

Abstract
Yes1 kinase has been implicated as a potential therapeutic target in a number of cancers including melanomas, breast cancers, and rhabdomyosarcomas. Described here is the development of a robust and miniaturized biochemical assay for Yes1 kinase that was applied in a high throughput screen (HTS) of kinase-focused small molecule libraries. The HTS provided 144 (17% hit rate) small molecule compounds with IC50 values in the sub-micromolar range. Three of the most potent Yes1 inhibitors were then examined in a cell-based assay for inhibition of cell survival in rhabdomyosarcoma cell lines. Homology models of Yes1 were generated in active and inactive conformations, and docking of inhibitors supports binding to the active conformation (DFG-in) of Yes1. This is the first report of a large high throughput enzymatic activity screen for identification of Yes1 kinase inhibitors, thereby elucidating the polypharmacology of a variety of small molecules and clinical candidates.
Keywords: Yes1, kinase, screening, HTS, small molecule, inhibitor, rhabdomyosarcoma
The Src family of non-receptor protein tyrosine kinases contains nine members including Yes1, c-Src, Fyn, Lyn, and Lyk. These kinases have important roles in a variety of cellular functions, such as cell proliferation, survival, and differentiation.1 c-Src, the most well characterized member of this family, has been previously identified as a proto-oncogene,2 and a number of antagonists have been influential for validating c-Src as a therapeutic target.3,4 Like c-Src, Yes1 kinase activity has been shown to be upregulated in a variety of cancers, including colon carcinomas,5 melanoma, head and neck, renal, lung, and stomach cancers.6,7 In brain-metastatic melanomas8 and malignant mesothelioma,9 it has been shown that Yes1, and not other family members, such as c-Src, is functionally involved in the malignant phenotype. A recent study examining the downregulation of Yes1 by shRNA found significant effects on cell survival and growth for basal-like and Her2-positive breast cancers.10 In a similar manner, the knock down of Yes1 expression with shRNA in both embryonal and alveolar rhabdomyosarcoma cell lines (RD and RH30) was found to significantly inhibit cell growth in vitro, thereby implicating Yes1 as a potential therapeutic target for this aggressive cancer.11 Despite the potential for Yes1 to be a target for the above-described cancers, there are very few reports on the identification of potent and selective inhibitors of Yes1 kinase.12,13 Antagonists of Yes1 would further elucidate the biology and help to confirm this kinase as a viable target for therapeutic intervention in a variety of cancers.
The known inhibitors of Yes1 kinase include dasatinib (1)14 and saracatinib (2)15 (Figure 1). Saracatinib, currently in phase II/III clinical trials for the treatment of metastatic breast cancer, is an orally bioavailable tyrosine kinase inhibitor with nanomolar IC50 values for c-Src, Yes1, Lck, and Bcr-Abl among other kinases.16 Saracatinib is an ATP competitive, reversible inhibitor of the Src family of kinases that is known to bind the ATP binding site of Yes1 in the active conformation. Preclinical models of saracatinib have shown only modest anti-proliferative effects with more signficiant effects on invasion and migration.15,16 Dasatinib, an FDA-approved sub-nanomolar inhibitor of the Src family of kinases and Bcr-Abl, is used for the treatment of chronic myelogenous leukemia (CML)17 and acute lymphoblastic leukemia (ALL).18 Preclinical data with dasatinib show significant anti-proliferative activity against solid tumor cell lines.19 Importantly, the differing activity profiles of these two compounds, despite their similar targets, may be a consequence of their off-target inhibitory effects.13,20 Anti-cancer agents with polypharmacological profiles can possess enhanced in vivo efficacy and fewer resistance mechanisms, and the design of drugs with multiple targets is proving to be a new paradigm in drug discovery.20,21 Through the discovery and investigation of additional inhibitors of Yes1 kinase with either distinct polypharmacologies or high Yes1 selectivity, we hope to better understand the role Yes1 kinase plays in cancer.
Figure 1.

Known Yes1 kinase inhibitors, dasatinib and saracatinib.
In an effort to identify novel, potent and more selective Yes1 kinase inhibitors, we employed a high throughput screening (HTS) approach utilizing an in vitro biochemical assay. The preparation of compound libraries for quantitative high throughput screening (qHTS) has been previously described.22 Three kinase-focused small molecule libraries were screened for Yes1 kinase inhibition including the GlaxoSmithKline (GSK) Published Kinase Inhibitor Set (367 compounds) attained from GSK through a public-private partnership,23,24 a collection of purchased kinase inhibitors with diverse targets (40 compounds), and an in-house library of compounds with annotated biological target information called the Mechanism Interrogation PlatE (MIPE) (465 compounds). The combined scope of these libraries include preclinical and clinical candidates and a number of approved drugs, the majority of which had not been identified as inhibitors of Yes1 kinase previously in the literature. The MIPE library alone consists of 73 approved drugs, 168 clinical candidates, and 207 preclinical candidates. Utilizing focused libraries with clinically advanced small molecules provides a pharmacological context to the hit compounds derived from a HTS. Accounting for compound overlap between these three libraries, a total of 845 small molecules were examined for Yes1 kinase inhibitory activity and 144 (17%) of these were discovered to be sub-micromolar hits.
Yes1 kinase activity was measured via a ADP-Glo™ Kinase Assay that quantifies the kinase—dependent enzymatic production of ADP from ATP using a coupled luminescence-based reaction.25 The kinase activity was evaluated with an 11-point dose curve (1.3 nM to 76.9 µM) in a 1536-well format for each compound (PubChem AID 686947). Each plate that was screened had a positive control (dasatinib), neutral control (DMSO + enzyme), and a no enzyme control (DMSO only) allowing for comparison of data between multiple plates. Screening data were corrected and normalized, and concentration-response curves were derived using in-house algorithms.22 The averaged statistical parameters for the screen (Z’ = 0.76±0.05, S/B = 23.7±1.95, %CV(DMSO) = 6.9±1.8) provided confidence in the quality of the assay and the hits. Upon measuring Yes1 kinase IC50 values, the compounds were sorted based on their efficacy (>50% inhibition) and curve classification22,26 (curve classes = −1.1, −1.2, and −2.1) to determine the percentage of high-quality actives. With these stipulations, the hit rate for the focused libraries was calculated to be 41% (348/845). These hits were then sorted by their IC50 values, and 41% (144/348) exhibited sub-micromolar inhibition, including 53 compounds with IC50s below 100 nM (Figure 2). The high hit rate for this screen can be attributed to both the use of kinase-focused libraries and the relative promiscuity of Yes1.13,27 A previous study profiling 72 kinase inhibitors against 442 kinases reported that Yes1 interacts with 26% of the tested inhibitors at 300 nM and up to 45% at a concentration of 3 µM.13
Figure 2.

Number of high-quality actives sorted by inhibitory activity (IC50) in a Yes1 kinase HTS biochemical assay.
Compounds that showed greater than 50% inhibition at 1.3 nM and select additional cherry-picked compounds, were re-examined in a follow-up assay using a broader 22-point dose curve with a concentration range of 7.3 fM to 76.9 µM (PubChem AID 686948).28 Notably, all high-quality actives were investigated for potential reactivity with the assay detection components by running a counter screen with all of the assay components except for the Yes1 kinase (PubChem AID 686950).29 There was no observed cross reactivity of the high-quality active small molecules with the assay components or the coupling enzymes used for the quantitation of ADP (e.g., luciferase; data not shown) with all exhibiting curve classes = 4.0, indicating inactive. Figure 3 shows representative nanomolar inhibition of Yes1 kinase by the small molecules saracatinib, AMG-Tie-2-1, and AZ-23. The dose-response curves and Yes1 IC50 values of all tested compounds in the biochemical assays have been deposited in PubChem and are available free of charge (http://pubchem.ncbi.nlm.nih.gov/ AID 686946).
Figure 3.

Inhibition of Yes1 kinase in a biochemical assay for saracatinib (A, IC50 = 6.2 nM), AMG-Tie-2-1 (B, IC50 = 8.7 nM), AZ-23 (C, IC50 = 39.1 nM).
With a significant number of potent inhibitors, we then turned our attention to examining the reported selectivity and pharmacological properties of a subset of these hits. The high-quality actives were manually curated to eliminate any compounds that contained covalent modifiers (e.g., Michael acceptors), highly reactive functional groups, or promiscuous moieties. It is important to note that the known Yes1 kinase inhibitors dasatinib (1) and saracatinib (2) were both components in the screen and were identified as potent hits (Figure 3, Table 1). Table 1 additionally shows newly identified potent Yes1 kinase inhibitors, their current clinical trial status,30,31 known biological targets30 and Yes1 IC50 values. These compounds were specifically selected for further investigation because of the diversity of chemical scaffolds represented and their relatively advanced pharmacological profiles as a result of their clinical advancement. Although not discussed in detail, many of the other active hits from these libraries may prove to be useful tools and amenable to SAR studies for medicinal chemistry optimization. To confirm the results of the biochemical assay described herein, these ten compounds, including dasatinib (1) and saracatinib (2), were sent to the commercial vendor Reaction Biology Corporation for 10-point dose inhibition curves of Yes1 using a [γ-33P]-ATP radiolabeled enzyme activity assay. The IC50 values obtained from Reaction Biology were consistent with the measured values in our assay. Differences for the values measured by Reaction Biology can be accounted for by the 10-fold lower ATP concentrations used in their assay along with general assay variability.
Table 1.
Select Yes1 kinase inhibitors from a HTS and their corresponding clinical phase, known targets, and IC50 values.
| Compound Name and NCGC ID |
Structure | Clinical Phase |
Known Targets |
Yes1 IC50 (nM) |
|---|---|---|---|---|
| Dasatinib (1) NCGC00181129 |
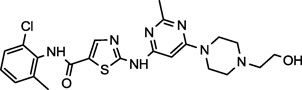 |
Approved | Lyn, PDGFR, KIT, Lck, BTK, Bcr-Abl, Fyn, Yes1, c-Src | 0.5 (< 1.0)a |
| Saracatinib (2) NCGC00241099 |
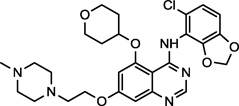 |
Phase II/III | c-Src, Bcr-Abl, Yes1, Lck | 6.2 (0.70)a |
| AEE-788 (3) NCGC00263149 |
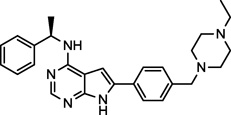 |
Phase I/II | EGFR, HER-2, VEGFR-2 | 17.5 (13.1)a |
| Dovitinib (4) NCGC00249685 |
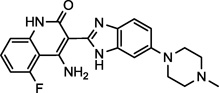 |
Phase III | FGFR, EGFR, PDGFR, VEGFR-1,2 | 31 (1.4)a |
| DCC-2036 (5) NCGC00263172 |
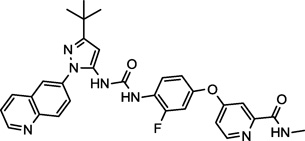 |
Phase I/II | Bcr-Abl, Tie-2, Lyn, FLT3, VEGFR-2 | 2.5 (1.5)a |
| SGI-1776 (6) NCGC00263186 |
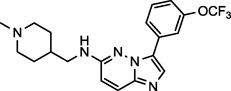 |
Discontinued | Pim-1, FLT3 | 2670 (240)a |
| AMG-Tie-2-1 (7) NCGC00263199 |
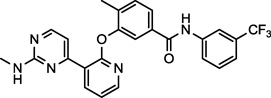 |
Preclinical | Tie-2 | 8.7 (22.0)a |
| AZ-23 (8) NCGC00250381 |
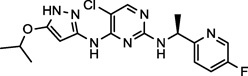 |
Preclinical | Trk | 39.1 (3.0)a |
| Dorsomorphin (9) NCGC00165869 |
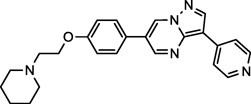 |
Preclinical | AMPK, BMPR, TGFβ Receptor | 195.9 (29.8)a |
| AZ-628 (10) NCGC00250380 |
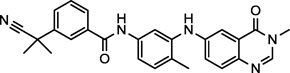 |
Preclinical | Raf Kinase B,C | 348.3 (51.2)a |
Data in parentheses were gathered by Reaction Biology Corp. using a [γ-33P]-ATP radiolabeled enzyme activity assay at an ATP concentration of 10 µM (www.reactionbiology.com)
Aside from dasatinib (1) and saracatinib (2), Table 1 shows three compounds (AEE-788, dovitinib, and DCC-2036, 3−5) that have been examined clinically.31 While there is one report of Yes1 inhibition by dovitinib,27 this is the first report of AEE-788 and DCC-2036 demonstrating nanomolar inhibitory activity for Yes1 kinase. Interestingly, the known primary targets of all of these compounds include vascular endothelial growth factor receptors (VEGFR). The compounds AEE-788 (3) and dovitinib (4) are both angiogenesis inhibitors and are in clinical trials for solid tumors and glioblastomas among other metastatic cancers.31,32 The compound DCC-2036 (5), on the other hand, is primarily a Bcr-Abl inhibitor in clinical trials for chronic myeloid and acute lymphocytic leukemias.33,34
DCC-2036 (5) also shows nanomolar activity against receptor tyrosine kinase FLT3 (Fms-like tyrosine kinase 3) and Tie-2 (tyrosine kinase with immunoglobulin-like and EGF-like domains),34 both of which are the primary targets for the compounds SGI-1776 (6)35 and AMG-Tie-2-1 (7),36 respectively. Overexpression and mutations of FLT3 that lead to constitutive activation and intracellular signal transduction of this receptor have been implicated in a number cancers, including, chronic and acute myeloid leukemias.37,38 Similarly, Tie-2 is an endothelial cell specific receptor tyrosine kinase that upon binding angiopoietin initiates signal transduction and in this manner plays an important role in angiogenesis.39 The preclinical candidate AZ-23 (8) is a selective tropomyosin-related kinase (Trk) inhibitor that exhibits low nanomolar inhibition in cell-based assays and tumor growth inhibition in a neuroblastoma mouse model.40,41 Trks are activated by soluble growth factors, including neurotrophins, and thereby induce signal transduction pathways.42 Interestingly, the altered expression of Yes1 is thought to play a significant role in the progression of melanomas to the brain-metastatic phenotype, and once in the brain, neurotrophins enchance the activity of Yes1.8 The inhibitory activity of AZ-23 (8) for both Yes1 and Trk may be responsible for its tumor growth inhibition in preclincal models. Potent Yes1 kinase inhibition may play a significant role in the biological activity of each of these compounds.
A few of the most potent Yes1 inhibitors in the biochemical assay, were subsequently investigated for cell growth inhibition in both the RD (embryonal) and RH30 (alveolar) rhabdomyosarcoma cell lines. Both of these cell lines have recently been shown to demonstrate significant growth inhibition in the presence of multiple Yes1 targeting shRNA sequences.11 Furthermore, this recent study showed a significant growth inhibition of these cell lines in the presence of the known Yes1 inhibitor dasatinib (1).11 Dasatinib also exhibited in vivo efficacy in rhabdomyosarcoma xenograft mouse models of both RD and RH30 tumors.11 The cell-based inhibition curves for the Yes1 inhibitors saracatinib (2), AMG-Tie-2-1 (7), and AZ-23 (8) were measured using a CellTiter-Glo Luminescent Cell Viability Assay at compound concentrations ranging from 0.01 to 20 µM (Figure 4).43
Figure 4.

Inhibition of cell survival in RD and RH30 rhabdomyosarcoma cell lines for potent Yes1 kinase inhibitors saracatinib (A, IC50[RH30] = 10.1 µM), AMG-Tie-2-1 (B, IC50[RD] = 10.5 µM, IC50[RH30] = 6.0 µM), and AZ-23 (C, IC50[RD] = 5.3 µM, IC50[RH30] = 1.8 µM).
Saracatinib (2), one of the most potent Yes1 inhibitors in the biochemical assay (Figure 3, Table 1), showed only moderate activity for the inhibition of cell growth in the RH30 cell line (IC50 = 10.1 µM) and did not reach the IC50 in the RD cell line with the concentrations tested (Figure 4, A). Notably, saracatinib has been reported to possess significant anti-metastatic activity and only moderate anti-proliferative activity in preclinical models, and our results in this 48-hour cell viability assay support these observations. AMG-Tie-2-1 (7) and AZ-23 (8) are known potent inhibitors of Tie-2 and Trk, respectively, both of which are targets that have yet to be implicated in rhabdomyosarcomas. Nonetheless, AMG-Tie-2-1 (7) and AZ-23 (8) were found to inhibit cell growth of the RH30 cell line with IC50 values of 6.0 µM and 1.8 µM, showing moderate efficacy in the assay (Figure 4, B and C). The RD cell line was also moderately inhibited by 7 and 8, with IC50 values of 10.5 µM and 5.3 µM, respectively. The IC50 offset between the biochemical and cell-based assays for compounds 7 and 8 are consistent with the previously published data for the known Yes1 kinase inhibitor dasatinib (1).11,13 Furthermore, the cell activities for 2, 7, and 8 are expected to be governed by the pharmacological profiles of these compounds and cell permeability and transport. Specific polypharmacology may be necessary for small molecule inhibitors to induce the anti-proliferative phenotypes for rhabdomyosarcoma cell lines. Kinase inhibitors with defined polypharmacologies have been successful for the treatment of cancers, specifically solid tumors, with the advantage of fewer resistance mechanisms.20 Additional studies would be necessary to delineate these factors.
To elucidate the possible binding modes of the potent Yes1 inhibitors from our high throughput screen, homology models of Yes1 were generated. These homology models of Yes1 kinase were built using publicly available c-Src crystal structures (88% identity to Yes1) as templates. The high-resolution crystal structures of c-Src in the active (PDB ID: 1Y57)44 and inactive (PDB ID: 3U4W)45 conformations were selected to construct the active and inactive conformations of Yes1.46 Because of the high sequence identity (88%) of Yes1 to c-Src and the availability of high-resolution c-Src crystal structures, the Yes1 homology models are expected to reasonably reflect the binding pocket of Yes1, and are appropriate for docking small molecules.
The docking of the compounds dovitinib (4) and AZ-23 (8) among others (not presented here) were examined to both the active (DFG-in) and inactive (DFG-out) Yes1 conformations. Both compounds were unable to bind in the inactive conformation of the homology model due to significant steric interactions with the Lys38 sidechain. Nonetheless, the compounds did bind to the ATP-binding pocket of the active conformation Yes1 homology model with little energy minimization required (Figure 5). The docking model of AZ-23 (8) to Yes1 was generated by superimposing the crystal structure of Trk kinase with AZ-23 bound (PDB ID: 4AOJ),47 followed by a series of energy minimizations with the heavy atoms and backbone atoms fixed or tethered. Neither the protein nor the ligand experienced significant changes to reach the energetically minimized complex structure (Figure 5, A). For the docking of dovitinib (4), a close analogue of this compound in complex with checkpoint kinase 1 (Chk1) was placed into the ATP binding site of Yes1 through protein superposition. The ligand was then transformed into dovitinib by addition of a piperizine group and deletion of an aminoalkylamino group.48 The energy minimization was carried out similar to that described above (Figure 5, B). Both compounds show similar binding modes and are within range for hydrogen bonding with the hinge region amino acids Glu82 and Met84.
Figure 5.

The interactions of AZ-23 (A) and dovitinib (B) in the ATP-binding pocket of a Yes1 homology model in an active (DFG-in) conformation with hinge region hydrogen bonds highlighted.
In an effort to identify potent Yes1 kinase inhibitors with enhanced selectivity relative to those previously reported, we developed a biochemical assay amenable to high throughput screening. This assay was used for the identification of potent Yes1 kinase inhibitors from three kinase-focused libraries including the GSK Published Kinase Inhibitor Set, a collection of purchased kinase inhibitors, and an in-house mechanism annotated MIPE library. From these focused libraries, a significant number of potent Yes1 kinase inhibitors were discovered, and this is the first report of the inhibition of this biological target by most of these compounds. Included among these nanomolar inhibitors, are clinical candidates such as AEE-788 and dovitinib, along with preclincial candidates, such as AZ-23 and AMG-Tie-2-1. The small molecules AZ-23 and AMG-Tie-2-1 showed good activity for the inhibition of cell survival in two rhabdomyosarcoma cell lines. Finally, the binding modes of dovitinib and AZ-23 in a Yes1 homology model provided evidence for binding to the ATP-pocket in the active (DFG-in) conformation and key interactions were examined. The results described herein provide evidence for the inhibition of Yes1 kinase as a part of the polypharmacology of a number of known compounds. With the high hit rate from the libraries screened here, Yes1 inhibition may be an important contributor to the in vivo activity for a number of kinase inhibitors. The identification of even non-selective inhibitors with diverse polypharmacologies may help to elucidate the biological role of Yes1 and its role in disease. Furthermore, the discovery of new potent inhibitors of Yes1 and examination of their binding modes, may enable the design of more selective Yes1 kinase inhibitors for use as molecular probes for this therapeutically relevant biological target.
Acknowledgments
The authors would like to thank Paul Shinn, Danielle VanLeer, Crystal McKnight, and Misha Itkin for assistance with compound management. Lesley Mathews, Marc Ferrer, Rajarshi Guha, and Scott Martin are thanked for helpful discussions. We would like to thank William J. Zuercher and David Drewry for valuable discussions and GlaxoSmithKline for access to the GSK Published Kinase Inhibitor Set. This work was supported by the Division of Preclinical Innovation, National Center for Advancing Translational Sciences; the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research; the Intramural Research Program of the National Human Genome Research Institute; the Intramural Research Program of the National Cancer Institute, Center for Cancer Research and the Frederick National Laboratory for Cancer Research, National Institutes of Health including contract HHSN261200800001E and grant # U54CA143930. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Abbreviations
- S/B
signal to background
- PDGFR
platelet-derived growth factor receptor
- BTK
Bruton’s tyrosine kinase
- EGFR
epidermal growth factor receptor
- HER-2
human epidermal growth factor 2
- AMPK
AMP activated protein kinase
- BMPR
bone morphogenetic protein receptor
- TGF
transforming growth factor
- Raf
rapidly accelerated fibrosarcoma
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Summy JM, Sudol M, Eck MJ, Monteiro AN, Gatesman A, Flynn DC. Front. Biosci. 2003;8:185. doi: 10.2741/1011. [DOI] [PubMed] [Google Scholar]
- 2.Irby RB, Yeatman TJ. Oncogene. 2000;19:5636. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 3.Kim LC, Song L, Haura EB. Nature Rev. Clin. Oncol. 2009;6:587. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 4.Zhang S, Yu D. Trends Pharmacol. Sci. 2012;33:122. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pena SV, Melham MF, Meisler AI, Cartwright CA. Gastroenterology. 1995;108:117. doi: 10.1016/0016-5085(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 6.Sugawara K, Sugawara I, Sukegawa J, Akatsuka T, Yamamoto T, Morita M, Mori S, Toyoshima K. Br. J. Cancer. 1991;63:508. doi: 10.1038/bjc.1991.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Summy JM, Sudol M, Eck MJ, Monteiro AN, Gatesman A, Flynn DC. Front. Biosci. 2003;8:185. doi: 10.2741/1011. [DOI] [PubMed] [Google Scholar]
- 8.Marchetti D, Parikh N, Sudol M, Gallick GE. Oncogene. 1998;16:3253. doi: 10.1038/sj.onc.1201877. [DOI] [PubMed] [Google Scholar]
- 9.Sato A, Sekine M, Virgona N, Ota M, Yano T. Oncol. Rep. 2012;28:1889. doi: 10.3892/or.2012.2010. [DOI] [PubMed] [Google Scholar]
- 10.Bilal E, Alexe G, Yao M, Cong L, Kularni A, Ginjala V, Toppmeyer D, Ganesan S, Bhanot G. Genes & Cancer. 2011;1:1063. doi: 10.1177/1947601910395583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeung CL, Ngo VN, Grohar PJ, Arnaldez FI, Asante A, Wan X, Khan J, Hewitt SM, Khanna C, Staudt LM, Helman LJ. Oncogene. 2013 doi: 10.1038/onc.2012.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan H, Laird AD, Blake RA, Tang C, Liang C. Bioorg. Med. Chem. Lett. 2004;14:187. doi: 10.1016/j.bmcl.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 13.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Nat. Biotechnol. 2011;29:1046. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 14.Rix U, Hantschel O, Durnberger G, Rix LLR, Planyavsky M, Fernback NV, Kaup I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G. Blood. 2007;12:4055. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 15.Green TP, Fennell M, Whittaker R, Curwen J, Jacobs V, Allen J, Logie A, Hargreaves J, Hickinson DM, Wilkinson RW, Elvin P, Boyer B, Carragher N, Ple PA, Bermingham A, Holdgate GA, Ward W, Hennequin LF, Davies BR, Costello GF. Mol. Oncol. 2009;3:248. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gucalp A, Sparano JA, Caravelli J, Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang C, Theodoulou M, Massague J, Norton L, Hudis C, Traina TA. Clin. Breast Cancer. 2011;11:306. doi: 10.1016/j.clbc.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cortes JE, Jones D, O'Brien S, Jabbour E, Ravandi F, Koller C, Borthakur G, Walker B, Zhao W, Shan J, Kantarjian H. J. Clin. Oncol. 2010;28:398. doi: 10.1200/JCO.2009.25.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foa R, Vitale A, Vignetti M, Meloni G, Guarini A, De Propris MS, Elia L, Paoloni F, Fazi P, Cimino G, Nobile F, Ferrara F, Castagnola C, Sica S, Leoni P, Zuffa E, Fozza C, Luppi M, Candoni A, Iacobucci I, Soverini S, Mandelli F, Martinelli G, Baccarani M. Blood. 2011;118:6521. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 19.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius L, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen M, Wityak J, Borzilleri RM. J. Med. Chem. 2004;47:6658. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 20.Fojo T. Oncologist. 2008;13:277. doi: 10.1634/theoncologist.2007-0090. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins AL. Nat. Chem. Biol. 2008;4:682. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 22.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Proc. Nat. Acad. Sci. 2006;103:11473. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knapp S, Arruda P, Blagg J, Burley S, Drewry DH, Edwards A, Fabbro D, Gillespie P, Gray NS, Kuster B, Lackey KE, Mazzafera P, Tomkinson NC, Willson TM, Workman P, Zuercher WJ. Nat. Chem. Biol. 2013;9:3. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 24.Dranchak P, MacArthur R, Guha R, Zuercher WJ, Drewry DH, Auld DS, Inglese J. PLoS One. 2013 doi: 10.1371/journal.pone.0057888. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For the in vitro kinase assay 2 µL of Yes1 enzyme was dispensed into 1536-well white solid-bottom plates (Greiner) with a Flying Reagent Dispenser (Beckman Coulter). DMSO solutions of compounds, which were arrayed in 1536-well polypropylene plates in dose response, were transferred by pintool to the assay plate (23 nL; Kalypsys Systems). The reaction was initiated by the addition of 1 µL of substrate and allowed to progress for 1 hour at room temperature. All reagents were purchased from Sigma unless otherwise specified. The final concentration of reagents in the reaction were 4 nM Yes1 (Millipore), 0.1 mM ATP, 100 µM EGTA, 10 mM MgCl2, 0.3 mg/mL poly(E4Y), 50 mM Tris pH 7.5, 1 mM DTT, 150 mM NaCl, 0.01% Brij-35 and 5% glycerol. The production of ADP was quantitated by measuring the luminescence (Perkin-Elmer, Viewlux) from the two-step ADP-Glo kit (Promega) per the manufacturer’s protocol. Data were normalized to DMSO-treated control columns with (maximum signal) and without (minimum signal) enzyme.
- 26.Class −1.1 and −1.2 are the highest-confidence complete concentration response curves (CRCs) containing upper and lower asymptotes with efficacies ≥ 80% and < 80%, respectively. Class −2.1 and −2.2 are incomplete CRCs having only one asymptote with efficacies ≥ 80% and < 80% respectively. Class −3 CRCs show activity at only the highest concentration or have a poor fit. Class 4 CRCs are inactive having a curve-fit of insufficient efficacy or lack a fit altogether.
- 27.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Nat. Biotechnol. 2011;29:1039. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Statistical parameters for the follow-up 22-point dose curve assay were: Z’ = 0.63±0.08, S/B = 4.4±1.7, %CV(DMSO) = 7.9±2.8.
- 29.The counterscreen assay was run as above but without enzyme and poly(E4Y) and with 0.08 mM ATP/0.02 mM ADP (representing 20% conversion) replacing the 0.1 mM ATP in the enzyme assay.
- 30.https://integrity.thomson-pharma.com
- 31.www.clinicaltrials.gov
- 32.Reardon DA, Conrad CA, Cloughesy T, Prados MD, Friedman HS, Aldape KD, Mischel P, Xia J, DiLea C, Huang J, Mietlowski W, Dugan M, Chen W, Yung WK. Cancer Chemother. Pharmacol. 2012;69:1507. doi: 10.1007/s00280-012-1854-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haak T, Hood MM, Jones J, Lord JW, Lu WP, Miller D, Patt WC, Smith BD, Petillo PA, Rutkoski TJ, Telikepalli H, Vogeti L, Yao T, Chun L, Clark R, Evangelista P, Gavrilescu LC, Lazarides K, Zaleskas VM, Steward LJ, Van Etten RA, Flynn DL. Cancer Cell. 2011;19:556. doi: 10.1016/j.ccr.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eide CA, Adrian LT, Tyner JW, Mac Partlin M, Anderson DJ, Wise SC, Smith BD, Petillo PA, Flynn DL, Deininger MW, O'Hare T, Druker BJ. Cancer Res. 2011;71:3189. doi: 10.1158/0008-5472.CAN-10-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pastor J, Oyarzabal J, Saluste G, Alvarez RM, Rivero V, Ramos F, Cendon E, Blanco-Aparicio C, Ajenjo N, Cebria A, Albarran MI, Cebrian D, Corrionero A, Fominaya J, Montoya G, Mazzorana M. Bioorg. Med. Chem. Lett. 2012;22:1591. doi: 10.1016/j.bmcl.2011.12.130. [DOI] [PubMed] [Google Scholar]
- 36.Hodous BL, Geuns-Meyer SD, Hughes PE, Albrecht BK, Bellon S, Bready J, Caenepeel S, Cee VJ, Chaffee SC, Coxon A, Emery M, Fretland J, Gallant P, Gu Y, Hoffman D, Johnson RE, Kendall R, Kim JL, Long AM, Morrison M, Olivieri PR, Patel VF, Polverino A, Rose P, Tempest P, Wang L, Whittington DA, Zhao H. J. Med. Chem. 2007;50:611. doi: 10.1021/jm061107l. [DOI] [PubMed] [Google Scholar]
- 37.Gu TL, Nardone J, Wang Y, Loriaux M, Villen J, Beausoleil S, Tucker M, Kornhauser J, Ren J, MacNeill J, Gygi SP, Druker BJ, Heinrich MC, Rush J, Polakiewicz RD. PLoS One. 2011;6:e19169. doi: 10.1371/journal.pone.0019169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meshinchi S, Appelbaum FR. Clin. Cancer Res. 2009;15:4263. doi: 10.1158/1078-0432.CCR-08-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters KG, Kontos CD, Lin PC, Wong AL, Rao P, Huang L, Dewhirst MW, Sankar S. Recent Prog. Horm. Res. 2004;59:51. doi: 10.1210/rp.59.1.51. [DOI] [PubMed] [Google Scholar]
- 40.Wang T, Lamb ML, Scott DA, Wang H, Block MH, Lyne PD, Lee JW, Davies AM, Zhang HJ, Zhu Y, Gu F, Han Y, Wang B, Mohr PJ, Kaus RJ, Josey JA, Hoffmann E, Thress K, Macintyre T, Omer CA, Yu D. J. Med. Chem. 2008;51:4672. doi: 10.1021/jm800343j. [DOI] [PubMed] [Google Scholar]
- 41.Thress K, Macintyre T, Wang H, Whitston D, Liu ZY, Hoffmann E, Wang T, Brown JL, Webster K, Omer C, Zage PE, Zeng L, Zweidler-McKay PA. Mol. Cancer Ther. 2009;8:1818. doi: 10.1158/1535-7163.MCT-09-0036. [DOI] [PubMed] [Google Scholar]
- 42.Kaplan DR, Stephens RM. J. Neurobiol. 1994;25:1404. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 43.Rhabdomyosarcoma cell lines (RD and RH30) were grown in DMEM (Quality Biological) with 10% FBS (Hyclone), 1% penicillin/strepomycin (Quality Biological), and 2 mM L-glutamine (Quality Biological). In a white, flat-bottom 96-well plate, 4,000 cells were seeded in each well with 100 µL of culture media for overnight. Then 100 µL of culture media with inhibitors was added to reach final concentrations of 0, 0.1, 5, 10, 15, and 20 µM with a constant DMSO concentration of 0.2%. After 48 hours, CellTiter-Glo (Promega) was used to measure the relative cell number per the manufacturer’s guidelines. Each data point is the mean reading of sextuplicate.
- 44.Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. Structure. 2005;13:861. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Georghiou G, Kleiner RE, Pulkoski-Gross M, Liu DR, Seeliger MA. Nat. Chem. Biol. 2012;8:366. doi: 10.1038/nchembio.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The protein homology modeling module in MOE (version 2011, Chemical Computing Group, Montreal, Canada) was employed for homology model construction. The force field for homology modeling was Amber99 and for docking was PFROSST.
- 47.Wang T, Lamb ML, Block MH, Davies AM, Han YX, Hoffmann E, Ioannidis S, Josey JA, Liu ZY, Lyne PD, MacIntyre T, Mohr PJ, Omer CA, Sjogren T, Thress K, Wang B, Wang H, Yu D, Zhang HJ. ACS Med. Chem. Lett. 2012;3:705. doi: 10.1021/ml300074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni ZJ, Barsanti P, Brammeier N, Diebes A, Poon DJ, Ng S, Pecchi S, Pfister K, Renhowe PA, Ramurthy S, Wagman AS, Bussiere DE, Le V, Zhou Y, Jansen JM, Ma S, Gesner TG. Bioorg. Med. Chem. Lett. 2006;16:3121. doi: 10.1016/j.bmcl.2006.03.059. [DOI] [PubMed] [Google Scholar]