

Abstract
A crucial component of the analysis of shotgun proteomics datasets is the search engine, an algorithm that attempts to identify the peptide sequence from the parent molecular ion that produced each fragment ion spectrum in the dataset. There are many different search engines, both commercial and open source, each employing a somewhat different technique for spectrum identification. The set of high-scoring peptide-spectrum matches for a defined set of input spectra differs markedly among the various search engine results; individual engines each provide unique correct identifications among a core set of correlative identifications. This has led to the approach of combining the results from multiple search engines to achieve improved analysis of each dataset. Here we review the techniques and available software for combining the results of multiple search engines and briefly compare the relative performance of these techniques.
The most commonly used proteomics approach, shotgun proteomics, has become an invaluable tool for the high-throughput characterization of proteins in biological samples (1). This workflow relies on the combination of protein digestion, liquid chromatography (LC)1 separation, tandem mass spectrometry (MS/MS), and sophisticated data analysis in its aim to derive an accurate and complete set of peptides and their inferred proteins that are present in the sample being studied. Although many variations are possible, the typical workflow begins with the digestion of proteins into peptides with a protease, typically trypsin. The resulting peptide mixture is first separated via LC and then subjected to mass spectrometry (MS) analysis. The MS instrument acquires fragment ion spectra on a subset of the peptide precursor ions that it measures. From the MS/MS spectra that measure the abundance and mass of the peptide ion fragments, peptides present in the mixture are identified and proteins are inferred by means of downstream computational analysis.
The informatics component of the shotgun proteomics workflow is crucial for proper data analysis (2), and a wide variety of tools have emerged for this purpose (3). The typical informatics workflow can be summarized in a few steps: conversion from vendor proprietary formats to an open format, high-throughput interpretation of the MS/MS spectra with a search engine, and statistical validation of the results with estimation of the false discovery rate at a selected score threshold. Various tools for measuring relative peptide abundances may be applied, dependent on the type of quantitation technique applied in the experiment. Finally, the proteins present, and their abundance in the sample, are inferred based on the peptide identifications.
One of the most computationally intensive and diverse steps in the computational analysis workflow is the use of a search engine to interpret the MS/MS spectra in order to determine the best matching peptide ion identifications (4), termed peptide-spectrum matches (PSMs). There are three main types of engines: sequence search engines such as X!Tandem (5), Mascot (6), SEQUEST (7), MyriMatch (8), MS-GFDB (9), and OMSSA (10), which attempt to match acquired spectra with theoretical spectra generated from possible peptide sequences contained in a protein sequence list; spectral library search engines such as SpectraST (11), X!Hunter (12), and Bibliospec (13), which attempt to match spectra with a library of previously observed and identified spectra; and de novo search engines such as PEAKS (14), PepNovo (15), and Lutefisk (16), which attempt to derive peptide identifications based on the MS/MS spectrum peak patterns alone, without reference sequences or previous spectra (17). Additionally, elements of de novo sequencing (short sequence tag extraction) and database searching have been combined to create hybrid search engines such as InSpecT (18) and PEAKS-DB (19).
The goal of this review is to evaluate the potential improvement made possible by combining the search results of multiple search engines. On their own, most of the common search engines perform well on typical datasets, with the results having significant overlap between the algorithms (20); and yet, the degree to which there is divergence in the results of different search engines remains quite high. Disagreement between search engines, where multiple different peptide sequences are identified with high confidence, is quite rare. It is much more common to observe different engines being in agreement on the correct identification, yet with neither of the identifications having a probability high enough to allow it to pass the selected error criterion when analyzed independently. When the results are analyzed together, the agreement on the identification might propel the PSM to pass the same error criterion. In cases when only one engine scores the PSM highly enough that it passes an acceptance threshold, these identifications are reported within the acceptable error rate. Also, some engines use unique methods to consider peptides or modifications not considered by other engines. Even if the experimenter is careful to choose similar search parameters when running multiple tools, different search engines will allow one to set non-identical search parameters, which contributes to reduced overlap between the search results. Spectral library search engines tend to be far more sensitive and specific than sequence search engines, but only for peptide ions for which there is a spectrum in the library.
Given that different search engines excel at identifying different subsets of PSMs, it seems natural to combine the power of multiple search engines to achieve a single, better result. Many algorithms and software tools have emerged that combine search results (21–28), each demonstrating an improved final result over any individual search engine alone. Such improved results come at the cost of the increased complexity of managing multiple searches in an analysis pipeline, as well as a several-fold increase in computational time in what is already the most computationally expensive step. However, with the ever-growing availability of fast computers, computing clusters, and cloud computing resources, researchers now have within reach the ability to quickly search their MS/MS data using several of the still-growing number of search engine algorithms. In some cases the open-source search engines are quite similar to their commercial alternatives; for example, Comet (29) is very similar to SEQUEST. Given the significant amount of time the average researcher takes to design an experiment, process the samples, and acquire the data, it is natural that a researcher would wish to maximize the number and confidence of peptide and protein identifications in each dataset with a rigorous computational analysis. Furthermore, when using label-free spectral counting for abundance analysis, maximizing the number of PSMs increases the dynamic range and accuracy of the quantitative approach (23). Therefore, the demand to use multiple search engines and integrate their results with the goal of maximizing the amount of information gleaned from each dataset is expected to continue growing.
Also emerging are software tools that use several iterative database searching passes of the same data, combining multiple database search tools, searches with different post-translational modifications, and searches against different databases in an attempt to use each specific tool under ideal conditions, utilizing each for its specific strengths and integrating the results (30). Some relevant aspects of such strategies are discussed by Tharakan et al. (31).
In the following sections, we review the various approaches and software programs available to assist with the merging of results from different search engines. We also provide a performance comparison of the various approaches described here on a test dataset to assess the expected performance gains from the various described methods.
MATERIALS AND METHODS
The following software was used in the analysis of the data presented in this review: search engines InsPecT, version 20090202; Mascot, version 2.2.04; OMSSA, version 2.1.1; SEQUEST, version 27; MyriMatch, version 1.5.7; and X!Tandem (k-score) 2007.07.01.3. The Trans-Proteomic Pipeline (TPP) version used in the analysis was 4.6.2. MSBlender was downloaded and built from source using code available on February 15, 2012. PepArML searches and analysis were completed through the PepArML website, and the results were downloaded on July 15, 2012.
The searches done locally were completed in parallel on a Linux cluster running CentOS release 5.9. Upon search completion, the search results were combined in all possible combinations using iProphet, without rerunning the searches. MSBlender was also done on the same search results and on our local cluster. In the case of PepArML, we uploaded our data to the PepArML website and performed the searches and PepArML analysis on their systems. When these were completed, we were able to download and process the results. Because the formats generated by MSBlender and PepArML are different than iProphet-generated pepXML output, tools for parsing and processing the MSBlender and PepArML results had to be written; these were based on the TPP scripts for performing decoy-based error rate calculations, reusing as much codebase as possible while adapting them to the unique tables and pepXML flavors generated by the non-TPP tools analyzed.
Overview of Approaches
In order to provide a multi-tool result, there are several notable obstacles that need to be cleared before one is able to perform search engine merging. First, nearly every search engine writes its output data in a different file format (32). Second, running multiple search engines requires bookkeeping to transfer and collate the output files for jobs farmed out to computing clusters that require dedicated nodes to perform the searches (e.g. Mascot). Third, nearly every search engine produces a different score parameter to indicate the quality of a PSM. Finally, the PSMs are sometimes given different identifiers by different search engines, which can make it difficult to match the PSMs corresponding to the same spectrum. The techniques by which these obstacles are overcome vary depending on the approach and are discussed below.
To overcome the problem of different output file formats for each search engine, the combiners must either use custom parsers for each supported search engine or require that the native search engine output is first converted to a common format, such as the community-developed HUPO-PSI mzIdentML (33) format or pepXML (21). Once the issues of differing output formats have been surmounted, one of the easiest approaches is to filter a final list of PSMs based on search engine congruency. The results from each of the search engines are merged in such a way that only those PSMs for which there is a minimum threshold of congruency for the supplied search results are considered. An example of such criteria is the requirement that all search engines be in complete agreement, or at least a majority consensus such as two out of three or three out of five, depending on the number of engines used. Decoys can be propagated through such a simple approach to estimate false discovery rates (FDRs) for the output. Although implementing such an algorithm is easy, spectra that may be very confidently identified by only one search engine are lost. Instead, a more sophisticated combination scheme is desired, one that can take advantage of multiple search engines without inflating the FDR while incorporating as many correct peptide assignments as possible.
Keller et al. (21) demonstrated the earliest mechanism for combining the output of three search engines, SEQUEST, Mascot, and COMET, within ProteinProphet (34) by merging results based on probabilities uniformly calculated in the different search results by PeptideProphet (35). The overlap between different engine results was shown to be incomplete, and an improved result was achievable via the SearchCombiner within ProteinProphet. This functionality has since been moved out of ProteinProphet and into iProphet (25), which is described in detail below.
The concept was expanded by Searle et al. (24) in the calculation of an agreement score for each spectrum, and it was implemented in Scaffold (Proteome Software, Portland, OR). Scaffold considers multiple top-scoring matches from each search engine. It uses a Bayesian approach implemented in an expectation maximization algorithm to combine the probabilities of the individual search engine results into a single probability for each PSM. Another commercial tool is the inChorus software tool (Bioinformatics Solutions, Inc., Waterloo, ON, Canada), a multi-engine search tool that incorporates results from SEQUEST, Mascot, OMSSA, X!Tandem, and the PEAKS de novo search engine.
PepArML (36) is described as an unsupervised, model-free, machine-learning meta-search engine wherein one submits a dataset to their Web interface for analysis. Multiple search jobs for Mascot, X!Tandem, OMSSA, X!Tandem with the K-score plug-in (37), SScore, MyriMatch, and InSpecT are queued to their own clusters for these submitted data, and the combined result is made available to the submitter free of charge after processing. A significant advantage of this system is that there is no reliance on local computer power to run multiple searches. It is open-source software and is freely available. However, throughput is limited to whatever is provided by the system, and one cannot upload custom databases without contacting the support team, which to date has been very responsive. The output of PepArML is a compressed archive containing the results of the individual searches and their combinations in pepXML and protXML formats, along with images estimating the FDR performance of the analysis.
MSblender (23) uses a probabilistic approach to combining results from multiple search engines, first computing probabilities of correct identification based on individual search engine scores and then combining results on this basis. This was shown by Kwon et al. (23) to dramatically improve the completeness of identifications in several datasets, performing as well as or better than previous approaches. The increased completeness improved the dynamic range and accuracy of a spectral counting quantitative approach, and MSblender was shown to compute an accurate estimation of the FDR in the combined result. MSblender is open-source software and is freely available online, and at present it is compatible with SEQUEST, X!Tandem, OMSSA, MyriMatch, MS-GFDB, and InSpecT, although it is claimed the approach could be applied to additional search engines. MSblender generates .tsv files as output and requires writing custom parsers to work with the results.
FDRAnalysis (38) is a recently published tool for combining the results of Mascot, X!Tandem, and OMSSA. FDRAnalysis is distributed as a stand-alone application that can be downloaded and run on a local computer or as a Web application that can be accessed by anyone with a Web browser. The input to FDRAnalysis is mzIdentML formatted results, although it is also able to process Mascot's dat format, OMSSA's csv format, and X!Tandem's native xml output. The FDRAnalysis algorithm counts decoy matches in the results and produces output consisting of a collection of tables in csv format and images showing distributions of identified peptide attributes for decoy and non-decoy proteins in the database.
The ConsensusID tool (28), a component of the OpenMS toolkit (39), forms a probabilistic consensus on the top-scoring PSM results of several search engines utilizing all the high-ranking scoring matches output by each search engine. A key feature of this tool is its sequence similarity scoring mechanism, which is a method to estimate the scores for PSMs in cases when the peptide is missing from the high-ranking results of a search engine. ConsensusID explicitly computes peptide match scores for all peptides and search engines involved by utilizing the highest sequence similarity peptide sequences in the other searches, thereby including all potential peptides reported by each search in a consensus-based rank order in the result. ConsensusID is open-source software and is freely available within the OpenMS suite.
The iProphet tool (25) is a component of the TPP (21, 40) that is used between PeptideProphet (35) and ProteinProphet (34). PeptideProphet uses a mixture model to discriminate between correct and incorrect assignments, assigning a probability of being correct to every PSM and calculating global FDRs at several thresholds. Any search engine for which the output can be written out in or converted to the pepXML format (21) is supported by PeptideProphet, including the spectral library search engine, SpectraST. PeptideProphet output is also written in pepXML. If a dataset has been searched with multiple search engines, each of the separate PeptideProphet modeling results can be combined with the iProphet tool. The iProphet algorithm recomputes the PSM probabilities and calculates peptide-level probabilities via several other mixture models, taking into account corroborating evidence of other PSMs, and it writes the resulting information in pepXML format. ProteinProphet then takes the results of iProphet pepXML to perform its protein inference calculation and writes the result in protXML. The iProphet software is open-source and is freely available within the TPP suite. Although iProphet is assessed here in the context of combining multiple search engines, it should be noted that the model for combining multiple searches used by iProphet (the Number Sibling Searches model) is only one out of a set of six models that can be applied to improve the identification rates on the analysis of even a single search engine.
Uses of Decoy Matches
It is a common practice to include decoy proteins in the database search step. PeptideProphet's semi-supervised (41) and semi-parametric (42) modes require the presence of decoy sequences in the database for estimating the mixture models. PSMs matching to peptides present only in decoy sequences are true negative results. In the iProphet paper, we used two independent sets of decoys in the database. One set of decoys was used to run PeptideProphet in the semi-parametric mode (which is currently the only way to run search engines other than Mascot, SEQUEST, Comet, and X!Tandem through the TPP). The second set of decoys was used to show that PeptideProphet, iProphet, and ProteinProphet generated accurate probabilities when compared against an independent set of true negative PSMs not known to be decoys to the TPP. This was a clean cross-validated approach unsusceptible to over-fitting of the model. As we showed in the iProphet paper, this approach provided accurate probabilities for PSMs, distinct peptides, and protein levels.
In practice, however, generating two independent decoy sets creates additional bookkeeping overhead and can be avoided. If there is only one set of decoys in the database, the user can specify the option called DECOYPROBS during the PeptideProphet modeling step, which allows PeptideProphet to compute probabilities among the known decoy PSMs using the mixture model learned during the last iteration of the algorithm. When the same decoy sequences are used in the model and their probabilities are computed, the FDRs based on the mixture models' probabilities should match very closely to the decoy-estimated FDRs. Because the iProphet search engine combining tool depends on accurate PSM-level probabilities computed by PeptideProphet for each search engine, any bias after the PeptideProphet step, seen as disagreement between decoy-estimated FDRs and model-estimated FDRs, should be addressed prior to running iProphet. One way to address potential bias in the PeptideProphet models used by TPP is to selectively disable all but one model at a time until the model causing disagreement between PeptideProphet-estimated FDRs and decoy-estimated FDRs is identified and disabled. Another way to deal with PeptideProphet bias is to apply the CLEVEL option in PeptideProphet. CLEVEL controls the number of standard deviations away from the mean of the negative f-value model that PeptideProphet may draw positive results from. By default, CLEVEL is set at 0; setting a higher CLEVEL ensures that only higher quality matches can make it past PeptideProphet with non-zero probability.
Comparison of Approaches
It is expected that methods combining results should demonstrate improved performance over any one individual contributing search engine, but certain sets of search engines might compare more favorably than others. Further, there will likely be a point of diminishing returns, after which including additional search engine results will not be worth the increase in computation time required in order to generate them. Finally, some search engine result combining tools might perform better than others. To address these questions, we analyzed a common dataset using various combination approaches and compared the results.
We selected a dataset from Shteynberg et al. (25) as a reference dataset with which to compare the tools. This dataset was taken from a study on Human Jurkat A3 T leukemic cells (43). The cells were lysed, the lysate was separated using one-dimensional SDS-PAGE, and sections of the gel were digested with trypsin. Digestion was followed by analysis using a Thermo-Fisher Scientific (San Jose, CA) LTQ linear ion trap mass spectrometer. For this article, we used a single complete replicate of the whole cell lysate experiment (replicate 1; 19 gel bands). It comprises a set of 19 MS files containing 161,425 MS/MS spectra in total. All raw data and search results used for this comparison are available online in Peptide Atlas.
We used the original search engine results from Shteynberg et al. (25). Six search engines were used on the data: InSpecT, MyriMatch, OMSSA, Mascot, SEQUEST, and X!Tandem + K-score (37). The iProphet tool was then applied to each possible unique combination of one to six search engines, and the results were compared using matches to decoy proteins to establish estimates of both the error rate and the number of correct results. In each analysis, all potential iProphet models were used to obtain the best possible result set each time. Fig. 1 provides a comparison of the performance on the PSM level of all combinations of one to six search engines combined with iProphet. The spline function in R was used to evaluate the receiver operating characteristic curves at decoy-estimated error rates of 0%, 0.5%, 1%, 1.5%, and 2%, where the decoys did not always yield an exact value.
Fig. 1.

Comparison of the number of PSMs for all combinations of one through six search engines combined with iProphet. Each bar represents the correct PSMs that can be recovered from a given dataset using the computed scores to rank and filter the results while requiring decoy-estimated error rates of 0%, 0.5%, 1.0%, 1.5%, and 2.0%. Also shown are MSblender results combining InSpecT, MyriMatch, OMSSA, SEQUEST, and X!Tandem, as well as PepArML results combining Mascot, X!Tandem, X!Tandem + K-score, MyriMatch, OMSSA, SScore, and InSpecT.
The results in Fig. 1 indicate that combining larger and larger groups of search engines yields generally higher identification rates. In fact, when any superset and subset of iProphet combinations are compared (see Fig. 1), the superset yields performance that is at least as good as that of any subset. In other words, one can safely add more search engines to the iProphet combination without degrading performance. On this dataset there does appear to be a ceiling that is reached by the iProphet combination of the best performing set of five search engines, which attains results on par with the iProphet combination of all six search engines, but this does not mean that on another dataset generating larger and larger combinations will result in the same ceiling.
We installed and applied all the search engine combiners introduced above on this dataset. We were successful in applying PepArML and MSblender, and the results from those tools are depicted in Fig. 1. We also aimed to use FDRAnalysis and ConsensusID for comparison. However, FDRAnalysis is a Web-page-based tool that works on a single searched MS/MS run at a time, and its use was not feasible for high-throughput proteomics of this type. Our attempt to run one MS/MS file through the FDRAnalysis Web page generated errors, and the run did not finish. The ConsensusID tool is part of the OpenMS pipeline, and we were able to successfully deploy this software on our computers. Unfortunately, the results generated by ConsensusID for our dataset had very high independently estimated error rates, and therefore we are unable to report the results from this tool.
From Fig. 1 it appears that MSblender might have under-performed relative to some individual search results. However, the individual search results shown here were processed through PeptideProphet and iProphet, which apply statistical models to significantly improve the classifier and, consequently, the number of correct PSMs that can be recovered at a given FDR; this step is currently not performed by the MSblender pipeline.
It is clear from Fig. 1 that the benefit afforded by combining multiple search engines depends significantly on which engines are used for the combination. Intuitively, one might expect that combining the best performing single search engines would yield the best performing combinations; however, this was not always observed. Different search engines identify different overlapping subsets of spectra and peptides, which might complement each other differently. Specific examples of this include spectra for which one search engine yielded a low-scoring incorrect assignment and another one matched the correct peptide with a high score, or instances in which both searches agreed on the peptide for a given spectrum but neither one could pass the FDR threshold in the individual analyses. Although SEQUEST and X!Tandem performed first and third best in the individual search engine results, their combination performed ninth among all 15 combinations of two search engines. This is likely because the K-score scoring function used in the X!Tandem search is inherently similar to SEQUEST's scoring algorithm, so the two results tend to agree more closely. MyriMatch seemed to contribute the most to combinations of two searches when combined with SEQUEST, X!Tandem+ K-score, or OMSSA, perhaps because its scoring model differs significantly from those. Although on its own InSpecT performed lower on the scale than most engines, in combinations of three, four, and five search engines it contributed a good share of correct results.
Search Engine Combination Improvement Measure
We define a measure for comparing the contribution of correct results by a given search engine combination at a certain FDR—for instance, the ubiquitous 1%.
Assume En represents a combined set of n search engines {e1, e2, …, en}.
Assume R(En) represents the number of correct results at an FDR of 1% for search engine combination En.
Assume En is a superset of Em; then let G(En, Em) = R(En) − R(Em) represent the gain in correct results observed from using the combination En instead of Em.
Now we can define the best gain in results that can be observed by expanding any starting set of search engines Es by exactly i new search engines as follows:
Finally, we define the set of Ei(Es) as the search engine set of size i that results in the largest gain when expanding the initial set Es.
Table I shows the additional number of correct PSMs (and the percentage) that can be gained by starting with one search engine and incrementally adding between one and four more search engines to maximize the gain with iProphet. Certainly, increasing the number of search engines used in an analysis will require additional computational resources both in storage space for the additional data produced by each search algorithm and in computational time required to execute the searches. Table I shows the requirements for storing pepXML-formatted search results of multiple search engines (prior to combining). Supplemental Table S1 shows the approximate computational speed requirements in terms of the number of PSMs generated per second for each of the sequence search engines used. The total search time for multiple files depends on the number of computers allocated to searching the data in parallel, which is limited by the size of the computer cluster. With the emergence of cloud-based computing, time on multiple cloud computers can be purchased, and searches can be done in a highly parallel fashion. In the cloud, for a cost and assuming no failures, a search of multiple files and multiple search algorithms can be performed in the amount of time it takes to do the longest search, plus the overhead that is required to upload the spectrum data to the cloud and download the results from the cloud.
Table I. iProphet Search Engine Combinations to Maximize PSM Gains.
iProphet PSM Gains (at 1% error) observed by starting with one search engine and adding the best performing 1 to 4 search engines to the combination. Also displayed are the total storage requirements for the resulting data files.
| Engine | Additional engines | Best PSM gain at 1% error | Best PSM percent gain (%) | Best to add | Total storage size (GB) |
|---|---|---|---|---|---|
| InSpecT | 1 | 10,306 | 46.4 | SEQUEST | 1.2 |
| Mascot | 1 | 8761 | 38.1 | SEQUEST | 0.5 |
| OMSSA | 1 | 5938 | 22.3 | SEQUEST | 0.4 |
| X!Tandem | 1 | 5060 | 17.8 | MyriMatch | 1.7 |
| MyriMatch | 1 | 4936 | 16.9 | SEQUEST | 1.6 |
| SEQUEST | 1 | 3603 | 11.8 | MyriMatch | 1.6 |
| InSpecT | 2 | 12,516 | 56.3 | SEQUEST, MyriMatch | 2.6 |
| Mascot | 2 | 11,217 | 48.8 | SEQUEST, MyriMatch | 1.9 |
| OMSSA | 2 | 8379 | 31.4 | SEQUEST, MyriMatch | 1.8 |
| X!Tandem | 2 | 6490 | 22.8 | SEQUEST, MyriMatch | 1.9 |
| MyriMatch | 2 | 5801 | 19.8 | OMSSA, SEQUEST | 1.8 |
| SEQUEST | 2 | 4468 | 14.6 | MyriMatch, OMSSA | 1.8 |
| InSpecT | 3 | 13,358 | 60.1 | SEQUEST, MyriMatch, X!Tandem | 2.9 |
| Mascot | 3 | 12,255 | 53.3 | SEQUEST, MyriMatch, X!Tandem | 2.2 |
| OMSSA | 3 | 8889 | 33.3 | SEQUEST, MyriMatch, X!Tandem | 2.1 |
| X!Tandem | 3 | 7176 | 25.3 | SEQUEST, MyriMatch, InSpecT | 2.9 |
| MyriMatch | 3 | 6349 | 21.7 | SEQUEST, X!Tandem, InSpecT | 2.9 |
| SEQUEST | 3 | 5017 | 16.4 | MyriMatch, X!Tandem, InSpecT | 2.9 |
| InSpecT | 4 | 13,850 | 62.3 | SEQUEST, MyriMatch, X!Tandem, OMSSA | 3.1 |
| Mascot | 4 | 12,813 | 55.7 | SEQUEST, MyriMatch, X!Tandem, InSpecT | 3.1 |
| OMSSA | 4 | 9412 | 35.3 | SEQUEST, X!Tandem, MyriMatch, InSpecT | 3.1 |
| X!Tandem | 4 | 7667 | 27.0 | SEQUEST, MyriMatch, InSpecT, OMSSA | 3.1 |
| MyriMatch | 4 | 6841 | 23.4 | SEQUEST, X!Tandem, InSpecT, OMSSA | 3.1 |
| SEQUEST | 4 | 5508 | 18.0 | MyriMatch, X!Tandem, InSpecT, OMSSA | 3.1 |
Search Engine Complementarity Measure
We also defined a measure for computing similarity between pairs of search engines. Given two sets of individual search engine results, A = {ei } and B = {ej}, and a set of two search engines AB = {ei, ej}, let their complementarity be defined as
 |
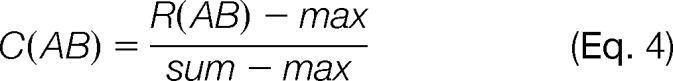 |
The idea behind this calculation is that if the sets of correct results between two search engines are complementary to each other, they should each contribute sets of spectra that overlap significantly less than between non-complementary search engines. We work under the assumption that the number of correct results in the combination is maximally the sum of correct results from each individual search. Thus, if the number of correct results in the combination equals the sum of correct results from the individual searches, the resulting complementarity value will be 1. If there is no increase in the number of correct results in the combination from the best performing individual search, the complementarity is zero.
Fig. 2 presents the complementarity scores for the sequence search engine results analyzed here as a scatter plot of total combined identifications versus complementarity score. The numerical values for these data points are listed in supplemental Table S1. For this dataset, Mascot and InSpecT were the most complementary (i.e. had the least overlap in results), yet their combined result was not as good as the combined results of other pairs. Clearly, complementarity is not the only factor of importance when considering which search engines to merge into a combined result. We reiterate that these results are derived from this single dataset, and there will be some variability among scores. To be fair to the less well-performing algorithms in this review, it is well known in the field that different database search algorithms perform best on different datasets and under different conditions. So although in our evaluation some search engines performed worse, the reader must not take this to mean that any search engine is necessarily and unequivocally better than another based on the analysis of this one dataset.
Fig. 2.

Scatter plot showing the combined number of correctly identified PSMs at a 1% error rate plotted against the complementarity scores of all pairs of search engine combinations. Combining the two most complementary search engines does not necessarily lead to the best overall result, and combining the two least complementary search engines does not necessarily lead to the worst overall result.
To help elaborate on the question of why certain search engine combinations perform better than others, we take a simple example that would never occur in the real world: combining a search with itself. Obviously, such a combination would not be expected to yield results any more correct than those of the original search, regardless of how well the initial search performed. In fact, it is reasonable to assume that the more similar the scoring function that two search engine algorithms apply, the greater the likelihood that they will match the same spectra with the same peptides; this would apply to PSMs that are correctly identified as well as to those that are wrong. Therefore, combining the search results of two very similar search engines is not likely to add to the ability of the classifier to separate the correct from the incorrect identifications and will not significantly improve performance. This is the likely reason that the SEQUEST and X!Tandem (k-score) combination performs less well than the SEQUEST and InSpecT combination, even though alone X!Tandem (k-score) performs better than InSpecT.
We further dissect this analysis by comparing the set of spectra identified by the iProphet combination of SEQUEST and InSpecT at a 1% error rate to the set of spectra identified by the iProphet combination of SEQUEST and X!Tandem (k-score) at a 1% error rate. There are 29,952 spectra identified by both combinations within the accepted error threshold. The InSpecT and SEQUEST combination yields an additional 2927 spectra not identified by the SEQUEST and X!Tandem (k-score) combination within the error threshold, of which 105 spectra are unique to InSpecT, 1738 are matched to the same peptide outside the error threshold by the SEQUEST and X!Tandem (k-score) combination, and the remaining 1084 are matched to a different peptide outside the error threshold by the SEQUEST and X!Tandem (k-score) combination. In contrast, the SEQUEST and X!Tandem (k-score) combination provides fewer unique identifications: 1739 spectra not identified by the InSpecT and SEQUEST combination within the error threshold, of which only 8 spectra are unique to X!Tandem (k-score), 934 are matched to the same peptide outside the error threshold by the InSpecT and SEQUEST combination, and the remaining 797 are matched to a different peptide outside the error threshold by the InSpecT and SEQUEST combination.
Additional manual exploration of the discrepancies in peptide assignments by exploring identifications contributed by the InSpecT algorithm alone revealed chimeric spectra containing fragments from differently charged precursors of similar m/z values, with both peptides having strong probabilities of being correct. When spectra are chimeras in the same charge, iProphet relies on consensus and picks one best-scoring peptide assignment in that charge state. However, when the chimeric precursors are of different charges, iProphet reports the best-scoring peptide match in each of the charge states searched, yielding an increase in the number of spectra that can be correctly assigned, because a multiply charged chimeric spectrum can now be identified in one of several charge states. The spectrum charge state is unknown in an LTQ dataset of this type, and the search engine must make an assumption of the charge, potentially leading to missed identifications where assuming a different charge on the spectrum would otherwise yield a high-scoring identification. Each search engine makes its own charge assumptions for each spectrum, accounting for the 105 identifications unique to InSpecT and 8 unique to X!Tandem (k-score). The remaining spectra are identified in both combinations in the same charge states, but not within the same error threshold, and not necessarily matching the same peptide sequence. When this occurs, a probable cause would be a noisy or chimeric spectrum where one or more of the three search engines identified different peptides (or having very low PSM probabilities) and iProphet reduced the probability of that spectrum in the combination. However, when one of the search engines in the combination is able to correctly match the peptide with a high probability, it will often be sufficient to rescue that identification, even when the other search in the combination matches the same (or different) peptide with a low probability. In the comparison of these two combinations, InSpecT is more successful than X!Tandem (k-score) at rescuing the identifications missed by SEQUEST.
Search Engine Unique Contributions
We next evaluated the unique contributions of PSMs made by each of the search engines in this dataset. To perform this task, PSM lists returned by each search engine (at a 1% error rate) were compared against the PSM lists returned from the combination of the other five search engines (at a 1% error rate). The PSMs identified only by the single search engine analysis, and not by the combination of the other five, were counted as a percentage of the total PSMs identified by the single search engine. From Fig. 3, we see that MyriMatch was the best at identifying the greatest number of unique PSMs, whereas SEQUEST identified the greatest total number of PSMs. One can also observe that InSpecT, while identifying the fewest PSMs in total, identified the third proportionally largest set of unique PSMs missed by the combination of the other five search engines. It should be noted that some of Mascot's relatively poor performance could be attributed to Mascot being handicapped by searching only fully tryptic peptides to save time; however, the identification of semi-tryptic peptides in this dataset is less than 6%, indicating that the poor performance is not due to this issue alone.
Fig. 3.

Scatter plot showing the percentage of PSMs identified uniquely by a single search engine, out of the total PSMs identified by a single search engine, relative to the combination of the other five search engines. The x-coordinate shows the total number of PSMs identified by a single search engine analyzed with the TPP.
Distinct Peptide Sequence Level Performance
Next we examine the performance at the distinct peptide sequence level of the three combiners that generated usable results in our analysis: iProphet, MSblender, and PepArML. The distinct peptide sequence level accounts for each distinct peptide sequence observed in the data exactly once, taking the probability of the highest scoring PSM to match to that peptide sequence. The maximum combiner (e.g. iProphet) probability for each distinct peptide sequence, as computed, represents the distinct peptide sequence level probability for that peptide sequence.
For this test, and MSblender used a combination of five searches: InSpecT, MyriMatch, X!Tandem, OMSSA, and SEQUEST. PepArML also adds X!Tandem-Native, SScore, and Mascot into the mix. Fig. 4 summarizes these results, showing the distinct peptide sequence level comparison of the three approaches at decoy-estimated error rates of 0%, 0.5%, 1%, 1.5%, and 2%. MSblender seemed to generate the fewest correct unique peptide sequences relative to the other two combiners, whereas the results of PepArML and iProphet were rather similar.
Fig. 4.

Comparison of the distinct peptide sequence identifications for three search engine combiners (MSblender, PepArML, and iProphet) at decoy-estimated error rates of 0%, 0.5%, 1.0%, 1.5%, and 2.0%. These error rates are denoted by the bottom whisker, bottom of the box, center line, top of the box, and top whisker, respectively. Also depicted on the right are the results of a spectral library search with SpectraST using the latest NIST spectral library, as well as the result when iProphet was used to combine SpectraST results with the combined sequence search engine results. Data points were inferred using the spline function, as not all of the tools allowed filtering at error rates below 1% as estimated by the decoys.
Fig. 4 also shows the performance of the SpectraST spectral library search tool on the same dataset. The spectral library used for the search was the NIST 2010 human spectral library, plus decoy spectra to evaluate the error rate beyond the estimate provided by the TPP mixture modeling. As can be seen from Fig. 4, SpectraST on its own, processed through PeptideProphet and iProphet, yielded a ∼16% increase in the number of correctly identified distinct peptides at an error rate of 1% over the iProphet combination of six search engines. The reason for such a large number of identifications made by SpectraST alone is that it is a very sensitive search tool that is able to identify a large portion of spectra on a standard dataset such as this, for which high-quality spectral libraries exist. However, one cannot always expect the spectral library to contain all of the peptides in the sample, and combining spectral library search results with sequence database search results should yield even more identifications. In fact, when SpectraST results were included in the combination of all seven search engine algorithms and combined with iProphet, the number of correctly identified distinct peptides increased by ∼26% over the iProphet combination of six sequence search engines. Fig. 5 displays the full receiver operating characteristic curves for each of the considered search engines, as well as the iProphet combined results as a function of FDR at the distinct peptide sequence level.
Fig. 5.

Receiver operating characteristic curves on the distinct peptide sequence level showing performance of iProphet on single search engine results versus the combination of all search engines. The spectral library search results are also included for comparison.
CONCLUSION
We have reviewed the approaches and tools available for improving dataset analysis via combining multiple search engine results, and we compared different combinations of search engines, using iProphet, applied to the same dataset. We also processed the same dataset through several other search combining algorithms and compared the peptide-level results among the approaches. We were not able to make some of the search engine result combining tools work properly, and therefore we could not include those results. Further effort in providing user-friendly and well-designed tools is encouraged to provide the field with effective alternatives.
It is clear that combining search engine results with one of the available tools improves the results of a single search engine. PepArML and iProphet appeared to perform the best among the combiners we were able to test successfully. An important consideration regarding which tool to use is which one matches best with the available expertise and data format handling capability. However, we also recommend that error rates be monitored using decoy matches to establish that the method of choice can perform precisely and accurately for estimating error rates with or without an independent set of decoys contained in the search space. When combining search results, imprecision of the method can be overcome by reliance on decoy searches and decoy counting in the sorted and filtered result sets.
The combination of a few search engines showed a dramatic improvement. However, the difference between combinations of five engines and six engines showed very small gains in the test dataset, and thus the benefit from merging more than five engines must be considered in the context of cost derived from the added computational expense. However, we have also demonstrated that various search engines and algorithms differ in their similarity to each other, and selecting the engines with the most complementary scoring functions is most beneficial. Combining search engines allows one to utilize each search engine for its specific strengths and can complement the identification of the whole analysis to generate complete and maximal results from each proteomic dataset.
In general, using as many search engines as possible with a combiner will serve to improve the classifier and increase the confidence of identified PSMs, distinct peptide sequences, and proteins. It will also maximize the coverage of identified proteins by identifying peptides that would not otherwise be identified in a single search engine result with high confidence. Additionally, search engine combining might have significant benefits for specific types of datasets. For instance, spectra of phosphorylated peptides collected in collision-induced dissociation tend to be of lower quality, and under such conditions search engine combining is of great benefit (44). However, because of the computational hurdles and expertise required to install and run multiple search engines, some will still choose not to use such methods until they become easy to install, use, and troubleshoot and can be relied on to generate meaningful results under all conditions.
Supplementary Material
Footnotes
* This work has been funded in part with federal funds from NIGMS National Institutes of Health under Grant No. R01 GM087221 (to E.W.D.), by Grant Nos. R01-CA-126239 and R01-GM-094231 (to A.I.N.), by American Recovery and Reinvestment Act (ARRA) funds through Grant No. R01 HG005805 (to R.L.M.) from the NHGRI, by NIGMS National Institutes of Health (Center for Systems Biology Grant No. 2P50 GM076547 to R.L.M.), by the National Science Foundation MRI (Grant No. 0923536 to R.L.M.), and by the Luxembourg Centre for Systems Biomedicine and the University of Luxembourg (to R.L.M.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are::
- FDR
- false discovery rate
- MS/MS
- tandem mass spectrometry
- PSM
- peptide-spectrum match.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Deutsch E. W., Lam H., Aebersold R. (2008) Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol. Genomics 33, 18–25 [DOI] [PubMed] [Google Scholar]
- 3. Nesvizhskii A. I. (2010) A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 73, 2092–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eng J. K., Searle B. C., Clauser K. R., Tabb D. L. (2011) A face in the crowd: recognizing peptides through database search. Mol. Cell. Proteomics 10, R111.009522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 6. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 7. Eng J., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 8. Tabb D. L., Fernando C. G., Chambers M. C. (2007) MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 6, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim S., Mischerikow N., Bandeira N., Navarro J. D., Wich L., Mohammed S., Heck A. J., Pevzner P. A. (2010) The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 9, 2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 11. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 12. Craig R., Cortens J. C., Fenyo D., Beavis R. C. (2006) Using annotated peptide mass spectrum libraries for protein identification. J. Proteome Res. 5, 1843–1849 [DOI] [PubMed] [Google Scholar]
- 13. Frewen B. E., Merrihew G. E., Wu C. C., Noble W. S., MacCoss M. J. (2006) Analysis of peptide MS/MS spectra from large-scale proteomics experiments using spectrum libraries. Anal. Chem. 78, 5678–5684 [DOI] [PubMed] [Google Scholar]
- 14. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 15. Frank A., Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 16. Taylor J. A., Johnson R. S. (2001) Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal. Chem. 73, 2594–2604 [DOI] [PubMed] [Google Scholar]
- 17. Pevtsov S., Fedulova I., Mirzaei H., Buck C., Zhang X. (2006) Performance evaluation of existing de novo sequencing algorithms. J. Proteome Res. 5, 3018–3028 [DOI] [PubMed] [Google Scholar]
- 18. Tanner S., Shu H., Frank A., Wang L. C., Zandi E., Mumby M., Pevzner P. A., Bafna V. (2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 19. Zhang J., Xin L., Shan B., Chen W., Xie M., Yuen D., Zhang W., Zhang Z., Lajoie G. A., Ma B. (2012) PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 11, M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kapp E. A., Schutz F., Connolly L. M., Chakel J. A., Meza J. E., Miller C. A., Fenyo D., Eng J. K., Adkins J. N., Omenn G. S., Simpson R. J. (2005) An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics 5, 3475–3490 [DOI] [PubMed] [Google Scholar]
- 21. Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alves G., Wu W. W., Wang G., Shen R. F., Yu Y. K. (2008) Enhancing peptide identification confidence by combining search methods. J. Proteome Res. 7, 3102–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon T., Choi H., Vogel C., Nesvizhskii A. I., Marcotte E. M. (2011) MSblender: a probabilistic approach for integrating peptide identifications from multiple database search engines. J. Proteome Res. 10, 2949–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Searle B. C., Turner M., Nesvizhskii A. I. (2008) Improving sensitivity by probabilistically combining results from multiple MS/MS search methodologies. J. Proteome Res. 7, 245–253 [DOI] [PubMed] [Google Scholar]
- 25. Shteynberg D., Deutsch E. W., Lam H., Eng J. K., Sun Z., Tasman N., Mendoza L., Moritz R. L., Aebersold R., Nesvizhskii A. I. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 10, M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sultana T., Jordan R., Lyons-Weiler J. (2009) Optimization of the use of consensus methods for the detection and putative identification of peptides via mass spectrometry using protein standard mixtures. J. Proteomics Bioinform. 2, 262–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dagda R. K., Sultana T., Lyons-Weiler J. (2010) Evaluation of the consensus of four peptide identification algorithms for tandem mass spectrometry based proteomics. J. Proteomics Bioinform. 3, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nahnsen S., Bertsch A., Rahnenfuhrer J., Nordheim A., Kohlbacher O. (2011) Probabilistic consensus scoring improves tandem mass spectrometry peptide identification. J Proteome Res 10, 3332–3343 [DOI] [PubMed] [Google Scholar]
- 29. Eng J. K., Jahan T. A., Hoopmann M. R. (2012) Comet: an open source tandem mass spectrometry sequence database search tool. Proteomics 13, 22–24 [DOI] [PubMed] [Google Scholar]
- 30. Ning K., Fermin D., Nesvizhskii A. I. (2010) Computational analysis of unassigned high-quality MS/MS spectra in proteomic data sets. Proteomics 10, 2712–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tharakan R., Edwards N., Graham D. R. (2010) Data maximization by multipass analysis of protein mass spectra. Proteomics 10, 1160–1171 [DOI] [PubMed] [Google Scholar]
- 32. Deutsch E. W. (2012) File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteomics 11, 1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones A. R., Eisenacher M., Mayer G., Kohlbacher O., Siepen J., Hubbard S. J., Selley J. N., Searle B. C., Shofstahl J., Seymour S. L., Julian R., Binz P. A., Deutsch E. W., Hermjakob H., Reisinger F., Griss J., Vizcaino J. A., Chambers M., Pizarro A., Creasy D. (2012) The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 11, M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 35. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 36. Edwards N., Wu X., Tseng C.-W. (2009) An unsupervised, model-free, machine-learning combiner for peptide identifications from tandem mass spectra. Clin. Proteomics 5, 23–36 [Google Scholar]
- 37. MacLean B., Eng J. K., Beavis R. C., McIntosh M. (2006) General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 22, 2830–2832 [DOI] [PubMed] [Google Scholar]
- 38. Wedge D. C., Krishna R., Blackhurst P., Siepen J. A., Jones A. R., Hubbard S. J. (2011) FDRAnalysis: a tool for the integrated analysis of tandem mass spectrometry identification results from multiple search engines. J. Proteome Res. 10, 2088–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sturm M., Bertsch A., Gropl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choi H., Nesvizhskii A. I. (2008) Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. J. Proteome Res. 7, 254–265 [DOI] [PubMed] [Google Scholar]
- 42. Choi H., Ghosh D., Nesvizhskii A. I. (2008) Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res. 7, 286–292 [DOI] [PubMed] [Google Scholar]
- 43. Wu L., Hwang S. I., Rezaul K., Lu L. J., Mayya V., Gerstein M., Eng J. K., Lundgren D. H., Han D. K. (2007) Global survey of human T leukemic cells by integrating proteomics and transcriptomics profiling. Mol. Cell. Proteomics 6, 1343–1353 [DOI] [PubMed] [Google Scholar]
- 44. Saleem R. A., Rogers R. S., Ratushny A. V., Dilworth D. J., Shannon P. T., Shteynberg D., Wan Y., Moritz R. L., Nesvizhskii A. I., Rachubinski R. A., Aitchison J. D. (2010) Integrated phosphoproteomics analysis of a signaling network governing nutrient response and peroxisome induction. Mol. Cell. Proteomics 9, 2076–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.