

Abstract
Astrocytes have important physiological roles in CNS homeostasis, and serve as a bridge between the CNS and the immune system. IL-17 and IL-6 are important in many CNS disorders characterized by neuroinflammation. We examined the role of IL-17 on the IL-6 signaling cascade in primary astrocytes. IL-17 functioned in a synergistic manner with IL-6 to induce IL-6 expression in astrocytes. The synergistic effect involved numerous signaling pathways including NF-κB, JNK MAPK and p38 MAPK. The NF-κB pathway inhibitor BAY-11, JNK inhibitor JNKiII, and p38 inhibitor SB203580 suppressed the synergistic effect of IL-6 and IL-17 on IL-6 expression. IL-17 synergized with IL-6 to enhance the recruitment of activated NF-κB p65, c-Fos, c-Jun, and the histone acetyltransferases CBP and p300 to the IL-6 promoter in vivo to induce IL-6 transcription. This was accompanied by enhanced acetylation of Histones H3 and H4 on the IL-6 promoter. Moreover, we elucidated an important role for SOCS3 in IL-17 enhancement of IL-6 signaling in astrocytes. SOCS3 siRNA knockdown and SOCS3 deletion in astrocytes augmented the synergistic effect of IL-6 and IL-17, due to an enhancement of activation of the NF-κB and MAPK pathways. These results indicate that astrocytes can serve as a target of Th17 cells and IL-17 in the CNS, and SOCS3 participates in IL-17 functions in the CNS as a negative feedback regulator.
Introduction
Astrocytes are the major glial cell type within the CNS and are critical for CNS homeostasis (1-3). Astrocytes regulate neuronal function by releasing neurotrophic factors, guiding neuronal development, contributing to neurotransmitter metabolism, and participating in the formation of the blood-brain-barrier (4-6). Astrocytes can serve as a bridge between the CNS and the immune system (5). In particular, astrocytes produce a wide array of chemokines and cytokines that act as immune mediators in cooperation with those produced by microglia (2, 6). Aberrant expression of chemokines and cytokines accompanies CNS disorders such as Multiple Sclerosis (MS), Alzheimer's Disease, HIV-1-Associated Dementia, and brain injury/trauma (2). However, the mechanisms by which astrocytes contribute to these disorders remains unclear.
IL-6 is a regulator of inflammatory and immunological responses and acts on target cells through a receptor complex composed of an IL-6-binding subunit, the IL-6 receptor (IL-6R), and the signal transducing receptor gp130. Initiation of IL-6 signaling occurs when IL-6 binds to the IL-6R, leading to an association with gp130. This leads to activation of gp130-associated Janus Kinases (JAKs), and various signaling pathways such as JAK/STAT, MAPK, and NF-κB (7). The IL-6R is found in both membrane-bound and soluble (sIL-6R) forms (8). Astrocytes are the major source of IL-6 in CNS injury and neuroinflammation (9). We previously determined that the IL-6/sIL-6R complex plays an important role in regulation of IL-6 expression by astrocytes, particularly in conjunction with the proinflammatory cytokines TNF- α and IL-1β (10-12).
Th17 cells produce IL-17A (IL-17), IL-17F, IL-21 and IL-22, and are required for the induction of several autoimmune diseases, including collagen-induced arthritis, experimental autoimmune encephalomyelitis (EAE), and inflammatory bowel disease (13-17). Th17 cells are generated as a discrete lineage following priming in the presence of TGF-β and IL-6, and expansion in the presence of IL-23 and IL-1β (18-20). IL-6 not only functions upstream of IL-17 but also acts as a critical downstream target of IL-17, and IL-17 together with IL-6 triggers a positive-feedback loop of IL-6 expression through the activation of NF-κB and STAT-3 in fibroblasts (21). IL-17 signals through a heteromeric receptor complex consisting of IL-17 receptor A (IL-17RA) and IL-17RC, which are single-pass transmembrane proteins expressed by a variety of cells (22-25). Evidence indicates that NF-κB and MAPK pathways are involved in IL-17RA and IL-17RC signaling (26), however, little is known about its signaling in cells of the CNS.
Suppressor Of Cytokine Signaling (SOCS) proteins function in a negative feedback loop to terminate signal transduction through the JAK/STAT pathway (27). One member, SOCS3, is expressed by immune cells and cells of CNS, and regulates inflammatory cytokine and chemokine production, activation of microglia, macrophages and astrocytes, immune cell infiltration and autoimmunity (27). The predominant function of SOCS3 is inhibition of signaling by the IL-6 family of cytokines (28). However, SOCS3 exerts a much broader effect on immune responses by inhibiting signaling of additional mediators such as LPS, type I and type II IFNs, IL-2 and IL-12 (29-32). Furthermore, SOCS3 inhibits the NF-κB pathway (33), antagonizes cAMP-mediated signaling (34) and enhances signaling through the Ras pathway (35). We recently found that TGF-β inhibits IL-6- and IL-21-induced SOCS3 expression, thus enhancing as well as prolonging STAT-3 activation in naive CD4+CD25- T cells and promoting Th17 cell development (36). These results suggest that SOCS3 may participate in the regulation of Th17 cell differentiation and IL-17 functions.
An important question is how IL-17 contributes to autoimmune diseases and/or inflammation in the CNS. Thus, identification of the downstream targets of IL-17 in the CNS should advance our understanding of the mechanisms underlying IL-17-mediated autoimmune diseases. In this study, we demonstrate that astrocytes are a target of IL-17. IL-17 enhances the IL-6/sIL-6R (IL-6/R) signaling cascade and positive-feedback loop of IL-6 expression in astrocytes. SOCS3 participates in the regulation of IL-6/R plus IL-17 enhanced IL-6 expression in astrocytes, and plays an important role in down-regulating the IL-6 signaling cascade, indicating that SOCS3 participates in IL-17 functions in the CNS as a negative feedback regulator.
Materials and Methods
Recombinant Proteins and Reagents
Recombinant human IL-6, recombinant human soluble IL-6 Receptor (sIL-6R) and recombinant mouse IL-17 were from R&D Systems (Minneapolis, MN). Abs against phospho-p65 Ser536, p65, phospho-ERK1/2 Thr202/Tyr204, ERK1/2, phospho-JNK Thr183/Tyr185, JNK, phospho-p38 MAPK Thr180/Tyr182 and p38 were purchased from Cell Signaling Technology (Beverly, MA). Ab against GAPDH was from Abcam (Cambridge, MA), and Abs against CREB-binding protein (CBP), p300, c-Jun and c-Fos were from Santa Cruz Biotechnology (Santa Cruz, CA). Abs against acetylated histones H3 (AcH3) and H4 (AcH4) were from Upstate Biotechnology (Billerica, MA), whereas the Ab against RNA polymerase II (Pol II) was from Covance (Princeton, NJ). Actinomycin D was purchased from Sigma-Aldrich (St. Louis, MO). The pharmacological inhibitors for NF-κB (BAY-11), ERK (U0126), JNK (JNK inhibitor II), and p38 (SB203580) were purchased from Calbiochem Biochemicals (La Jolla, CA).
Mice
C57BL/6 wild-type (WT) mice were bred in the animal facility at the University of Alabama at Birmingham (Birmingham, AL). SOCS3 floxed transgenic mice (37) were the generous gift of Dr. Warren Alexander (Cancer and Haematology Division, The Walter and Eliza Hall Institute of Medical Research and the Cooperative Research Centre for Cellular Growth Factors), and were bred in the animal facility at the University of Alabama at Birmingham. All experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Primary Astrocyte Cultures and Cell Lines
Primary astrocytes cultures from C57BL/6 WT mice and SOCS3 floxed mice were established from neonatal cerebra as described previously (38). After 2 weeks in culture, oligodendrocytes and microglia were separated from astrocytes by mechanical dislodgment. Astrocytes were monitored for purity by immunofluorescence, and were routinely > 97% positive for glial fibrillary acidic protein. The murine macrophage cell line RAW264.7 was maintained as described previously (39).
Adenoviral-Cre-GFP Construct and In Vitro Excision of SOCS3
Adenovirus expressing GFP and Cre recombinase (GFP-Cre) or GFP alone under the control of CMV promoter was provided by Dr. Rosa Serra (University of Alabama at Birmingham, Birmingham, AL). For in vitro deletion of SOCS3 (37), astrocytes containing floxed SOCS3 alleles were cultured to 70% confluence and then, in the absence of serum, were infected with adenovirus-encoding GFP-Cre or GFP alone. After 4 h, culture medium containing 10% FBS was added, and the cells were allowed to recover for the next 48 h. Digestion of genomic DNA coupled with primers of the SOCS3 gene distinguishes full-length (fl) and excised alleles (Δ) (37). The following primers and cycling conditions were used to distinguish wild-type and excised SOCS3 alleles: 5′-TCT TGT GTC TCT CCC CAT CC-3′, 5′-ACG TCT GTG ATG CTT TGC TG-3′, 35 cycles at 55°C for 30 s, 72°C for 80 s and 94°C for 30 s. Greater than 90% SOCS3 deletion was confirmed by PCR for every experiment.
RNA Isolation, RT-PCR and TaqMan Gene Expression Assays
Total cellular RNA was isolated from unstimulated, IL-6/R, IL-17 or IL-6/R plus IL-17 treated astrocytes, and RT reactions were performed as previously described (36). The concentration and purity of total RNA was assessed using a NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). One μg of RNA was used to reverse transcribe mRNA into cDNA and subjected to PCR with primers specific for mouse IL-6 and GAPDH for 30 cycles of amplification. Primer sequences were as follows: IL-6 (sense), 5′-AGT TGC CTT CTT GGG ACT GA-3′; IL-6 (antisense), 5′-CAG AAT TGC CAT TGC ACA AC-3′; IL-17RA (sense), 5′-TCC AGT TTC TGT CCA TGC TG-3′; IL-17RA (antisense), 5′-GCT CAC GCA TGA GGT AGT CA -3′; IL-17RC (sense), 5′-GAG CTC AAC CTC ACA CAG CA-3′; IL-17RC (antisense), 5′-GCA GAA TTC GAC CCT CTC AG-3′; GAPDH (sense), 5′-AAC TTT GGC ATT GTG GAA GG-3′; and GAPDH (antisense), 5′- CCC TGT TGC TGT AGC CGT AT-3′. The ABI Prismo 7500 Sequence Detection System, TaqMan Gene Expression Master Mix and TaqMan Gene Expression Assay probes were from Applied Biosystems (Foster City, CA) and were used for quantitative real-time PCR (QRT-PCR) to determine the levels of IL-6 and SOCS3 mRNA. For the TaqMan method, each 20 μl reaction contained 1 μl of TaqMan Gene Expression Assay probes (5 μM), 10 μl of TaqMan Gene Expression Master Mix, and 9 μl of template cDNA. The reactions were performed with the following conditions: 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. The data were analyzed using the comparative Ct method to obtain relative quantitation value, as previously described (40).
Immunoblotting
Thirty μg of total cell lysate was separated by electrophoresis on 10% SDS-polyacrylamide gels and probed with antibodies against phospho-p65 p65,, phospho-ERK1/2, ERK1/2, phospho-p38 MAPK, p38, phospho-JNK MAPK, JNK or GAPDH as described previously (41).
ELISA
Supernatants were collected from unstimulated, IL-6/R, IL-17 or IL-6/R plus IL-17-stimulated astrocytes and assayed by ELISA for secretion of murine IL-6 (BioLegend, San Diego, CA). IL-6 levels were normalized to total protein levels.
RNA Interference
RNA interference was performed as previously described (38). DharmaFECT 1 small interfering RNA (siRNA) transfection reagent, SMARTpool siRNAs specific for murine SOCS3, and siCONTROL nontargeting siRNAs were purchased from Dharmacon. Primary astrocytes (5 × 105) in 6-well plates were transfected with 100 nM of siCONTROL or SOCS3 specific siRNAs using the DharmaFECT 1 reagent following the manufacturer's recommendations. After 48 h of transfection, levels of SOCS3 mRNA expression was determined by QRT-PCR.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP analysis was done following a protocol provided by Upstate Biotechnology with modifications as described previously (29, 39). Primary astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for up to 90 min, fixed with 1% formaldehyde for 15 min at room temperature, and nuclei were isolated. Chromatin was sheared by sonication and samples were precleared for 2 h at 4°C with salmon sperm DNA-saturated protein A/G Sepharose. Chromatin solutions were precipitated overnight at 4°C with 5 μg of Abs or isotype-matched control IgG. Input chromatin and immunoprecipitated chromatin were incubated at 65°C overnight to reverse crosslinks. After proteinase K digestion, DNA was extracted with the Qiagen Miniprep kit. Purified DNA was analyzed by PCR with Taq polymerase. The primer pair 5′- GCA CAC TTT CCC CTT CCT AGT TGT G-3′ and 5′-AGT CTA TCG TTC TTG GTG GGC TCC AGA G -3′ was used to amplify a 230-bp region in the mouse IL-6 promoter containing the distal AP-1 element and the NF-κB elements. Densitometry was used to quantify the PCR results, and all results were normalized by the respective input values.
Statistical Analysis
All experiments were repeated a minimum of three times. Levels of significance for comparison between samples were determined by the Student's t test distribution. All results are shown as mean ± Standard Deviation (mean ± SD). A value of p ≤ 0.05 was considered to be statistically significant.
Results
IL-17 Enhances IL-6-induced IL-6 Expression in Primary Astrocytes
We hypothesized that astrocytes are a target of Th17 cells and IL-17 in the CNS. IL-17RA and IL-17RC are expressed in most tissues examined to date, and functional IL-17RA is expressed in CNS glia, however, little is known about the expression of IL-17RC in glial cells (26, 42). We examined whether IL-17RA and IL-17RC genes are expressed in primary astrocytes, using RAW264.7 cells as a positive control. IL-17RA and IL-17RC mRNA was expressed in both astrocytes and RAW264.7 cells (Fig.1A). We have previously shown that IL-6, in conjunction with the sIL-6R, induces its own expression in astrocytes (10). We wished to determine the influence of IL-17 on this process. Astrocytes were stimulated with medium (UN), IL-6 plus sIL-6R (IL-6/R), IL-17 or IL-6/R plus IL-17 for up to 24 h, and expression of IL-6 mRNA analyzed by RT-PCR and QRT-PCR. IL-6 mRNA expression was induced at 2 h after the addition of IL-6/R (Fig.1B, lane 2), peaked at 4 h (lane 5), and then decreased slightly at 8 and 24 h (lanes 8 and 11). IL-17 alone modestly induced IL-6 expression (lanes 3, 6, 9 and 12), but when astrocytes were treated with both IL-6/R and IL-17, expression of IL-6 was significantly enhanced at all time points (Fig. 1B, lanes 4, 7, 10 and 13). To determine the effect of different concentrations of IL-17 on IL-6 expression, primary astrocytes were incubated with medium (UN), IL-6/R, different concentrations of IL-17 (1-50 ng/ml), or IL-6/R plus IL-17 for 4 h, and analyzed for IL-6 mRNA expression by QRT-PCR. IL-17 enhanced IL-6/R-induced IL-6 mRNA expression in a dose-dependent manner, with optimal induction using 25 ng/ml (Fig. 1C). Previously, we have shown that astrocytes express low levels of the IL-6R and require the addition of the sIL-6R for optimal IL-6-mediated responses (10). To address the necessity of sIL-6R in this response, astrocytes were treated with medium (UN), IL-6, sIL-6R or IL-17 in various combinations for 4 h, and levels of IL-6 mRNA expression determined by QRT-PCR. Stimulation with IL-6, sIL-6R or IL-17 alone induced a modest increase in IL-6 mRNA expression at 4 h (Fig. 1D). Addition of IL-6 plus sIL-6R increased IL-6 mRNA compared with IL-6 or sIL-6R alone. Inclusion of IL-6/R plus IL-17 induced a synergistic increase in IL-6 mRNA expression (Fig. 1D). These results confirm IL-17 enhancement of IL-6 induction, and also provide evidence that the sIL-6R is required to complex with IL-6 to initiate IL-6 expression. IL-6 protein expression was induced at 4 h after the addition of IL-6/R. IL-17 had a modest effect on IL-6 protein expression, but when added with IL-6/R, the expression of IL-6 protein was significantly enhanced at all time points (Fig. 1E). Taken together, these results indicate that IL-17 enhances IL-6/R-induced IL-6 expression at the mRNA and protein levels in astrocytes.
Figure 1. IL-17 Enhances IL-6/sIL-6R-mediated IL-6 Expression in Primary Astrocytes.

A, IL-17RA and IL-17RC mRNA expression was determined by RT-PCR in RAW264.7 cells (positive control), and primary astrocytes, in duplicate. B, Primary astrocytes were treated with medium (UN), IL-6/R (IL-6, 10 ng/ml and sIL-6R, 25 ng/ml), IL-17 (25 ng/ml) or IL-6/R plus IL-17 for up to 24 h, and levels of IL-6 and GAPDH mRNA expression were determined by RT-PCR and QRT-PCR. C, Primary astrocytes were treated with medium (UN), IL-6/R, different concentrations of IL-17 (1-50 ng/ml) or IL-6/R + IL-17 (1-50 ng/ml) for 4 h, and levels of IL-6 mRNA expression were determined by QRT-PCR. D, Primary astrocytes were treated with medium (UN), IL-6, sIL-6R or IL-17 in various combinations for 4 h, and levels of IL-6 mRNA expression were determined by QRT-PCR. E, Primary astrocytes were treated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 2 - 48 h, and supernatants were analyzed for IL-6 protein using ELISA. Experiments shown are representative of at least three experiments. All data are the mean ± SD of three experiments. *, p ≤0.05; **, p ≤0.01.
IL-17 Synergizes with IL-6 for Activation of the NF-κB and MAPK Pathways
Astrocytes and microglia express IL-17RA in vitro, and IL-17A treatment induces chemokine production by these cells (42). Although the signaling pathways for IL-17 are not completely understood, evidence indicates that the NF-κB and MAPK pathways can be activated upon IL-17 stimulation (26). We evaluated the effect of IL-6/R, IL-17, and IL-6/R plus IL-17 on the NF-κB, ERK, p38 and JNK MAPK pathways in astrocytes. IL-6/R or IL-17 treatment degraded IκBα and led to p65 serine phosphorylation, and IL-6/R plus IL-17 resulted in enhanced activation of NF-κB (Figs. 2A and 2B). IL-6/R or IL-17 alone led to rapid ERK1/2 phosphorylation at 15 and 30 min, and this phosphorylation was enhanced by treatment with IL-6/R plus IL-17 (Fig. 2C, lanes 4 and 7). IL-6/R activated p38 MAPK at 30 min (Fig. 2D, lane 2), which decreased at 60 min (lane 5). IL-17 activated p38 at 60 min (Fig. 2D, lane 6), and IL-6/R plus IL-17 enhanced p38 activation at 30 and 60 min (Fig. 2D, lanes 4 and 7). IL-6/R activated JNK MAPK signaling at 30 min (Fig. 2E, lane 2), while IL-17 alone had very little effect (lanes 3 and 6). The combination of IL-6/R and IL-17 enhanced activation of JNK at 30 and 60 min (Fig. 2E, lanes 4 and 7). These results indicate the IL-17 alone induces activation of the NF-κB, ERK1/2 and p38 pathways, and synergizes with IL-6/R for enhancement of NF-κB, ERK1/2, p38 and JNK signaling.
Figure 2. IL-17 Synergizes with IL-6/R for Activation of the NF-κB and MAPK Pathways.

A, Astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 15 and 30 min, and then cell lysates were immunoblotted with antibodies against IκBα and GAPDH. The basal level of the untreated sample was set at 100, and the percentage change of IκBα upon IL-6/R, IL-17 or IL-6/R + IL-17 treatment compared with the basal value. B, Astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 30 and 60 min, and then cell lysates were immunoblotted with antibodies against phospho-p65 Ser 536, p65 and GAPDH. C, Astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 15 and 30 min, and then cell lysates were immunoblotted with antibodies against phospho-ERK1/2, ERK1/2 and GAPDH. D, Astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R and IL-17 for 30 min and 60 min, and then cell lysates were immunoblotted with antibodies against phospho-p38, p38 and GAPDH. E, Astrocytes were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 30 and 60 min, and then cell lysates were immunoblotted with antibodies against phospho-JNK, JNK and GAPDH. The basal level of the untreated sample was set at 1.0 and fold activation upon IL-6/R, IL-17 or IL-6/R + IL-17 treatment was compared with that value (B, C, D and E). Representative of at least three experiments.
IL-17-enhanced IL-6 Gene Expression Involves the NF-κB, JNK MAPK, and p38 MAPK Pathways
Several reports have shown that IL-17 positively regulates cytokine gene expression by enhancing mRNA stability (43, 44). To determine the mechanism of IL-17-enhanced IL-6 gene expression, we analyzed the effect of IL-17 on IL-6 mRNA stability. The results shown in Fig. 3A demonstrate that the half-life of IL-6 mRNA induced by IL-6/R was approximately 90 min, and the half-life was unaffected by IL-17. These results indicate that the IL-17 effect is not mediated by stabilization of IL-6 mRNA. To evaluate the role of the NF-κB and MAPK pathways in IL-17-enhanced IL-6 expression, specific pharmacological inhibitors for NF-κB, ERK1/2, p38, and JNK were utilized. We have previously used the inhibitors, and have demonstrated their specificity of action (29, 39). Inclusion of the NF-κB inhibitor BAY-11 significantly suppressed IL-6/R plus IL-17 induction of IL-6 mRNA (Fig. 3B). Interestingly, the inclusion of ERK1/2 inhibitor U0126 did not affect IL-6/R plus IL-17 induction of IL-6 expression (Fig. 3B), suggesting that ERK1/2 is not involved in this response. Blockage of the p38 MAPK pathway with SB203580 or JNK MAPK with JNK inhibitor II partially inhibited the expression of IL-6 (Fig. 3C). Taken together with data presented in Fig. 2, these results indicate that in astrocytes, IL-6/R plus IL-17 induction of IL-6 gene expression occurs by a transcriptional mechanism that involves NF-κB, p38 and JNK signaling.
Figure 3. IL-17 and IL-6/R Induction of IL-6 Depends on NF-κB, p38 and JNK MAPK Activity.

A, Astrocytes were cultured in absence or presence of IL-17 followed by IL-6/R treatment for 4 h. Actinomycin D (5 ng/ml) was then added, and cells harvested at 0, 30, 60, 120, and 240 min after addition. The abundance of IL-6 mRNA was determined by QRT-PCR. B, C, DMSO vehicle, BAY 11 (5 μM), U0126 (10 μM), SB203580 (10 μM) or JNKi II (10 μM) were added to cultures 1 h before cytokine addition, and then astrocytes were incubated with medium, IL-6/R, IL-17 or IL-6/R plus IL-17 for 4 h. Levels of IL-6 mRNA expression were determined by QRT-PCR. All data are the mean ± SD of three experiments. **, p ≤ 0.01; NS = not significant.
Recruitment of Phospho-p65, c-Jun, c-Fos, RNA Pol II, and the Coactivators CBP and p300 to the IL-6 Promoter and Modification of H3 and H4 Acetylation in Response to IL-6/R and IL-17
We have shown that activation of the NF-κB, p38, and JNK MAPK pathways are involved in IL-6/R plus IL-17 induction of IL-6 gene transcription. However, it is still unknown whether NF-κB and the AP-1 transcription factors, which are downstream targets of p38 and JNK MAPK, are recruited to the IL-6 promoter, and how this is coupled to IL-6 gene transcription. To monitor transcription factor binding to the endogenous IL-6 promoter, astrocytes were incubated in medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 90 min and ChIP assays were performed. PCR analysis of the positive control (input) indicated that soluble chromatin samples obtained from each treatment had equal amounts of chromatin fragments containing the IL-6 promoter (Figs. 4A and 4B). NF-κB p65 was weakly associated with the IL-6 promoter in untreated astrocytes, and increased recruitment in response to IL-6/R, IL-17 or IL-6/R plus IL-17 was observed (Fig. 4A, lanes 1-4). We next examined the activated form of p65, phospho-p65 ser536 at the IL-6 promoter. NF-κB P-p65 ser536 was also weakly associated with the IL-6 promoter in untreated astrocytes, and increased binding was observed after IL-6/R or IL-17 treatment (lanes 1, 2 and 3). Upon treatment with IL-6/R plus IL-17, there was an additive effect on the recruitment of P-p65 ser536 to the IL-6 promoter (lane 4). c-Fos was not associated with the IL-6 promoter in untreated cells, while an increase in binding was observed after IL-6/R or IL-17 treatment (Fig. 4A, lanes 1, 2 and 3). Enhanced recruitment of c-Fos to the IL-6 promoter was observed upon treatment with IL-6/R plus IL-17 (Fig. 4A, lane 4). A similar trend for c-Jun was observed. Increased binding of c-Jun was observed after IL-6/R or IL-17 treatment (Fig. 4A, lanes 2 and 3), and recruitment was enhanced after IL-6/R plus IL-17 treatment (lane 4).
Figure 4. IL-6/R plus IL-17 Enhances Recruitment of p65, P-p65, c-Jun, c-Fos, CBP, p300, and RNA Pol II to the IL-6 Promoter.

A, Primary astrocytes were treated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 90 min, and then cells were cross-linked with formaldehyde. Soluble chromatin was subjected to immunoprecipitation with Abs against p65, P-p65, c-Fos, c-Jun, or normal rabbit IgG. PCR analysis of the positive control (input) indicates that soluble chromatin samples obtained from each time point had equal amounts of chromatin fragments containing the IL-6 promoter. B, Primary astrocytes were treated as above. Soluble chromatin was subjected to immunoprecipitation with Abs against histone acetylation (Ac-H3 and Ac-H4), p300, CBP, RNA Pol II or normal rabbit IgG. The basal level of the untreated sample was set at 1.0 and fold activation upon IL-6/R, IL-17 or IL-6/R plus IL-17 treatment was compared with that value. Representative of at least three experiments.
Covalent histone modifications have functional roles in gene transcription (45). The N-terminal tails of core histones are subject to modifications such as acetylation, methylation, phosphorylation, and ubiquitination (46). The acetylation of histone H3 was increased in an additive fashion by treatment of astrocytes with IL-6/R plus IL-17 (Fig. 4B, lanes 2, 3 and 4). For acetylation of H4, IL-17 treatment induced this modification, which was synergistically enhanced upon inclusion of IL-6/R (Fig. 4B, lanes 2, 3 and 4). Histone acetylation of lysine residues requires the activities of histone acetyltransferases (HATs) such as CBP and/or p300, which are important in relaxing the compact structure of nucleosomes (47). CBP and p300 were weakly associated with the IL-6 promoter in untreated astrocytes (Fig. 4B, lane 1), and IL-6/R and IL-17 stimulation promoted recruitment of CBP and p300 after treatment (lanes 2 and 3). Enhanced recruitment of CBP and p300 to the IL-6 promoter was observed after treatment with IL-6/R plus IL-17 (Fig. 4B, lane 4). RNA Pol II is present at the IL-6 promoter in the absence of cytokine stimulation; however, increased recruitment was observed after IL-6/R, IL-17 or IL-6/R plus IL-17 addition (Fig. 4B, lanes 1-4). These results suggest that IL-6/R plus IL-17 enhance the acetylation of histones H3 and H4 on the IL-6 promoter, concurrent with CBP, p300 and RNA Pol II recruitment to initiate transcription of the IL-6 gene.
SOCS3 is a Negative Regulator of the Synergistic Effect of IL-6/R and IL-17
SOCS3 is expressed in astrocytes in response to various cytokines (27), and its predominant function is inhibition of JAK-mediated STAT-3 activation. However, the STAT-3 pathway is not involved in transcriptional regulation of IL-6 expression in our system (data not shown). SOCS3 has been shown to modulate other signaling pathways such as NF-κB and MAPK (27), which we demonstrated are involved in IL-6/R plus IL-17 induction of IL-6. Thus, we hypothesized that SOCS3 may affect this response. Primary astrocytes were treated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for up to 8 h, and SOCS3 expression analyzed by QRT-PCR. SOCS3 mRNA expression was induced at 2 h after the addition of IL-6/R alone and peaked at 4 h (Fig. 5A). SOCS3 mRNA expression was not induced by IL-17 alone, however, in the presence of IL-6/R and IL-17, the expression of SOCS3 mRNA was significantly enhanced at all time points, still increasing at 8 h (Fig. 5A). We next investigated whether SOCS3 was involved in the process of IL-17-enhancement of IL-6 gene expression in astrocytes. SOCS3 siRNA was utilized to knockdown SOCS3 in astrocytes as previously described (38). Following a 48 h transfection with siRNA control or siSOCS3, cells were treated in the absence or presence of IL-6/R plus IL-17 for 4 h, and analyzed for SOCS3 and IL-6 mRNA expression. SOCS3 mRNA induced by IL-6/R plus IL-17 was inhibited approximately 49% by siSOCS3 (Fig. 5B). We observed a significant enhancement of IL-6/R plus IL-17-induced IL-6 mRNA induction in astrocytes transfected with SOCS3 siRNA (Fig. 5C). Similarly, SOCS3 knockdown significantly enhanced the expression of IL-6 protein (Fig. 5D). These data suggest that SOCS3 is a negative regulator of IL-6/R plus IL-17 induction of IL-6 expression.
Figure 5. IL-17 Enhances IL-6-mediated SOCS3 Expression in Primary Astrocytes, and SOCS3 is a Negative Regulator of the Synergistic Effect of IL-6 and IL-17.
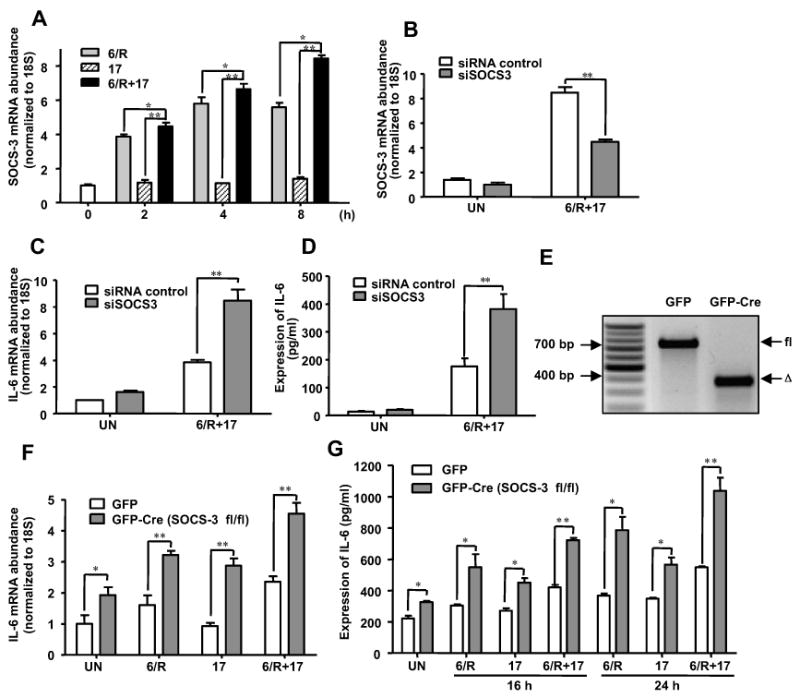
A, Astrocytes were treated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 2 - 8 h, and levels of SOCS3 mRNA expression were determined by QRT-PCR. B and C, Astrocytes were transfected with SOCS3 siRNA (100 nM) or siRNA control (100 nM) for 48 h. Transfected cells were then treated with medium (UN) or IL-6/R plus IL-17 for 4 h. Levels of SOCS3 (B) and IL-6 (C) mRNA expression was determined by QRT-PCR. D, Astrocytes were transfected with SOCS3 siRNA or siRNA control for 48 h. Transfected cells were then treated with medium (UN) or IL-6/R plus IL-17 for 24 h, and supernatants analyzed for IL-6 protein by ELISA. E, SOCS3 floxed astrocytes were infected with GFP as control or GFP-Cre for deletion. After 48 h in culture, cells were harvested. Digestion of genomic DNA distinguishes the full-length (fl) and excised alleles (Δ). F, SOCS3 floxed astrocytes were infected with GFP as control or GFP-Cre for deletion. After 48 h in culture, the cells were treated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 4 h, and levels of IL-6 mRNA expression determined by QRT-PCR. G, SOCS3 floxed astrocytes were infected with GFP as control or GFP-Cre for deletion. After 48 h in culture, cells were incubated with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 for 16 or 24 h, and supernatants analyzed for IL-6 protein by ELISA. All data are the mean ± SD of three experiments. *, p ≤ 0.05;**, p ≤ 0.01.
To confirm our hypothesis that SOCS3 is involved in IL-17-enhancement of IL-6 signaling, and negatively regulates the expression of IL-6 in astrocytes, we utilized SOCS3 floxed astrocytes to delete SOCS3. We assessed the efficiency of GFP-Cre-mediated deletion of the SOCS3 locus by infecting primary astrocytes from mice carrying the floxed SOCS3 allele, and distinguishing fl and excised (Δ) alleles (Fig. 5E). We then treated GFP infected and GFP-Cre infected SOCS3 floxed astrocytes with IL-6/R, IL-17 or IL-6/R plus IL-17 for 4 h, and IL-6 mRNA expression was examined. In GFP infected SOCS3 floxed astrocytes, IL-6 expression was enhanced by IL-6/R plus IL-17 (Fig. 5F). In GFP-Cre infected SOCS3 floxed astrocytes, we observed a significant increase in IL-6 mRNA expression for all conditions (Fig. 5F). We next examined the effect of SOCS3 knockout on IL-6 protein expression at two time points (16 and 24 h). IL-6 protein expression in GFP infected SOCS3 floxed astrocytes was enhanced by IL-6/R plus IL-17 (Fig. 5G). IL-6 protein expression was significantly increased in SOCS3 knockout astrocytes upon treatment with medium (UN), IL-6/R, IL-17 or IL-6/R plus IL-17 (Fig. 5G). These results indicate that SOCS3 negatively regulates IL-6 expression.
SOCS3 Deletion Enhances Activation of the NF-κB and MAPK Pathways in Astrocytes
As we have shown that IL-6/R and IL-17 induction of IL-6 gene expression occurs via activation of the NF-κB, JNK, and p38 pathways (Figs. 3 and 4), we analyzed activation of these pathways in GFP and GFP-Cre infected SOCS3 floxed astrocytes. In GFP infected astrocytes, treatment with IL-6/R plus IL-17 led to rapid p65 serine phosphorylation. Activation of NF-κB p65 peaked at 15 min and persisted up to 120 min (Fig. 6, lanes 2-5). In GFP-Cre infected SOCS3 floxed astrocytes, basal activation of NF-κB was observed, which was substantially enhanced by IL-6/R plus IL-17 treatment at all time points (Fig. 6, lanes 6-10). Total levels of p65 were not affected by SOCS3. In GFP infected SOCS3 floxed astrocytes, activation of p38 MAPK occurred at 15 min, peaked at 60 min and persisted up to 120 min (Fig. 6, lanes 2-5). In GFP-Cre infected SOCS3 floxed astrocytes, there was a striking increase in the basal levels of activated P-p38 (Fig. 6, lane 6), which was enhanced by IL-6/R plus IL-17 treatment at 15 min (Fig. 6, lane 7). We observed a similar trend for JNK MAPK. In GFP infected SOCS3 floxed astrocytes, activation of JNK MAPK was detected at 30 min, and peaked at 120 min (Fig. 6, lanes 3-5). In GFP-Cre infected SOCS3 floxed astrocytes, there was a substantial increase in the basal level of P-JNK, which was further enhanced with IL-6/R plus IL-17 treatment (Fig. 6, lanes 6-10). These results indicate that SOCS3 negatively regulates the expression of IL-6 via inhibition of NF-κB, p38 and JNK MAPK pathways.
Figure 6. Enhanced Activation of NF-κB and MAPK Pathways in SOCS3 Deficient Astrocytes.

To evaluate NF-κB and MAPK activation in the absence or presence of SOCS3, SOCS3 floxed astrocytes were infected with GFP as control or GFP-Cre for deletion. After 48 h in culture, cells were treated with medium (UN) or IL-6/R plus IL-17 for 15, 30, 60 or 120 min, and then cell lysates were immunoblotted with antibodies against phospho-p65 Ser 536, p65, phospho-p38, p38, phospho-JNK, JNK or GAPDH. The basal level of the untreated GFP infected sample was set at 1.0 and fold activation upon IL-6/R plus IL-17 treatment compared with that value. Representative of at least three experiments.
Discussion
In this study, we addressed the specific role of IL-17 on the IL-6 signaling cascade in astrocytes, and found that IL-17 enhanced a positive-feedback loop of IL-6 expression. The synergistic effect of IL-17 and IL-6/R on IL-6 gene expression involved numerous signaling pathways including NF-κB, p38 MAPK and JNK MAPK. Furthermore, we demonstrated that IL-17 synergized with IL-6/R to enhance the recruitment of activated NF-κB p65, c-Fos, c-Jun, and the histone acetyltransferases CBP and p300 to the IL-6 promoter. This was accompanied by enhanced acetylation of histones H3 and H4 on the IL-6 promoter (Fig. 7A). Moreover, we elucidated an important role for SOCS3 in the regulation of IL-17 plus IL-6/R induction of IL-6 in astrocytes. SOCS3 siRNA knockdown and SOCS3 deletion in astrocytes enhanced the synergistic effect of IL-6/R and IL-17 on IL-6 gene expression, which was due to enhanced activation of the NF-κB and MAPK pathways (Fig. 7A). These results indicate that in astrocytes, SOCS3 can function as a negative regulator of NF-κB, p38 MAPK and JNK signaling.
Figure 7. Proposed Model of IL-17 Enhancement of the IL-6 Signaling Cascade in Astrocytes.
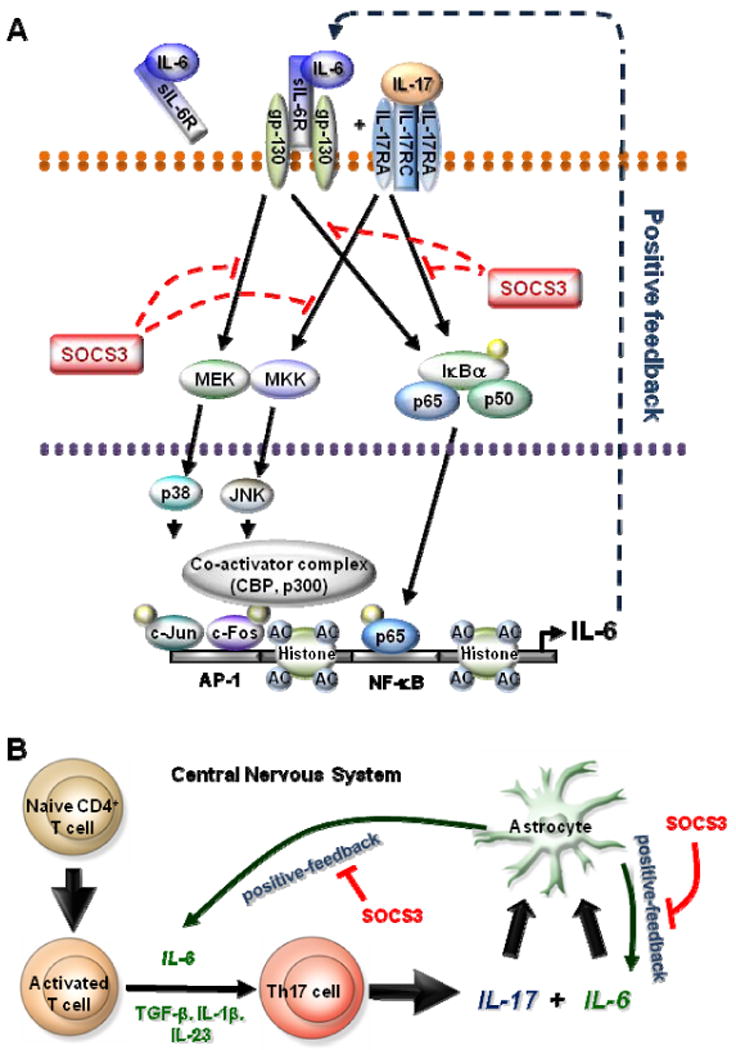
A, IL-6/R and IL-17 activate the NF-κB and MAPK pathway, which then induce IL-6 gene expression. The synergistic effect of these two mediators depends on NF-κB, p38 and JNK MAPK activity. Activated NF-κB p65, c-Fos and c-Jun bind to the IL-6 promoter. Concurrent with NF-κB and MAPK recruitment, IL-6/R and IL-17 leads to the recruitment of coactivators CBP and p300, modifications in AcH3 and AcH4, and recruitment of RNA Pol II to the IL-6 promoter, which results in transcriptional activation of the IL-6 gene. IL-17 can also enhance IL-6/R induced SOCS3 expression, and SOCS3 inhibits IL-6/R plus IL-17-induced NF-κB and MAPK activation, which results in a reduction of IL-6 gene expression in astrocytes. B, Naive CD4+ T cells, after activation by signaling through the T cell receptor and co-stimulatory molecules, can differentiate into Th17 cells in the presence of IL-6, TGF-β, IL-1 and IL-23. IL-17 together with IL-6/R triggers a positive-feedforward loop of IL-6 expression in astrocytes, which may also influence Th17 cell differentiation. SOCS3 participates in these processes as a negative feedback regulator. See text for details.
One mechanism by which IL-17 enhances gene expression is by enhancing mRNA stability (43, 44). However, our results showed that the half-life of IL-6 mRNA was unaffected by IL-17. IL-17 signals through IL-17RA and IL-17RC, and the NF-κB and MAPK pathways can be activated in response to IL-17 (26). The IL-6 promoter contains numerous regulatory elements, including AP-1, C/EBPβ and NF-κB binding sites. Previous results indicated that IL-6 gene transcription required the NF-κB and AP-1 binding sites (48). We speculated that IL-17 may enhance events on the IL-6 promoter. Our results demonstrate that IL-17 enhances IL-6/R-induced recruitment of activated p65, c-Fos and c-Jun, which in concert with increased recruitment of the coactivators CBP and p300, enhancement of permissive histone H3 and H4 modifications, and enhancement of RNA Pol II recruitment to the IL-6 promoter, leads to IL-6 gene transcription (Fig. 4). These results demonstrate that IL-6/R and IL-17 increase activation of NF-κB and MAPK signaling pathways and transcriptional events occurring at the IL-6 promoter.
We have previously shown that SOCS3 induction can occur by activation of numerous signaling pathways, including STAT3, NF-κB and MAPK (27). In this study, we demonstrate that IL-17 alone does not induce SOCS3, but can enhance IL-6/R induction of SOCS3 (Fig.5). We did not elucidate the mechanisms underlying this effect, but it is likely that the IL-17 enhancement occurs through the NF-κB and/or MAPK pathways, since IL-17 does not activate STAT3 in astrocytes (data not shown). Previous findings from our laboratory indicated that SOCS3 attenuates inflammatory events in macrophages/microglia, and SOCS3 exerts an inhibitory effect on chemokine expression in astrocytes (30, 38). Interestingly, knockdown of SOCS3 expression in astrocytes by siRNA enhanced IL-6/R plus IL-17 induction of IL-6. We confirmed these results in SOCS3-deficient astrocytes, and also demonstrated that SOCS3 knockout enhanced activation of the NF-κB, p38 MAPK and JNK MAPK pathways. As we have shown that IL-17-enhanced IL-6 gene expression depends on these pathways, we can conclude that SOCS3 negatively regulates the expression of IL-6 via inhibition of NF-κB and MAPK activation. SOCS3 can therefore regulate numerous signaling pathways of importance in astrocyte biology.
The physiological functions of IL-6 within the CNS are complex; IL-6 exerts neurotrophic and neuroprotective effects, and yet can also function as a mediator of inflammation, demyelination, and astrogliosis, depending on the cellular context (49). In the normal brain, IL-6 levels remain low. However, elevated expression occurs in injury, infection, stroke, and neuroinflammation. Our laboratory reviewed previous reports which determined that the predominant CNS source of IL-6 is the activated astrocyte (49). IL-6 has recently been implicated as a cytokine critical for promoting Th17 cell differentiation and expression, in conjunction with TGF-β and other cytokines (13). Th17 cells are involved in numerous neuroinflammatory responses (14), thus IL-17 enhancement of IL-6 expression in astrocytes can generate an autoamplification loop for Th17 cells in the CNS microenvironment. This may be one of the important functions of IL-6 in the CNS during the development of autoimmunity.
IL-17 is a proinflammatory cytokine that activates T cells and other immune cells to produce a variety of cytokines, chemokines, and cell adhesion molecules (50). IL-17 expression has been associated with many inflammatory diseases, such as rheumatoid arthritis, asthma, systemic lupus erythematosus and allograft rejection. In vitro, expression of the genes encoding several chemokines, including CC-chemokine ligand 2 (CCL2), CCL7, CCL20 and CXC-chemokine ligand 1 (CXCL1), as well as genes encoding matrix metalloproteinase 3 (MMP3) and MMP13, are significantly upregulated in fibroblasts after treatment with IL-17 (13, 51). Research on the function of IL-17 in the CNS is mainly focused on the disease of MS and the animal model EAE. In MS patients, IL-17 mRNA and protein are increased in both brain lesions and mononuclear cells isolated from blood and cerebrospinal fluid (17, 52-54).
Furthermore, the accumulation of Th17 cells in CNS MS lesions has been demonstrated (55). EAE was significantly suppressed in IL-17-/-□mice; these animals exhibited a delay in disease onset, reduced maximum severity scores, ameliorated histological changes, and early recovery (56-58). However, it should be noted that there is controversy regarding the involvement of IL-17 in autoimmune CNS disease (59, 60). Naive CD4+ T cells, after activation by signaling through the T cell receptor and co-stimulatory molecules, can differentiate into Th17 cells in the presence of IL-6, TGF-β, IL-1 and IL-23 (13). Our data suggest that IL-17, together with IL-6, triggers a positive-feedforward loop of IL-6 expression in astrocytes, which can contribute to Th17 cell differentiation in the context of CNS inflammation (Fig. 8B). SOCS3 participates in these processes as a negative feedback regulator, indicating that SOCS3 in astrocytes functions as an attenuator of inflammatory responses.
Acknowledgments
We thank Dr. Warren Alexander (Cancer and Haematology Division, The Walter and Eliza Hall Institute of Medical Research and the Cooperative Research Centre for Cellular Growth Factors) for the gift of SOCS3 floxed transgenic mice and Dr. Rosa Serra (UAB) for providing the adenoviral Cre constructs.
This work was supported in part by National Institutes of Health Grants NS-50665, NS-57563 and NS-50665-05WI (to E.N.B.), a Collaborative MS Research Center Award from the National Multiple Sclerosis Society (CA1059-A-13) to E.N.B., National Multiple Sclerosis Society Grant RG 3892-A-12 (to E.N.B.), National Institutes of Health Pilot and Feasibility Grant AR-48311 (to H.Q.), and the National Natural Science Foundation of China 30672163 (to X L). X.M. was supported by the China Scholarship Council (2008622086) and B.J.B was supported by NIH T32-NS-48039.
Abbreviations used in this paper
- MS
Multiple Sclerosis
- siRNA
small interfering RNA
- SOCS
Suppressor Of Cytokine Signaling
- WT
wild type
- Th17
T helper 17
References
- 1.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- 2.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 3.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams A, Piaton G, Lubetzki C. Astrocytes--friends or foes in multiple sclerosis? Glia. 2007;55:1300–1312. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- 5.Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- 6.Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86:342–367. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheller J, Ohnesorge N, Rose-John S. Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol. 2006;63:321–329. doi: 10.1111/j.1365-3083.2006.01750.x. [DOI] [PubMed] [Google Scholar]
- 9.De Keyser J, Mostert JP, Koch MW. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci. 2008;267:3–16. doi: 10.1016/j.jns.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 10.Van Wagoner NJ, Oh JW, Repovic P, Benveniste EN. Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J Neurosci. 1999;19:5236–5244. doi: 10.1523/JNEUROSCI.19-13-05236.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benveniste EN, Sparacio SM, Norris JG, Grenett HE, Fuller GM. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol. 1990;30:201–212. doi: 10.1016/0165-5728(90)90104-u. [DOI] [PubMed] [Google Scholar]
- 12.Benveniste EN, Kwon J, Chung WJ, Sampson J, Pandya K, Tang LP. Differential modulation of astrocyte cytokine gene expression by TGF-beta. J Immunol. 1994;153:5210–5221. [PubMed] [Google Scholar]
- 13.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 14.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–898. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 15.Lohr J, Knoechel B, Wang JJ, Villarino AV, Abbas AK. Role of IL-17 and regulatory T lymphocytes in a systemic autoimmune disease. J Exp Med. 2006;203:2785–2791. doi: 10.1084/jem.20061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flierl MA, Rittirsch D, Gao H, Hoesel LM, Nadeau BA, Day DE, Zetoune FS, Sarma JV, Huber-Lang MS, Ferrara JL, Ward PA. Adverse functions of IL-17A in experimental sepsis. Faseb J. 2008;22:2198–2205. doi: 10.1096/fj.07-105221. [DOI] [PubMed] [Google Scholar]
- 17.Graber JJ, Allie SR, Mullen KM, Jones MV, Wang T, Krishnan C, Kaplin AI, Nath A, Kerr DA, Calabresi PA. Interleukin-17 in transverse myelitis and multiple sclerosis. J Neuroimmunol. 2008;196:124–132. doi: 10.1016/j.jneuroim.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 19.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 20.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity. 2008;29:628–636. doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Inoue D, Numasaki M, Watanabe M, Kubo H, Sasaki T, Yasuda H, Yamaya M, Sasaki H. IL-17A promotes the growth of airway epithelial cells through ERK-dependent signaling pathway. Biochem Biophys Res Commun. 2006;347:852–858. doi: 10.1016/j.bbrc.2006.06.137. [DOI] [PubMed] [Google Scholar]
- 23.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- 25.Trajkovic V, Stosic-Grujicic S, Samardzic T, Markovic M, Miljkovic D, Ramic Z, Mostarica Stojkovic M. Interleukin-17 stimulates inducible nitric oxide synthase activation in rodent astrocytes. J Neuroimmunol. 2001;119:183–191. doi: 10.1016/s0165-5728(01)00391-5. [DOI] [PubMed] [Google Scholar]
- 26.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker BJ, Akhtar LN, Benveniste EN. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009;30:392–400. doi: 10.1016/j.it.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 29.Baker BJ, Qin H, Benveniste EN. Molecular basis of oncostatin M-induced SOCS-3 expression in astrocytes. Glia. 2008;56:1250–1262. doi: 10.1002/glia.20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin H, Wilson CA, Roberts KL, Baker BJ, Zhao X, Benveniste EN. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177:7761–7771. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- 31.Pauli EK, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, Ludwig S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4:e1000196. doi: 10.1371/journal.ppat.1000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS, Johnston JA. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 34.Bellezza I, Neuwirt H, Nemes C, Cavarretta IT, Puhr M, Steiner H, Minelli A, Bartsch G, Offner F, Hobisch A, Doppler W, Culig Z. Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am J Pathol. 2006;169:2199–2208. doi: 10.2353/ajpath.2006.060171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cacalano NA, Sanden D, Johnston JA. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat Cell Biol. 2001;3:460–465. doi: 10.1038/35074525. [DOI] [PubMed] [Google Scholar]
- 36.Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, Benveniste EN, Cong Y. TGF-beta promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 38.Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 40.Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D, Benveniste EN. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009;28:3735–3745. doi: 10.1038/onc.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin H, Wilson CA, Lee SJ, Benveniste EN. IFN-beta-induced SOCS-1 negatively regulates CD40 gene expression in macrophages and microglia. Faseb J. 2006;20:985–987. doi: 10.1096/fj.05-5493fje. [DOI] [PubMed] [Google Scholar]
- 42.Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, Fitzgerald DC, Shindler KS, Rostami A. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2009;6:14. doi: 10.1186/1742-2094-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 44.Lee JW, Wang P, Kattah MG, Youssef S, Steinman L, DeFea K, Straus DS. Differential regulation of chemokines by IL-17 in colonic epithelial cells. J Immunol. 2008;181:6536–6545. doi: 10.4049/jimmunol.181.9.6536. [DOI] [PubMed] [Google Scholar]
- 45.Horn PJ, Peterson CL. Molecular biology. Chromatin higher order folding--wrapping up transcription. Science. 2002;297:1824–1827. doi: 10.1126/science.1074200. [DOI] [PubMed] [Google Scholar]
- 46.Berger SL. An embarrassment of niches: the many covalent modifications of histones in transcriptional regulation. Oncogene. 2001;20:3007–3013. doi: 10.1038/sj.onc.1204324. [DOI] [PubMed] [Google Scholar]
- 47.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 48.Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell. 2005;16:4954–4966. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Wagoner NJ, Benveniste EN. Interleukin-6 expression and regulation in astrocytes. J Neuroimmunol. 1999;100:124–139. doi: 10.1016/s0165-5728(99)00187-3. [DOI] [PubMed] [Google Scholar]
- 50.Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. doi: 10.1111/j.1600-065X.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- 51.Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti-CD3: inducible CD4+CD25-LAP+ regulatory T cells control the expansion of IL-17+ follicular helper T cells. Lupus. 2009;18:586–596. doi: 10.1177/0961203308100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 53.Tompkins SM, Miller SD. An array of possibilities for multiple sclerosis. Nat Med. 2002;8:451–453. doi: 10.1038/nm0502-451. [DOI] [PubMed] [Google Scholar]
- 54.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, Rogan S, Armao D, Jewells V, Soulillou JP, Markovic-Plese S. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol. 2009;130:133–144. doi: 10.1016/j.clim.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 57.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 58.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, Waisman A. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119:61–69. doi: 10.1172/JCI35997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steinman L. Mixed results with modulation of TH-17 cells in human autoimmune diseases. Nat Immunol. 2010;11:41–44. doi: 10.1038/ni.1803. [DOI] [PubMed] [Google Scholar]