gene expression signature, called the radiosensitivity index (RSI), consists of 10 genes (AR, cJun, STAT1, PKC, RelA, cABL, SUMO1, CDK1, HDAC1, and IRF1) that associate with radiosensitivity within a collection of human cancer cell lines(96). This signature has been clinically validated in five independent clinical data sets, which include rectal, esophageal, head and neck, and breast cancers treated with chemoradiation or radiotherapy alone(97). A related approach identified a chemotherapy/radiation resistance signature, using tumors generated in mice. This interferon-related gene signature analysis was evaluated retrospectively in clinical breast cancer data sets, and it successfully predicted the efficacy of adjuvant chemotherapy and local-regional control after radiation(98).
An alternative approach is to look for associations between radiosensitivity and genetic alterations, using methods like comparative genomic hybridization (CGH) arrays or next generation sequencing. CGH and SNP (single-nucleotide polymorphism) arrays measure copy number variations and can quantify global levels of genomic instability. Bristow and co-workers have used CGH on prostate cancer biopsy specimens to identify deletions of chromosome regions that normally harbor DNA repair genes (including PARP1, ATM, DNA-PKcs, p53, Rb, and RAD17). Detection of these gene losses might help identify tumors susceptible to radiation or other specific treatments(99). Similar work showed NKX3.1 haploinsufficiency to predict risk for prostate cancer recurrence after radiotherapy, and it also predicted for recurrence after prostatectomy(100). These and other emerging methods will need to be validated more thoroughly in prospective clinical trials, but the findings are encouraging. Furthermore, next generation sequencing of tumor genomes will presumably revolutionize these types of approaches as we move forward.
While the excitement of these new methods has echoed throughout the entire oncology community, it remains unclear whether personalized medicine can live up to our collective expectations. These newer methods generally rely on ‘representative’ tumor biopsies, which can underestimate tumor cell heterogeneity. A recent cautionary study demonstrated alarmingly high levels of intra-tumor heterogeneity in renal cell cancers, both in terms of gene expression levels and DNA sequence(101). In fact, >60% of all somatic mutations were not detectable across all regions of a given tumor. Hopefully, many of the ‘driver mutations’ (which form the basis of many new targeted therapies) will prove to be better represented throughout tumors. These data suggest that our existing therapies, conventional chemotherapy drugs and radiotherapy, will continue to serve important roles.
IV. Advances in Radiotherapy: technical aspects and clinical applications
The majority of treatments with radiation are delivered through an external approach, in which radiation energy is produced by a device which can generate photons or particles, such as electrons, protons, carbon ions, or neutrons. High energy photons (6–20 MV) produced by commercially-available linear accelerators are by far the most common form of radiation therapy. Patients are reproducibly positioned on a fixed treatment table, and radiation is directed towards the patient’s tumor from multiple angles, as the linear accelerator gantry is free to rotate around the treatment table. Radiation can also be applied to close proximity of tissue using radioactive sources (termed brachytherapy). The wide array of radiotherapeutic approaches is outlined in Table 2. In some circumstances, advanced technology has led to the ability to treat tumors with a more favorable therapeutic ratio. The applicability of a new radiation technologies depends on several factors, including inherent tumor radioresponsiveness, susceptibility of adjacent normal tissue to radiation injury, and non-radiotherapeutic alternatives that are available for given cancer diagnoses.
Table 2.
Modalities in radiation therapy
| External beam Photons 2D, 3D, IMRT | External beam photons SRS, SBRT | External beam Particle therapy | Brachytherapy | |
|---|---|---|---|---|
| Common indications | Various types of cancer | Brain metastases Early stage lung cancer Oligometastatic disease |
Pediatric malignancies Uveal melanoma Skull base tumors |
Prostate cancer Cervical cancer |
| Illustrations |
The target volume (yellow) for anal cancer includes the anus and rectum, and lymph nodes in the mesorectum, internal and external iliac, and inguinal regions. The pelvic bones and bladder are adjacent to the target. With IMRT, the prescription isodose line (45 Gy, red) matches the shape of the target more closely, leading to improved sparing of normal tissue.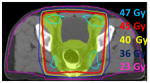 3D radiation: 45 Gy at 1.8 Gy/fx 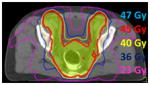 IMRT: 45 Gy at 1.8 Gy/fx |
Using specialized approaches to set up and deliver treatment, more aggressive doses can be safely delivered. Multiple beams angles are chosen to generate radiosurgery and stereotactic body radiation therapy plans, to treat localized targets in 1–5 days of treatment with ablative radiation doses.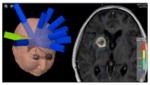 SRS for brain metastasis: 18 Gy in one fraction 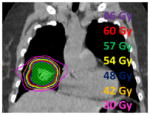 SBRT for lung cancer: 60 Gy at 12 Gy/fraction |
Proton therapy is distinguished from photon therapy by having a different dose distribution in tissue. In craniospinal radiation, proton therapy can reduce the amount of radiation which passes through the body, anterior to the target volume (the spinal column).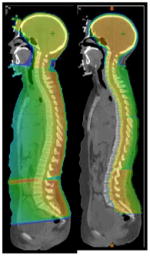 Craniospinal radiation: 36 Gy at 1.8 Gy/fraction |
Radioactive sources can be positioned within or near a target volume in order to limit radiation dose to surrounding tissue. Brachytherapy is an effective form of treatment for prostate cancer. Radioactive seeds are implanted using preloaded needles from a perineal approach, with transrectal ultrasound guidance.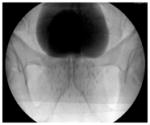 An x-ray after seed implantation of the prostate (contrast in the bladder, above the prostate)  Implant dosimetry: 145 Gy |
| Distinguishing features | Photon radiation delivered by linear accelerator Most widely available modality |
Photon radiation delivered by linear accelerator Steep dose gradients Heavy reliance on precise immobilization and pre- treatment imaging |
Proton radiation delivered by cyclotron Low integral radiation dose Higher cost for development and operation |
Varying radiation types deliver the dose Low integral radiation dose Candidacy dependent on anatomy and accessibility |
IMRT=intensity modulated radiation therapy; SBRT=stereotactic body radiation therapy; SRS=stereotactic radiosurgery.
Proton therapy illustration is courtesy of University of Florida Proton Therapy Institute.
Intensity-Modulated Radiation Therapy (IMRT)
Standard radiation planning is performed using axial CT imaging and 3D planning software, in which multiple beams are arranged to pass through the target volume. With IMRT, the intensity of each beam is modulated to provide a tighter approximation of high-dose radiation to the intended target, a characteristic known as conformality. IMRT planning utilizes an iterative computer algorithm approach which has input variables that specify dose coverage and normal tissue sparing goals, and output that defines the shape of the multiple beam “segments” which are delivered. This high degree of conformality is especially useful for irregularly shaped targets or disease in close proximity to critical structures which are sensitive to high doses of radiation (102).
Clinical use of IMRT has become widespread, and has enabled the escalation of dose and reduction of toxicity. IMRT has become fairly standard in the organ-sparing treatment of head and neck cancers with radiation therapy. The protection of parotid and submandibular salivary glands from radiation dose (sparing goal < 26 Gy) has led to improvements in quality of life through a reduction in treatment-related xerostomia (103)). IMRT improves the ability to treat irregularly shaped targets such as advanced nasopharynx tumors that can wrap around the brainstem or approximate the temporal lobes of the brain. Furthermore, since beam intensity can be modulated, IMRT can enable differential dosing within a singular volume to treat variable levels of disease risk. This technique of “dose painting” or “simultaneous integrated boost” planning offers the oncologist more latitude to individualize plans by tailoring dose to match the appropriate burden of disease ((104)). In tandem with concurrent chemotherapy (often including 5-FU or cisplatin) and altered fractionation (such as twice daily treatment with smaller fraction sizes), the therapeutic window can potentially be further widened (105).
Despite the many predicted advantages of IMRT, there are only a few instances in which IMRT has been proven to reduce morbidity compared to conventional RT in a randomized setting(106), and potential disadvantages exist. Related to the conformality of IMRT planning, accurate treatment setup including immobilization becomes more critical to avoid under-dosing of the target volume. Target volumes that are subject to motion or deformation over the course of treatment require more advanced technology for daily target localization, and in some cases may be more appropriately treated with a 3D plan. Additionally, IMRT requires more resources to plan and deliver treatment, and it results in significantly more low-dose exposure to tissues outside the target area due to the increased number of beam angles and cumulative “beam on” time required to deliver IMRT. There exists some concern that the exposure of more normal tissue to low doses of radiation might increase the risk of normal tissue complications, specifically radiation-induced malignancy years after RT(107). These concerns, along with additional costs, explain why IMRT has not entirely supplanted more traditional forms of radiation therapy for all disease sites.
Imaged guided radiation therapy (IGRT), Hypofractionated therapy, and ablative therapy (SRS, SBRT)
Although CT-based RT planning provides a more precise ability to target radiation to soft tissue areas, inaccuracies in the reproducibility of daily set-up and organ motion (such as respiratory motion) force the radiation oncologist to treat a volume larger than the actual tumor. Advancements in technology have addressed both positional error and physiologic motion to allow this setup margin to be reduced. Linear accelerators are now commonly fitted with various on-board imaging devices, which can provide diagnostic quality kV x-rays or cone beam CT. Soft tissue targets which are not easily visible on an x-ray can potentially be implanted with radio-opaque “fiducial” markers to better guide treatment delivery with pre-treatment imaging(108). With the regular implementation of any of these imaging modalities, known as image guided radiation therapy (IGRT), positioning setup error can be reduced, thereby enabling the use of small treatment volumes. Finally, commercially available software/hardware packages have been developed to account for physiologic motion such as respiratory motion.
Improvements in treatment accuracy have inspired a paradigm shift within radiation oncology in favor of shorter treatment courses that utilize higher daily doses of radiation (termed hypofractionated RT). This reduction in overall treatment time offers favorable potential biological effects against tumors, in part because it counteracts the proliferation of tumor cell clones (termed accelerated repopulation) that can occur in tumors over the course of RT(109). Prostate cancer, which is commonly treated with external beam radiation over 8 weeks, is a model site to showcase the potential impact of IGRT and hypofractionation. Several randomized studies support the use of higher doses of radiation (such as 78 Gy compared to 70 Gy) to treat prostate cancer, although this improvement in disease control comes at the expense of heightened rectal toxicity (110) when more advanced technology is not used. These and other data suggest that the sigmoidal dose response curve (see illustration in Figure 1) for rectal complication is steep over the 70–78 Gy dose range, which limits the therapeutic window for 2D or 3D radiation. Although IMRT can be used to observe strict sparing goals on the normal tissues at risk, an 8 week RT course to treat a relatively small, mobile target area in the pelvis is particularly subject to interfraction and intrafraction setup uncertainty. Several image guided approaches are available, such as on-board cone beam CT, or intraprostatic gold markers which are easily seen on a pre-treatment kV x-ray. With accurate target localization, uncertainty margins can be reduced in order to better protect normal tissues. In addition, larger daily doses can be safely administered, thereby shortening treatment time. For prostate cancer, this may hold a particular biological advantage, related to the notion that the radiosensitivity of prostate cancer increases with higher daily doses of treatment more so than for other cancers (111). A randomized trial has completed accrual comparing standard fractionation (73.8 Gy over 41 days) with hypofractionation (70 Gy over 28 days), and results are maturing.
Extreme versions of hypofractionated RT include stereotactic radiosurgery (SRS) and stereotactic body radiation therapy (SBRT). SRS (112) has been an effective modality of treating primary brain tumors, brain metastases, and benign brain tumors using a single large ablative dose of radiation. The accurate targeting of intracranial lesions traditionally requires a fixed immobilization system, in which a metal frame is affixed to the patient’s skull to maximally reduce setup uncertainty. SBRT is “an external beam RT method used to very precisely deliver a high dose of radiation to an extra-cranial target within the body, using either a single dose or a small number of fractions” (113). While SBRT is a relatively new technology, it has become more commonly used following the widespread implementation of IGRT and IMRT. The use of stereotaxy, which implies the use of a precise coordinate system, has been relaxed somewhat for SBRT in favor of on-board image guidance, in part due to the difficulties in establishing a rigid system of immobilization for extra-cranial targets. SRS and SBRT rely on the concept that improved accuracy of delivery enables the use of smaller target volumes, which in turn expose smaller volumes of normal tissues to radiation. Clinical outcomes have been favorable across multiple disease sites. For example, local control rates with a single treatment of 20 Gy for small brain metastases, or 20 Gy × 3 for liver metastases have been shown to result in local control rates of >80–85%. These high local control rates are appreciably higher than those observed with conventional fractionation schedules and compare favorably to surgical resection data. In the case of non-small cell lung cancer, the traditional approach of 60–66 Gy over six weeks with concurrent chemotherapy yields median survival times of 14–18 months for patients with non-metastatic, locally advanced disease. Local failure with this approach is unfortunately common, and therapy must be tailored to minimize the dose to normal lung, in order to reduce the risk of radiation pneumonitis (118). SBRT in this setting could be expected to alter the risk benefit ratio, particularly for cases in which disease is still localized. Indeed, short courses of therapy (3–5 fractions of 12–20 Gy) have resulted in high local control rates and low radiation pneumonitis risks in select early-stage tumors (114). This approach had initially been reserved for patients that were medically unfit to undergo surgery; however the very favorable outcomes have prompted clinical trials to directly compare SBRT and surgical resection. While these technical advances have clearly translated to clinical progress for early stage cancers, the same has not been so in locally-advanced disease. Techniques to account for tumor localization and respiratory motion have facilitated the use of higher doses, however, a recent trial of 74 Gy offered no survival advantage compared with the traditional 60 Gy dosing (115). This disappointing observation suggests that future improvements in locally-advanced non-small cell lung cancer may depend on new biologic-based options.
Whereas the development of IMRT after 3D-CRT could be considered evolutionary, in that IMRT was an incremental advancement in planning which arose from hardware (multi-leaf collimator) and software (inverse-planning) innovations, perhaps the implementation of SRS and SBRT could be considered more revolutionary. IMRT is typically utilized to achieve maximal conformality of the high-dose region, especially in cases where the target volume is irregularly shaped. On the other hand, SBRT typically prioritizes the steepness of the dose gradient between the target area and adjacent critical normal structures. The use of multiple beam angles, sometimes arranged with the treatment table rotated off axis, can help position this dose gradient in a favorable location. SBRT techniques allow for ‘hot-spots’ of increased dose within the target volume, which can significantly exceed the intended prescription dose; in certain cases the hot spot within the target can be >50% the prescription dose. This dose inhomogeneity further contributes to the steepness of the dose gradient. Notably, IMRT and SBRT techniques are not mutually exclusive – IMRT techniques can be used to deliver more favorable and homogeneous dose distributions. However, a combined approach comes at the expense of added complexity of planning and delivery, and an increase in treatment time could compromise the reproducibility of setup due to infraction patient movement.
The success of hypofractionated, image-guided radiation therapy to date has challenged conventional paradigms in radiation oncology. SBRT and SRS shift priority towards tumor ablation rather than normal tissue preservation, along the same lines of a surgical approach. As such, the emphasis of hypofractionated RT is increasingly focused on local rather than local-regional therapy. Clinical data to date have provided reason for cautious optimism, given the high rates of local control and reasonably low rates of severe toxicity that have been reported (116). However, the best use of SBRT remains to be defined. Perhaps most importantly, appropriate dose limits to normal tissues need to be redefined with clinical outcome data, given that the bulk of the existing dose-volume analyses of normal tissue complications are derived from the conventional fractionation era (117). It is important to note that only a small subset of tumors is amenable to SBRT. Factors such as tumor volume (118), method of daily localization, and proximity and nature of adjacent normal tissue (119) may play critical roles in determining whether SBRT or traditional fractionated radiotherapy techniques are more appropriate.
Brachytherapy
Brachytherapy represents a modality of treatment which could potentially outperform external beam RT for particular clinical situations. By varying the strength of the radioactive sources and manipulating the location and exposure time, it is possible to achieve highly conformal and dose-intense radiation distributions within tumors, while generating relatively low exposures to adjacent normal organs. Source placement can be interstitial, in which sources are placed directly into the target tissue. Alternatively in intra-cavitary brachytherapy, sources are placed in a space next to the target tissue, such as a body cavity or a body lumen. Due to the direct visualization of sources within the treatment site, concerns with setup uncertainty and organ motion are minimal.
Despite the dosimetric advantages of brachytherapy, this modality of radiotherapy is not used as commonly as external beam radiation. This is because only a small subset of cases present with well-defined areas of risk, which can be accessed in a relatively non-invasive fashion. Given this practical restriction, cancers of the cervix, prostate, breast, and skin are the most common sites to be treated with brachytherapy. In some situations, brachytherapy is combined with a course of external beam radiation therapy, which precedes, follows, or interdigitates with brachytherapy. Cervical cancer, for example, may include a course of conventional fractionation radiation and concurrent chemotherapy, in order to treat the pelvic lymph nodes, cervical parametria, and cervix over six weeks of daily treatment. Additional intra-cavitary brachytherapy procedures are done to deliver dose to the primary cervical tumor. In prostate cancer, brachytherapy presents an alternative approach to the narrow therapeutic window. Some studies suggest that disease outcomes after brachytherapy are superior to those after dose-escalated external beam RT (120), although this hypothesis has not been formally tested. It should be noted that many anatomic locations are not suitable for brachytherapy, such that the therapeutic ratio for external beam radiation exceeds that of brachytherapy. Additionally, the high doses delivered by brachytherapy carry the potential risk of increased morbidity if sources are misplaced or migrate (121), so a certain level of technical proficiency is required.
Particle therapy
Particle therapy (122)is a form of external beam radiotherapy in which particles are delivered rather than x-rays. Beams of electrons, protons, carbon ions, or neutrons are generated by a particle accelerator and delivered to the target volume. The use of proton therapy in particular has grown in recent years, because of its unique interaction in tissue. The underlying physics is characterized by a dose distribution known as the Bragg peak, whereby radiation is deposited immediately before the proton comes to rest in tissue, at a depth that can be manipulated by varying its energy. Photons, by comparison, deposit a trail of in-transit dose as the x-ray beams enter and exit the body, typically requiring multiple beams to be arranged so that their paths intersect over the target area. This exposure of tissue within each beam path can have negative clinical consequences for normal tissues. In clinical practice, to treat an entire tumor at a given depth, protons of different energies must be used. This “spread out bragg peak” offers an improved ability to minimize radiation exposure to normal tissues situated adjacent to the target volume, especially for tissues behind or deeper to the tumor. These reductions in total body exposure (termed integral dose) can translate to reduced complication risks, including the risk of radiation-induced malignancies. Proton-beam therapy has had a rapidly growing role in pediatric malignancies, and particularly pediatric brain tumors. Tumors located near the spine, like chordomas or para-spinal chondrosarcomas, are a well-defined indication for proton beam therapy, since it allows these relatively radioresistant tumors to be aggressively treated while respecting the radiation tolerance of the spinal cord (123).
Another attribute that distinguishes particle therapies from x-ray beams is that some types of particle beams generate higher biologic effectiveness per unit of energy deposited. For example, the relative biologic effectiveness of protons is 1–1.2 times that of photons, whereas for neutrons or carbon ions it could be 4–10 times that of photons. The difference in biologic effect is related to the rate at which energy is deposited in tissue, which is a function of both mass and charge of the radiation. This radiobiological difference could decrease the dependency of treatment success on the oxygen levels and cell cycle phase of the tumor target. It is also possible that heavy ion particle therapy could have a more desirable effect on blood vessels that supply tumors. However, further research is necessary to determine whether these radiobiological differences will translate into a better therapeutic index relative to photon-based therapy. Neutron radiotherapy, for example, provided these theoretical advantages, but significant normal tissue damage has limited its clinical applicability.
A clear challenge to particle therapy is the high cost to build treatment centers, which require significantly more physical space and highly-trained support staff than do photon-based centers. As of 2012, the international Particle Therapy Cooperative Group (124)reported that only 33 centers are using proton therapy and only 6 centers are using carbon ion therapy worldwide. Between 1969 and 2011, particle therapy has compromised <1% of external beam radiation treatment. This may change in the future, however, since there are currently plans to build 25 more particle facilities worldwide within the next 2 years.
V. Conceptual and therapeutic bottlenecks, and future directions to improve radiotherapy
Radioresistance prevents local control of some tumor types
In tumors like glioblastoma, the probability of local tumor control remains miserably poor, despite the use of advanced radiotherapy technologies and very high doses of radiotherapy. This has been well illustrated by unsuccessful trials of dose escalation, including one study increasing doses to >200 Gy using brachytherapy (125). Many possible mechanisms have been proposed for this intrinsic treatment resistance, including prevalence of radioresistant tumor stem cell clones, hypoxia, and uncontrolled signaling by growth factor receptors and/or interferons. While some of these hypotheses may prove to be correct, the current supporting data are not compelling and a complete explanation is likely to be much more complex. For example, unknown mechanisms may stimulate repopulation of malignant clones to brain tumors from sanctuary sites, which are located outside the irradiated volume. Another example of unyielding radioresistance is observed in very large tumors, like the typical presentation of locally-advanced lung cancer. A recent trial for non-small cell lung cancer showed that radiation dose escalation by almost 25% above the standard dose offered no measurable improvement to patients (115). One probable explanation is that the tumor cell burden is simply too large to be controlled in these tumors, such that high enough radiation doses cannot be achieved without incurring major complications to adjacent normal tissues. Another likely explanation is that larger tumors have greater genomic heterogeneity, and hence a greater likelihood for harboring treatment-resistant clones.
Avenues to overcome this resistance will require a better understanding of the underlying biological problems. They will likely also require a better arsenal of drugs that can specifically sensitize tumor cells or protect normal tissues. It is critical to widen the therapeutic window for radiotherapy at the biological level, particularly in situations where the physical and technical advances could be nearing a plateau. Some of the newer drug approaches discussed earlier may hold promise in this respect, particularly if issues related to drug pharmacokinetics and timing of administration relative to radiation are solved. However, we are left with the impression that there are many uncoordinated and competing research efforts. This probably occurs in part because radiation oncology is a relatively small field, relative to the much larger field of cancer biology research. Relatively few radiotherapy-modifying agents have progressed to testing in clinical trials, and the interest by large drug companies to invest in this class of agents appears somewhat underwhelming. Most of the current trials testing radiosensitizers involve HDAC and PARP inhibitors, neither of which was developed with this specific use in mind.
Uncontrolled systemic disease can overshadow the importance of local control
For a malignancy to be cured, it must be controlled at both the local and systemic levels. However, the ability to prevent distant metastases remains an unmet challenge in many disease types, such as lung and pancreatic cancers. As systemic chemotherapies continue to improve outcomes in these diseases, the previously unnoticed importance of local control can become more obvious. Recent studies in breast cancer demonstrate this inter-dependent relationship well. Older studies showed that chest wall radiotherapy improved the regional control of locally advanced breast cancers after mastectomy; these improvements were often clinically dismissed since they did not translate to improvements in overall survival. However, more recent studies done in the context of more effective systemic chemotherapy have shown that post mastectomy chest wall radiation therapy does improve survival (126, 127). In fact, the magnitude of survival benefit from radiotherapy is similar or greater than that of chemotherapy. This highlights the fact that uncontrolled systemic disease can overshadow the importance of local control, such that oncologists may view local management as secondary. This, in turn, can reduce enthusiasm for improving local/regional modalities like radiotherapy.
An exciting new development is the use of SBRT to ablate sites of metastatic disease, which represents a 180° shift in paradigm. It is based on the observation that some cancers present with only a few sites of metastatic disease, a condition termed oligometastases (128),(129) It remains unclear which patients will benefit the most from SBRT to metastases. However there is evidence for the concept, based on surgical series that achieved cures after resection of limited liver or lung metastases in patients with colon cancer and sarcomas, respectively. It is probably unrealistic to believe that local therapies will ever be able to address micrometastases, so SBRT for metastases and systemic chemotherapies should continue to evolve together. Nonetheless, impressive outcomes have been reported for SBRT in this metastatic setting. One unmet challenge is the ability to determine which tumors will behave in an oligometastatic manner, and which are likely to develop widespread metastases. Advances in imaging would be helpful in this regard, so that oligometastases can be detected and treated when the disease burden is lowest. Recent microRNA signatures have begun to identify patients with oligometastasis who remain oligometastatic, and this might be useful to select optimal candidates for curative surgery or radiotherapy (130). Another exciting idea is the use of SBRT to augment anti-tumor immunotherapy. Although clinical data are limited (78) and this approach might be limited to particular cancers, this relatively unexplored strategy has a huge potential payoff if it is successful.
A more individualized approach is needed in radiotherapy
Treatment guidelines are available for most types of cancer, such as those put forth by the National Comprehensive Cancer Network(131). In most cases, the decision to deliver radiation and the key parameters of radiation (e.g. dose and volume) are primarily based on the stage of disease at presentation. Management decisions tend not to be personalized towards individual biology or sensitivity to treatment. As an example, locally advanced rectal cancer is typically treated with radiosensitizing chemotherapy and 50.4 Gy, followed by total mesorectal excision, and then adjuvant chemotherapy. Although 10–20% of tumors are essentially sterilized by the initial preoperative chemoradiotherapy, almost all patients complete the pre-planned course of therapy including surgical excision and full-dose adjuvant chemotherapy. If, however, the treatment-sensitive tumors could be identified early, this information could be used to guide subsequent management. For example, systemic therapy could possibly be de-intensified and perhaps surgical resection could be eliminated for some patients. By contrast, patients found to have treatment-refractory tumors could be offered even more intensive therapies.
The definition of such predictive markers remains yet another unmet challenge for many cancer types, but some promising leads are beginning to emerge. One example is carcinoma for the head and neck treated with chemoradiotherapy, where the presence of human papilloma virus (HPV) carries a favorable prognosis (132). An ongoing national trial by the RTOG is testing whether the intensity of chemoradiotherapy can be safely reduced in this sub-population. To move forward as a field, it will be important to start focusing on biomarkers that predict responsiveness to specific therapies, as opposed to simple prognostic markers. This concept has already been realized for some chemotherapies, like KRAS mutation status for predicting EGFR inhibitor sensitivity (133) or MGMT gene silencing for predicting temozolomide sensitivity (134). Similar markers that predict sensitivity to radiation are being developed, but these have not yet found widespread clinical use. Such innovations will help transition radiotherapy away from a ‘one-size fits all’ mentality and allow us to start the inevitable transition to personalized medicine.
The allocation of radiotherapy resources needs to be evidence based
Advances in radiation therapy generally involve biologic or technologic innovations, some of which will improve cancer cure rates or decrease the risk of normal tissue complication. In a perfect world, these advances would be developed with a rigorous scientific approach, designed to overcome the most stubborn clinical challenges, tested in objective clinical trials, and then allocated to the public in an evidence-based manner. However, this is not always how innovations are developed in a free market, and there are obvious practical challenges associated with delivering health care to large populations. Most of the innovations in radiation oncology over the past decade have come in the form of physics-based technology, much more so than biology. This explosion in new radiation delivery devices has been promoted in large part by for-profit companies that develop new products and directly market them to physicians and patients. For example, one radiation delivery product is advertised on highway billboards claiming to be “New hope for the toughest tumors” and “The Ultimate Cancer-Fighting Machine.” In some cases, but not all, these advances (e.g. IMRT, IGRT, and SBRT) have directly addressed unmet needs and benefit a broad range of cancer types. For other new modalities, the clinical indications remain unclear and the superiority over standard radiotherapy remains unproven.
This issue will pose an inevitable challenge in coming years, especially given the anticipated changes in health care policy on the national level in the US. Allocation of heath care dollars will increasingly require providers to justify the use of expensive treatments. As an example, proton-beam therapy may prove to be a major advance for treating some cancer types, such as pediatric brain tumors where this technology can reduce exposure to normal surrounding tissue of developing children. However, proton-beam therapy is also widely marketed to men with prostate cancer, with some advertisements claiming that it carries no risk for causing erectile dysfunction – an improbable assertion based on the lack of supportive biologic or dosimetric advantages. Though the cost of proton beam far exceeds that of IMRT in prostate cancer, its purported clinical advantage is unproven and fiercely debated (135). IMRT and protons will be prospectively compared by a multi-institutional clinical trial. However, this case highlights the importance of well-designed, multi-disciplinary clinical trials. The consolidation and integration of US cooperative groups (as opposed to specialty-specific cooperative groups) may also help in this regard, with the successful model of the Children’s Oncology Group for pediatric cancers to serve as a role model. This type of objective evidence-based decision making will elevate radiation oncology, as well as the field of oncology as a whole.
Acknowledgments
The authors apologize to colleagues whose contributions could not be included in this review due to space constraints.
Funding: This work was supported by funding from the National Institutes of Health [CA142642-02 2010–2015 (PPC), P50 CA090386 2001–2014 (RRW and SLL)], P01 CA071933 (RRW), the Ludwig Center for Metastasis Research, the Center for Radiation Therapy, the Chicago Tumor Institute, a Young Investigator Award from the Prostate Cancer Foundation (SLL), and a generous gift from The Foglia foundation.
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Conflicts of interest: SL and PC declare no competing interests. RRW has a commercial organizational interest in: Catherex, Magi, RefleXion, and Oncosenescence. He has consulted for Medimmune ($1,500 one-time consultant fee) and has a $100,000 grant to study the effects IL-6 neutralizing antibodies on radiation antitumor effects.
References and Notes
- 1.Connell PP, Hellman S. Cancer Res. 2009 Jan 15;69:383. doi: 10.1158/0008-5472.CAN-07-6871. [DOI] [PubMed] [Google Scholar]
- 2.Thompson LH, Schild D. Mutat Res. 2001;477:131. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- 3.Thompson LH, Schild D. Mutat Res. 2002 Nov 30;509:49. doi: 10.1016/s0027-5107(02)00224-5. [DOI] [PubMed] [Google Scholar]
- 4.Ma Y, Pannicke U, Schwarz K, Lieber MR. Cell. 2002;108:781. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 5.Dupre A, et al. Nat Chem Biol. 2008 Feb;4:119. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rass E, et al. Nat Struct Mol Biol. 2009 Aug;16:819. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 7.Finlay MR, Griffin RJ. Bioorg Med Chem Lett. 2012 Jul 1; doi: 10.1016/j.bmcl.2012.06.053. [DOI] [PubMed] [Google Scholar]
- 8.Hickson I, et al. Cancer Res. 2004 Dec 15;64:9152. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 9.Budke B, et al. Nucleic Acids Res. 2012 May 9; [Google Scholar]
- 10.Klein HL. DNA Repair (Amst) 2008 May 3;7:686. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hine CM, Seluanov A, Gorbunova V. Proc Natl Acad Sci U S A. 2008 Dec 30;105:20810. doi: 10.1073/pnas.0807990106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell JS, et al. Cancer Res. 2003 Nov 1;63:7377. [PubMed] [Google Scholar]
- 13.Ito M, et al. J Gene Med. 2005 Mar 8;7:1044. doi: 10.1002/jgm.753. [DOI] [PubMed] [Google Scholar]
- 14.Quanz M, et al. Clin Cancer Res. 2009 Feb 15;15:1308. doi: 10.1158/1078-0432.CCR-08-2108. [DOI] [PubMed] [Google Scholar]
- 15.Bryant HE, et al. Nature. 2005 Apr 14;434:913. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 16.Farmer H, et al. Nature. 2005 Apr 14;434:917. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 17.Calabrese CR, et al. Clin Cancer Res. 2003 Jul;9:2711. [PubMed] [Google Scholar]
- 18.Chalmers AJ, Lakshman M, Chan N, Bristow RG. Semin Radiat Oncol. 2010 Oct;20:274. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Powell C, et al. Cancer Treat Rev. 2010 Nov;36:566. doi: 10.1016/j.ctrv.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Horton JK, Wilson SH. DNA Repair (Amst) 2007 Apr 1;6:530. doi: 10.1016/j.dnarep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan N, et al. Cancer Res. 2010 Oct 15;70:8045. doi: 10.1158/0008-5472.CAN-10-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munshi A, et al. Clin Cancer Res. 2005 Jul 1;11:4912. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 23.Zhang F, et al. Cancer Biol Ther. 2009 May;8:823. doi: 10.4161/cbt.8.9.8143. [DOI] [PubMed] [Google Scholar]
- 24.Ree AH, et al. Lancet Oncol. 2010 May;11:459. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 25.Bao S, et al. Nature. 2006 Dec 7;444:756. [Google Scholar]
- 26.Eriksson D, Stigbrand T. Tumour Biol. 2010 Aug;31:363. doi: 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 27.Greenberger JS. In Vivo. 2009 Mar-Apr;23:323. [PMC free article] [PubMed] [Google Scholar]
- 28.Dent P, et al. Radiat Res. 2003 Mar;159:283. doi: 10.1667/0033-7587(2003)159[0283:sariao]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 29.Hallahan DE, Haimovitz-Friedman A, Kufe DW, Fuks Z, Weichselbaum RR. Important Adv Oncol. 1993;71 [PubMed] [Google Scholar]
- 30.Kolesnick R, Fuks Z. Oncogene. 2003 Sep 1;22:5897. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- 31.Linkous AG, Yazlovitskaya EM. Anticancer Res. 2012 Jul;32:2487. [PubMed] [Google Scholar]
- 32.Bonner JA, et al. Lancet Oncol. 2010 Jan;11:21. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 33.Mothersill C, Seymour C. Radiat Res. 2001 Jun;155:759. doi: 10.1667/0033-7587(2001)155[0759:ribeph]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 34.Turesson I, et al. Acta Oncol. 2003;42:92. doi: 10.1080/02841860310004959. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Barros M, et al. Science. 2003 May 16;300:1155. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, et al. Nature. 2012 Aug 23;488:522. [Google Scholar]
- 37.Beck B, et al. Nature. 2012 Oct 20;478:399. [Google Scholar]
- 38.Schepers AG, et al. Science. 2012 Aug 10;337:730. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 39.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Nature. 2011 Jul 7;475:53. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 40.Al-Assar O, et al. Cancer Biol Ther. 2011 Jun 15;11:1028. doi: 10.4161/cbt.11.12.15699. [DOI] [PubMed] [Google Scholar]
- 41.Gupta PB, et al. Cell. 2009 Aug 21;138:645. [Google Scholar]
- 42.Sachlos E, et al. Cell. 2012 Jun 8;149:1284. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 43.Somaiah N, et al. Clin Cancer Res. 2012 Aug 1; [Google Scholar]
- 44.Nanduri LS, et al. Radiother Oncol. 2011 Jun;99:367. doi: 10.1016/j.radonc.2011.05.085. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberg A, Knox S. Int J Radiat Oncol Biol Phys. 2006 Feb 1;64:343. doi: 10.1016/j.ijrobp.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Overgaard J, Horsman MR. Semin Radiat Oncol. 1996 Jan;6:10. doi: 10.1053/SRAO0060010. [DOI] [PubMed] [Google Scholar]
- 47.Janssens GO, et al. J Clin Oncol. 2012 May 20;30:1777. doi: 10.1200/JCO.2011.35.9315. [DOI] [PubMed] [Google Scholar]
- 48.Hoskin PJ, Rojas AM, Bentzen SM, Saunders MI. J Clin Oncol. 2010 Nov 20;28:4912. doi: 10.1200/JCO.2010.28.4950. [DOI] [PubMed] [Google Scholar]
- 49.Henke M, et al. Lancet. 2003 Oct 18;362:1255. doi: 10.1016/S0140-6736(03)14567-9. [DOI] [PubMed] [Google Scholar]
- 50.Choy H, et al. J Clin Oncol. 2005 Sep 1;23:5918. doi: 10.1200/JCO.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 51.Suh JH, et al. J Clin Oncol. 2006 Jan 1;24:106. doi: 10.1200/JCO.2004.00.1768. [DOI] [PubMed] [Google Scholar]
- 52.Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR. Lancet Oncol. 2005 Oct;6:757. doi: 10.1016/S1470-2045(05)70292-8. [DOI] [PubMed] [Google Scholar]
- 53.Brown JM, Wang LH. Anticancer Drug Des. 1998 Sep;13:529. [PubMed] [Google Scholar]
- 54.Rischin D, et al. J Clin Oncol. 2010 Jun 20;28:2989. [Google Scholar]
- 55.Williamson SK, et al. J Clin Oncol. 2005 Dec 20;23:9097. doi: 10.1200/JCO.2005.01.3771. [DOI] [PubMed] [Google Scholar]
- 56.Bindra RS, et al. Mol Cell Biol. 2004 Oct;24:8504. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mehta MP, et al. Int J Radiat Oncol Biol Phys. 2009 Mar 15;73:1069. doi: 10.1016/j.ijrobp.2008.05.068. [DOI] [PubMed] [Google Scholar]
- 58.Ito E, et al. Sci Transl Med. 2011 Jan 26;3:67ra7. doi: 10.1126/scitranslmed.3001922. [DOI] [PubMed] [Google Scholar]
- 59.Gupta VK, et al. Cancer J. 2002 Jan-Feb;8:47. [Google Scholar]
- 60.Gorski DH, et al. Cancer Res. 1999 Jul 15;59:3374. [PubMed] [Google Scholar]
- 61.Goel S, et al. Physiol Rev. 2011 Jul;91:1071. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kennecke H, et al. Eur J Cancer. 2012 Jan;48:37. doi: 10.1016/j.ejca.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 63.Spigel DR, et al. J Clin Oncol. 2009 Jan 1;28:43. [Google Scholar]
- 64.Lind JS, Senan S, Smit EF. J Clin Oncol. 2012 Mar 10;30:e104. doi: 10.1200/JCO.2011.38.4552. [DOI] [PubMed] [Google Scholar]
- 65.Crane CH, et al. J Clin Oncol. 2009 Sep 1;27:4096. doi: 10.1200/JCO.2009.21.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hallahan DE, et al. Nat Med. 1995;1:786. doi: 10.1038/nm0895-786. [DOI] [PubMed] [Google Scholar]
- 67.Weichselbaum RR, Kufe D. Cancer Gene Ther. 2009 Aug;16:609. doi: 10.1038/cgt.2009.37. [DOI] [PubMed] [Google Scholar]
- 68.Rainov NG. Hum Gene Ther. 2000 Nov 20;11:2389. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 69.Freytag SO, et al. Cancer Res. 2003 Nov 1;63:7497. [PubMed] [Google Scholar]
- 70.Harrington KJ, et al. Clin Cancer Res. 2010 Aug 1;16:4005. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- 71.Harrington KJ, et al. Clin Cancer Res. 2010 Jun 1;16:3067. doi: 10.1158/1078-0432.CCR-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kamrava M, Bernstein MB, Camphausen K, Hodge JW. Mol Biosyst. 2009 Nov;5:1262. doi: 10.1039/b911313b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Apetoh L, et al. Nat Med. 2007 Sep;13:1050. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 74.Lee Y, et al. Blood. 2009 Jul 16;114:589. [Google Scholar]
- 75.Gulley JL, et al. Clin Cancer Res. 2005 May 1;11:3353. doi: 10.1158/1078-0432.CCR-04-2062. [DOI] [PubMed] [Google Scholar]
- 76.Seung SK, et al. Sci Transl Med. 2012 Jun 6;4:137ra74. doi: 10.1126/scitranslmed.3003649. [DOI] [PubMed] [Google Scholar]
- 77.Ribas A. N Engl J Med. 2012 Jun 28;366:2517. doi: 10.1056/NEJMe1205943. [DOI] [PubMed] [Google Scholar]
- 78.Postow MA, et al. N Engl J Med. 2012 Mar 8;366:925. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mole RH. Br J Radiol. 1953 May;26:234. doi: 10.1259/0007-1285-26-305-234. [DOI] [PubMed] [Google Scholar]
- 80.Seetharam S, et al. Int J Oncol. 1999 Oct;15:769. doi: 10.3892/ijo.15.4.769. [DOI] [PubMed] [Google Scholar]
- 81.Demaria S, et al. Int J Radiat Oncol Biol Phys. 2004 Mar 1;58:862. doi: 10.1016/j.ijrobp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Meng Y, et al. Mol Ther. 2012 May;20:1046. doi: 10.1038/mt.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuhas JM, Yurconic M, Kligerman MM, West G, Peterson DF. Radiat Res. 1977;70:433. [PubMed] [Google Scholar]
- 84.Brizel DM, et al. J Clin Oncol. 2000 Oct 1;18:3339. doi: 10.1200/JCO.2000.18.19.3339. [DOI] [PubMed] [Google Scholar]
- 85.Movsas B, et al. J Clin Oncol. 2005 Apr 1;23:2145. doi: 10.1200/JCO.2005.07.167. [DOI] [PubMed] [Google Scholar]
- 86.Mell LK, et al. Int J Radiat Oncol Biol Phys. 2007 May 1;68:111. [Google Scholar]
- 87.Johnson SM, et al. J Clin Invest. 2010 Jul;120:2528. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Komarov PG, et al. Science. 1999 Sep 10;285:1733. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 89.Thotala DK, Hallahan DE, Yazlovitskaya EM. Cancer Res. 2008 Jul 15;68:5859. doi: 10.1158/0008-5472.CAN-07-6327. [DOI] [PubMed] [Google Scholar]
- 90.Rotolo J, et al. J Clin Invest. 2012 May 1;122:1786. doi: 10.1172/JCI59920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Atkinson J, et al. Nat Commun. 2011;2:497. doi: 10.1038/ncomms1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ozturk B, Egehan I, Atavci S, Kitapci M. Int J Radiat Oncol Biol Phys. 2004 Jan 1;58:213. doi: 10.1016/s0360-3016(03)01444-5. [DOI] [PubMed] [Google Scholar]
- 93.Cohen EP, et al. Int J Radiat Oncol Biol Phys. 2012 May 1;83:292. doi: 10.1016/j.ijrobp.2011.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Le QT, et al. J Clin Oncol. 2011 Jul 10;29:2808. doi: 10.1200/JCO.2010.32.4095. [DOI] [PubMed] [Google Scholar]
- 95.Levin VA, et al. Int J Radiat Oncol Biol Phys. 2011 Apr 1;79:1487. doi: 10.1016/j.ijrobp.2009.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eschrich S, et al. Int J Radiat Oncol Biol Phys. 2009 Oct 1;75:497. doi: 10.1016/j.ijrobp.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eschrich SA, et al. Clin Cancer Res. 2012 Sep 4; [Google Scholar]
- 98.Weichselbaum RR, et al. Proc Natl Acad Sci U S A. 2008 Nov 25;105:18490. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ishkanian AS, Zafarana G, Thoms J, Bristow RG. Acta Oncol. 2010 Oct;49:888. doi: 10.3109/0284186X.2010.499371. [DOI] [PubMed] [Google Scholar]
- 100.Locke JA, et al. Clin Cancer Res. 2012 Jan 1;18:308. doi: 10.1158/1078-0432.CCR-11-2147. [DOI] [PubMed] [Google Scholar]
- 101.Gerlinger M, et al. N Engl J Med. 2012 Mar 8;366:883. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bortfeld T. Phys Med Biol. 2006 Jul 7;51:R363. doi: 10.1088/0031-9155/51/13/R21. [DOI] [PubMed] [Google Scholar]
- 103.Lin A, et al. Int J Radiat Oncol Biol Phys. 2003 Sep 1;57:61. doi: 10.1016/s0360-3016(03)00361-4. [DOI] [PubMed] [Google Scholar]
- 104.de Arruda FF, et al. Int J Radiat Oncol Biol Phys. 2006 Feb 1;64:363. doi: 10.1016/j.ijrobp.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 105.Konski AA, Winter K, Cole BF, Ang KK, Fu KK. Head Neck. 2009 Feb;31:207. doi: 10.1002/hed.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Donovan E, et al. Radiother Oncol. 2007 Mar;82:254. doi: 10.1016/j.radonc.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 107.Hall EJ. Int J Radiat Oncol Biol Phys. 2006 May 1;65:1. doi: 10.1016/j.ijrobp.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 108.Shirato H, et al. Int J Radiat Oncol Biol Phys. 2003 May 1;56:240. doi: 10.1016/s0360-3016(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 109.Schmidt-Ullrich RK, et al. Radiat Oncol Investig. 1999;7:321. doi: 10.1002/(SICI)1520-6823(1999)7:6<321::AID-ROI2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 110.Kuban DA, et al. Int J Radiat Oncol Biol Phys. 2008 Jan 1;70:67. doi: 10.1016/j.ijrobp.2007.06.054. [DOI] [PubMed] [Google Scholar]
- 111.Miles EF, Lee WR. Semin Radiat Oncol. 2008 Jan;18:41. doi: 10.1016/j.semradonc.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 112.Leksell L. Acta Chir Scand. 1951 Dec 13;102:316. [PubMed] [Google Scholar]
- 113.Potters L, et al. Int J Radiat Oncol Biol Phys. 2010 Feb 1;76:326. doi: 10.1016/j.ijrobp.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 114.Grills IS, et al. J Clin Oncol. 2010 Feb 20;28:928. doi: 10.1200/JCO.2009.25.0928. [DOI] [PubMed] [Google Scholar]
- 115.Bradley PRJ, Komaki R, et al. [Google Scholar]
- 116.Lo SS, et al. Nat Rev Clin Oncol. 2010 Jan;7:44. doi: 10.1038/nrclinonc.2009.188. [DOI] [PubMed] [Google Scholar]
- 117.Marks LB, et al. Int J Radiat Oncol Biol Phys. 2010 Mar 1;76:S10. doi: 10.1016/j.ijrobp.2009.07.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hoyer M, et al. Radiother Oncol. 2005 Jul;76:48. doi: 10.1016/j.radonc.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 119.Timmerman R, et al. J Clin Oncol. 2006 Oct 20;24:4833. doi: 10.1200/JCO.2006.07.5937. [DOI] [PubMed] [Google Scholar]
- 120.Jabbari S, et al. Int J Radiat Oncol Biol Phys. 2010 Jan 1;76:36. doi: 10.1016/j.ijrobp.2009.01.029. [DOI] [PubMed] [Google Scholar]
- 121.Bogdanich W. The New York Times. Vol. 2012 New York City: Jun 20, 2009. [Google Scholar]
- 122.Durante M, Loeffler JS. Nat Rev Clin Oncol. 2010 Jan;7:37. doi: 10.1038/nrclinonc.2009.183. [DOI] [PubMed] [Google Scholar]
- 123.Schulz-Ertner D, Tsujii H. J Clin Oncol. 2007 Mar 10;25:953. doi: 10.1200/JCO.2006.09.7816. [DOI] [PubMed] [Google Scholar]
- 124.P. T. C.-O. Group. PTCOG. Vol. 2012 New York City: Jun 6, 2012. [Google Scholar]
- 125.Chen AM, et al. Int J Radiat Oncol Biol Phys. 2007 Nov 1;69:825. doi: 10.1016/j.ijrobp.2007.03.061. [DOI] [PubMed] [Google Scholar]
- 126.Overgaard M, et al. N Engl J Med. 1997 Oct 2;337:949. doi: 10.1056/NEJM199710023371401. [DOI] [PubMed] [Google Scholar]
- 127.Ragaz J, et al. J Natl Cancer Inst. 2005 Jan 19;97:116. [Google Scholar]
- 128.Weichselbaum RR, Hellman S. Nat Rev Clin Oncol. 2011 Jun;8:378. doi: 10.1038/nrclinonc.2011.44. [DOI] [PubMed] [Google Scholar]
- 129.Hellman S, Weichselbaum RR. J Clin Oncol. 1995 Jan;13:8. doi: 10.1200/JCO.1995.13.1.8. [DOI] [PubMed] [Google Scholar]
- 130.Lussier YA, et al. PLoS One. 2011;6:e28650. doi: 10.1371/journal.pone.0028650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.NCCN. NCCN. Vol. 2012 Fort Washington, PA: 2012. [Google Scholar]
- 132.Ang KK, et al. N Engl J Med. 2010 Jul 1;363:24. [Google Scholar]
- 133.Lievre A, et al. Cancer Res. 2006 Apr 15;66:3992. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 134.Hegi ME, et al. N Engl J Med. 2005 Mar 10;352:997. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 135.Emanuel E. The New York Times. Vol. 2012 New York City: Jan 2, 2012. [Google Scholar]