

Abstract
Nucleotide sugars are the donor substrates of various glycosyltransferases, and an important building block in N- and O-glycan biosynthesis. Their intercellular concentrations are regulated by cellular metabolic states including diseases such as cancer and diabetes. To investigate the fate of UDP-GlcNAc, we developed a tracing method for UDP-GlcNAc synthesis and use, and GlcNAc utilization using 13C6-glucose and 13C2-glucosamine, respectively, followed by the analysis of mass isotopomers using LC-MS.
Metabolic labeling of cultured cells with 13C6-glucose and the analysis of isotopomers of UDP-HexNAc (UDP-GlcNAc plus UDP-GalNAc) and CMP-NeuAc revealed the relative contributions of metabolic pathways leading to UDP-GlcNAc synthesis and use. In pancreatic insulinoma cells, the labeling efficiency of a 13C6-glucose motif in CMP-NeuAc was lower compared with that in hepatoma cells.
Using 13C2-glucosamine, the diversity of the labeling efficiency was observed in each sugar residue of N- and O-glycans on the basis of isotopomer analysis. In the insulinoma cells, the low labeling efficiencies were found for sialic acids as well as tri- and tetra-sialo N-glycans, whereas asialo N-glycans were found to be abundant. Essentially no significant difference in secreted hyaluronic acids was found among hepatoma and insulinoma cell lines. This indicates that metabolic flows are responsible for the low sialylation in the insulinoma cells. Our strategy should be useful for systematically tracing each stage of cellular GlcNAc metabolism.
Protein glycosylation, which is the most abundant post-translational modification, has important roles in many biological processes by modulating conformation and stability, whereas its dysregulation is associated with various diseases such as diabetes and cancer (1, 2). Glycosylation is regulated by various factors including glucose metabolism, the availability and localization of nucleotide sugars, and the expression and localization of glycosyltransferases (3, 4). Thus, ideally all of these components should be considered when detecting changes in a dynamic fashion; namely, it is necessary not only to take a snapshot but also to make movies of the dynamic changes in glycan metabolism.
Glucose is used by living cells as an energy source via the glycolytic pathway as well as a carbon source for various metabolites including nucleotide sugars (e.g. UDP-GlcNAc and CMP-NeuAc). These nucleotide sugars are transported into the Golgi apparatus, and added to various glycans on proteins. UDP-GlcNAc is the donor substrate for N-acetylglucosaminyl (GlcNAc)1 transferases; alternatively, it is used in the cytosol for O-GlcNAc modification (i.e. O-GlcNAcylation) of intracellular proteins (5). The UDP-GlcNAc synthetic pathway is complex as it is a converging point of glucose, nucleotide, fatty acid and amino acid metabolic pathways. Thus, the metabolic flow of glucose modulates the branching patterns of N-glycans via UDP-GlcNAc concentrations because many of the key GlcNAc transferases that determine the branching patterns have widely different Km values for UDP-GlcNAc ranging from 0.04 mm to 11 mm (6, 7). Indeed, it was demonstrated that the branching formation of N-glycans in T cells is stimulated by the supply from the hexosamine pathway, whereby it regulates autoimmune reactions promoted by T cells (8).
UDP-GlcNAc is also used for the synthesis of CMP-NeuAc, the donor substrate for sialyltransferases (9). The CMP-NeuAc concentration is controlled by the feedback inhibition of UDP-GlcNAc epimerase/ManNAc kinase by the final product CMP-NeuAc, and hence a high CMP-NeuAc level reduces metabolic flow in CMP-NeuAc de novo synthesis (10). However, there is still only limited information about how the levels of nucleotide sugars dynamically change in response to the environmental cues, and how such changes are reflected in the glycosylation of proteins.
Stable isotope labeling is a promising approach to quantify metabolic changes in response to external cues (11, 12). For example, the use of nuclear magnetic resonance to obtain isotopomer signals of metabolically labeled molecules has been applied to trace the flux in glycolysis and fatty acid metabolism (13). An approach based on the mass isotopomers of labeled metabolites with 13C6-glucose has been developed to monitor the UDP-GlcNAc synthetic pathway (13–15). The method based on the labeling ratio of each metabolite related to UDP-GlcNAc synthesis has clarified the contribution of each metabolic pathway (14). Moseley reported a novel deconvolution method for modeling UDP-GlcNAc mass isotopomers (15).
Previous studies into the use of nucleotide sugars in glycosylation have relied on the specific detection of metabolically radiolabeled glycans (16). It is possible not only to deduce the glycan structures but also to trace their relative contributions to glycan synthesis without MS. On the other hand, mass isotopomer analysis of glycans labeled with stable isotope provides the ratios of labeled versus unlabeled molecules from MS spectra and structural details of the glycans. However, there are only a limited number of publications reporting the application of stable isotope labeling of glycans for monitoring the dynamics of glycans (17). To date, there have been no reports describing a systematic method for tracing cellular GlcNAc biosynthesis and use based on mass isotopomer analysis.
The aim of this study was to extend our knowledge of the synthesis and metabolism of UDP-GlcNAc as well as its use in the synthesis of CMP-NeuAc, N- and O-glycans. We recently developed a conventional HPLC method for simultaneous determination of nucleotide sugars including unstable CMP-NeuAc (18). We first established an LC-MS method for isotopomer analysis of 13C6-glucose labeled nucleotide sugars for tracing UDP-GlcNAc metabolism from synthesis to use, because previous methods were not suitable for estimating UDP-GlcNAc use in CMP-NeuAc de novo synthesis (15). We also established a method for isotopomer analysis of labeled N- and O-glycan to monitor the metabolic flow of hexosamine into glycans. Using these two methods, we demonstrated the differences in the use of hexosamines between hepatoma and pancreatic insulinoma cell lines. Our approach may be useful for identifying a metabolic “bottleneck” that governs the turnover speed and patterns of cellular glycosylation, which may be relevant for various applications including glycoprotein engineering and discovery of disease biomarkers.
EXPERIMENTAL PROCEDURES
Materials
13C6-Glucose was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA), and [1,2-13C2]-glucosamine and [1,2-13C2]-GlcNAc were purchased from Omicron Biochemicals Inc. (South Bend, IN). UDP-Gal, UDP-Glc, UDP-GalNAc, UDP-GlcNAc, UDP-GlcA and CMP-NeuAc were purchased from Sigma Aldrich Japan (Tokyo, Japan). GDP-Fuc and GDP-Man were purchased from Calbiochem (San Diego, CA). Ammonium bicarbonate, acetonitrile, methanol, triethylamine, formic acid and distilled water were of LC-MS grade. The sources of enzymes used in this study were as follows: peptide N-glycoamidase F (Roche Diagnostics, Mannheim, Germany); benzonase nuclease (Merck, Germany); and neuraminidase derived from Arthrobacter ureafaciens (Nacalai Tesque, Kyoto, Japan). The sources of other materials were as follows: Dulbecco's modified Eagle's medium (DMEM) with high glucose (Sigma Aldrich Japan, Tokyo); fetal bovine serum (Bio West, France), glucose-free DMEM, penicillin and streptomycin sodium (Invitrogen, Carlsbad, CA); all other chemicals (Wako Chemicals, Osaka, Japan). The QnE hyaluronic acid ELISA was purchased from Biotech Trading Partners (Encinitas, CA).
Cells and Culture
The mouse hepatoma cell line, Hepa1–6, was obtained from the ATCC (Manassas, VA), and the mouse pancreatic insulinoma cell line, Min6 (19), was obtained from Dr. Jun-ichi Miyazaki (Osaka University, Japan). Cell culture vessels were kept at 37 °C in a humidified atmosphere containing 5% CO2. Hepa1–6 was cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin, and Min6 was also cultured in DMEM supplemented with 15% FBS, 60 μm 2-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin, until they reached ∼70% confluence.
For the preparation of 13C-labeled nucleotide sugars, the cells were plated on six-well plates cultured for 24 h in glucose-free DMEM supplemented with 5.5 mm natural glucose, and further cultured in glucose-free DMEM supplemented with 5.5 mm 13C6-glucose for up to 6 h. For the isotopomer analysis of glycans, the culture medium (containing natural glucose) was replaced with the glucose-free DMEM supplemented with 5.5 mm natural glucose and 1 mm 13C2-glucosamine, and the cells were cultured for 24 h.
Preparation of Labeled Nucleotide Sugars in Cells
Nucleotide sugars were prepared from the cultured cells according to the method we have previously reported (18).
Ion-pair Reversed-phase LC-ESI-MS for Isotopomer Analysis of Nucleotide Sugars
Ion-pair reversed-phase LC-ESI-MS was performed using an Agilent 1100 series HPLC system (Agilent Technologies, Santa Clara, CA) coupled to an Esquire HCT ion trap mass spectrometer (Bruker Daltonics, Germany). To separate the contaminant signal, which prevents the observation of UDP-HexNAc isotopomers, cellular extracts were separated on an Inertsil ODS-4 column (3 μm, 150 × 2.1 mm, GL Science, Japan). Elution buffers were: (A) 20 mm triethylammonium acetate buffer pH 6.0 and (B) buffer A containing 20% acetonitrile (20). Each cellular extract was dissolved in water (50 μl) and a 10-μl aliquot was injected onto the column equilibrated with buffer A. The elution gradient was as follows: 100% buffer A for 15 min; 0–20% linear gradient of buffer B over 20 min; 20–100% linear gradient of buffer B over 1 min; and 100% buffer B for 14 min. The flow rate was maintained at 0.2 ml/min. The eluate was continuously introduced into an electrospray ionization (ESI) source, and the negative ion mode was used for the ionization.
To calculate the ratios of labeled ions to total isotopomers based on the MS spectra of nucleotide sugars, we first calculated the theoretical isotopic patterns for the natural abundance of each nucleotide sugar using Smart Formula version 3.4. (Bruker Daltonics). Natural isotopic ions (X) were as follows: 13C, 2H, 17O, 18O, and 15N. The relative abundance of the natural isotopic ions of UDP-HexNAc (X0, X1, and X2; m/z = 606.1, 607.1 and 608.1, respectively) was 1:0.20:0.055. The amount of natural isotopic ion with 3 or more (m/z = 609.1 or higher) was negligible (Fig. 2A, panel 0 h) and thus disregarded in the analysis. The peak intensities of the isotopomers were extracted with Data Analysis version 3.1 (Bruker Daltonics). The distribution of isotopic ions after the 13C6-glucose labeling, as shown in Fig. 2A (panels 0.5 h, 2 h, and 6 h), was a result of the incorporation of 13C6-hexose and its metabolites (13C5-ribose and 13C2-acetyl moieties) into UDP-GlcNAc, in combination with the natural occurrence of X0 - X2 mass isotopomers (supplemental Table S1). For example, the isotopic ion of m/z = 614 contains 13C8-(hexose and N-acetyl) labeled UDP-GlcNAc, along with isotopic ions of X2[natural] + 13C6-hexose[labeled] and X1[natural] + 13C7-(ribose and N-acetyl)[labeled] UDP-GlcNAc. We have calculated the intensity of the 13C8-(hexose and N-acetyl) labeled ion by subtracting those for the second and third compounds (calculated based on the relative abundance of natural isotopic ions of UDP-HexNAc, i.e. X0/X1/X2 = 1:0.20:0.055, as mentioned above) from the observed intensity. To determine the combined intensity of 13C6-hexose labeled ions, we summed the total intensity of the 13C6-hexose labeled ions (m/z = 612, 614, 617 and 619) and those with natural isotopic replacement (X1: m/z = 613, 615, 618 and 620; X2: m/z = 614, 616, 619, and 621). Similarly, the intensities of the 13C5-ribose labeled motifs were calculated from the isotopic ions detected at m/z = 611, 613, 617, and 619. The intensities of the 13C2-acetyl residues were calculated from the isotopic ions detected at m/z = 608, 613, 614, and 619. The total intensity of all peaks between m/z = 606 and m/z = 621 was used as a denominator in calculating the replacement ratio of each labeled structural motif (Fig. 2C).
Fig. 2.

Time-dependent changes of mass isotopomers of 13C-UDP-HexNAc and 13C-CMP-NeuAc. Mass isotopomers of (A) 13C-UDP-HexNAc and (B) 13C-CMP-NeuAc. The mouse hepatoma cell line (Hepa1–6) was cultured in medium containing 5.5 mm 13C6-glucose for 0, 0.5, 2 and 6 h. Prepared nucleotide sugar fractions were dissolved in 50 μl water and the portions (20 μl) were injected onto a LC-MS system. 13C6-glucose was metabolized to various precursors used in UDP-GlcNAc synthesis, such as GlcN-6-P, UTP and acetyl-CoA, leading to the incorporation of various structural elements (“motifs”) labeled with 13C in UDP-HexNAc (UDP-GlcNAc plus UDP-GalNAc). The UDP-GlcNAc is a substrate for CMP-NeuAc biosynthesis. The UDP-HexNAc isotopomers contain combinations of 13C6-hexose (red), 13C5-ribose (blue) and a 13C2-acetyl residue (green), whereas the CMP-NeuAc isotopomers have a 13C3-pyruvate motif (orange) in addition to the motifs mentioned above. C, The replacement ratio of each labeled structural motif. The values for UDP-HexNAc were calculated for the sums of isotopomers except for the native abundance. The values for CMP-NeuAc are represented as possible ranges as described in the Experimental Procedures section.
The possible replacement patterns of the structural motifs in CMP-NeuAc were also calculated as the combination of isotopic ions labeled at 13C6-hexose, 13C5-ribose, 13C2-acetyl, and 13C3-pyruvate (Supplemental Table 1). To estimate the replacement ratio of the 13C6-hexose motif, we calculated the combined intensities of isotopic ions detected at m/z = 619, 622, 624, 626, 627, and 629. The ion at m/z = 621.1 had two possible patterns of labeling (13C6-hexose + 13C2-acetyl and 13C5-ribose + 13C3-pyruvate), and hence we represented the replacement ratios of 13C6-hexose as possible ranges (maximum: including this peak; minimum: not including this peak).
Determination of Nucleotide Sugars by Ion-pair Reversed-phase HPLC
Nucleotide sugars such as UDP-GlcNAc and CMP-NeuAc were determined by HPLC as previously reported (18). The portions of labeled nucleotide sugars prepared above were quantified with UV detection, and nucleotide sugar levels were normalized as pmol/mg protein. The protein contents were determined using a BCA protein assay reagents (Pierce, Rockford, IL).
Preparation of Whole Cellular Proteins
Whole cellular proteins were prepared according to a previous report (21). In brief, cells (5 × 106 cells) were extracted for 30 min at room temperature with 1 m dithiothreitol (16.7 μl) and 30 mm Tris-HCl buffer (pH 8.5) (267 μl) containing 7 m urea, 2 m thiourea, and 4% CHAPS. Benzonase (125 mU/5 μl) was also added to the extraction buffer to reduce the viscosity of the cell lysates. The extracted cell lysates were centrifuged at 15,000 × g for 15 min, and then the supernatants were collected in new 2.0 ml tubes. The cellular extracts were filled up with precipitation buffer (85% acetone, 5% triethylamine, and 5% acetic acid in water), and incubated for 30 min at −30 °C. Samples were centrifuged at 15,000 × g for 15 min, and then the pellets were twice washed with 75% ethanol (2.0 ml). After samples were dried under vacuum, the precipitated proteins were stored at −25 °C if not used immediately.
Enzymatic Release of Cellular N-glycans
N-Glycans were enzymatically released with PNGase F from the whole cellular protein prepared as described above and blotted onto PVDF membranes (22, 23). In brief, PVDF membranes were prewet with 100% methanol and water, and then dried. The whole cellular proteins were redissolved in 10% SDS (10 μl) and were dot-blotted (2.5 μl × 3) on the PVDF membranes. Protein spots were cut from the PVDF membranes and placed in separate wells of a 96-well microtiter plate. The spots were covered with 100 μl of 1% (w/v) poly(vinylpyrrolidone) 40,000 in 50% (w/v) methanol for 20 min, and washed with water (100 μl × 5 times). To release the N-glycans, PNGase F (3 U) in 10 μl in 20 mm phosphate buffer (pH 7.2) was added to each sample well and incubated at 37 °C for 15 min. An additional 10 μl of water was added to each well and incubated at 37 °C overnight. During the incubation, the sample wells were sealed with an adhesive plastic strip to prevent evaporation. After the sample plate was sonicated, released N-glycans (20 μl) were collected in new tubes and each sample well was washed with water (40 μl × 3), and the washing solution was combined. After the addition (24 μl) of 100 mm ammonium acetate buffer (pH 5.0), the samples were incubated for 1 h at room temperature, and were used as the N-glycan samples.
Preparation of Cellular O-glycan Alditols
After the removal of N-glycans, O-glycans were released from the proteins remaining on the PVDF membrane with 0.5 m NaBH4 in 50 mm KOH (20 μl) for 16 h at 50 °C. The samples were neutralized with 1 μl glacial acetic acid as described (23). O-Glycan samples were purified using AG-X8 cation exchange microcolumns.
Preparation of N-glycan alditols
N-Glycan samples were reduced with 1 m NaBH4/50 mm KOH (20 μl) at 50 °C for 3 h to prevent the separation of anomers by graphite carbon. The samples were neutralized with 1 μl glacial acetic acid and purified with carbon microcolumns as described (22). The N-glycan alditols were collected in 0.1% formic acid containing 40% acetonitrile and dried under vacuum.
Graphite Carbon LC-ESI-MS and ESI-MS/MS for Isotopomers Analysis of Glycan Alditols
Capillary LC-ESI-MS and ESI tandem MS (ESI-MS/MS) were performed using an Agilent 1100 series HPLC Cap LC system coupled to an Esquire HCT (23). N- and O-glycan alditols (5 × 105 cells) were applied to a HyperCarb column (5 μm, 0.32 mm I.D. × 100 mm; Thermo Hypersil, Runcorn, UK). Mobile phases were: (A) 10 mm NH4HCO3 and (B) acetonitrile. The elution gradient for N-glycans was slightly modified as follows: 2–16% (v/v) linear gradient of B over 45 min; 16–25% linear gradient of B over 20 min, 25–45% linear gradient of B over 15 min, 45% of B for 10 min and re-equilibrated with A for 20 min. O-glycans were separated using the following conditions: 0–25% (v/v) linear gradient of B over 25 min and 25–80% (v/v) linear gradient of B for 15 min. The flow rate was maintained at 5 μl/min.
The MS spectra were obtained in the negative ion mode over the mass range m/z 300 to 2500. The scan rate was 8100 amu for both the MS and MS/MS modes. Monoisotopic masses were assigned with possible monosaccharide compositions from m/z using the GlycoMod tool available on the ExPASy server (http://web.expasy.org/glycomod/), with parameters of ± 0.3 Da in mass tolerance. All glycan structures were described using the Essentials of Glycobiology symbolic drawing guidelines (24). Validation of the technical reproducibility was carried out using known glycans derived from fetuin before analyzing each sample.
To quantify the ratios of unlabeled ions to total isotopomers, 12C/(12C + 13C), for each glycan, the peak intensities of the isotopomers were extracted and then evaluated manually with Data Analysis version 3.1 (Bruker Daltonics). We have evaluated the theoretical natural isotopic patterns of each glycan using Smart Formula version 3.4. (Bruker Daltonics). The 13C-labeled ions were included not only in the expected mass (from the number of GlcNAc, GalNAc, or NeuAc) isotopomers but also in the high mass isotopomers. For example, for the high-mannose type Man9GlcNAc2, a series of ions from m/z = 941.3 to m/z = 942.8 was calculated as the 12C-unlabeled ions, and a series of ions from m/z = 943.3 to m/z = 945.8 was calculated as the 13C-labeled ions.
Quantitative Analysis of Pyridylaminated N-glycans
N-Glycans released from the PVDF membrane were pyridylaminated (25). Excess reagents in PA derivatization were removed by phenol-chloroform extraction (26), followed by gel filtration on a Sephadex G-15 column (1 × 30 cm). Purified N-glycans were quantitatively determined based on the peak areas by weak anion-exchange chromatography on a DEAE-5PW column (27). The neutral fraction (flow-through from the anion exchange column) was further separated by reversed-phase HPLC to quantify high mannose and asialo complex type N-glycans separately (28).
Hyaluronan ELISA Assay
Cells were cultured on 100-mm dishes until 70% confluent, and then washed with serum free DMEM three times and maintained in the serum-free medium for 24 h. The cells were counted and portions of the medium were used for ELISA, in accordance with the manufacturer's instructions.
RESULTS
Experimental Design
To trace the cellular biosynthesis and use of GlcNAc molecules, we designed a strategy based on mass isotopomer analysis of stable isotope-labeled nucleotide sugars and glycans (Fig. 1).
Fig. 1.

Overall strategy to trace UDP-GlcNAc metabolism by LC-MS. A, For isotopomer analysis of nucleotide sugars, cells were cultured in medium containing 5.5 mm 13C6-glucose and nucleotide sugars fractions were then extracted as described in the experimental procedures. The resulting isotopomer ratios of 13C-UDP-HexNAc (UDP-GlcNAc plus UDP-GalNAc) and 13C-CMP-NeuAc allows the measurement of the UDP-GlcNAc synthesis and use pathway, respectively. B, For isotopomer analysis of N- or O-glycans, cells were cultured in medium containing 5.5 mm glucose and 1 mm 13C2-glucosamine, and the glycan alditols were analyzed by LC-MS. The resulting isotopomers of each glycan reflect UDP-GlcNAc use in each step of glycan biosynthesis.
First, for tracing nucleotide sugar synthesis, we used 13C6-glucose (labeled at all six carbons), which allowed us to analyze the relative contributions of various metabolic pathways starting from glucose and eventually converging on nucleotide sugars, based on the replacement ratios of various structural elements or “motifs” in nucleotide sugars (Fig. 1A). For example, metabolites of 13C6-glucose are incorporated into UDP-glucose not only as the 13C6-hexose motif (i.e. glucose) but also as a 13C5-pentose motif (i.e. ribose); isotopomers of UDP-glucose showed signal shifts of five and six mass units reflecting the labeling at ribose and glucose, respectively, and also showed an 11-mass shift when labeled at both motifs. Since the separation of UDP-GlcNAc from UDP-GalNAc was incomplete using the experimental chromatographic conditions (20), the relative contribution of the UDP-GlcNAc synthetic pathway was monitored using the replacement ratios of each motif in 13C-UDP-HexNAc. The isotopomer ratio of 13C-CMP-NeuAc was also analyzed to quantify the degree of UDP-GlcNAc use in CMP-NeuAc de novo synthesis.
To monitor UDP-GlcNAc use in glycan synthesis, we established a second method that combined glycomic analysis with 13C2-glucosamine labeling instead of 13C6-glucose (Fig. 1B), to avoid the complications contained in the mass spectra of 13C6-glucose labeled glycans (supplemental Fig. S1B and C). Labeling with 13C2-glucosamine led to specific labeling of UDP-GlcNAc, UDP-GalNAc and CMP-NeuAc, and subsequent labeling of GlcNAc, GalNAc and sialic acid residues in glycans. It was expected that the isotopomers in each glycan reflect the turnover speed of each N- and O-glycan. In particular, the mass shifts in isotopomers of sialyl N-glycans reflect the efficiencies of sialylation and branching formation in N-glycan synthesis. By combining the data obtained from both techniques, we could extensively trace UDP-GlcNAc metabolism from glucose or glucosamine, eventually converging to N- and O-glycans.
Tracing of UDP-GlcNAc Synthesis Based on Mass Isotopomers of UDP-HexNAc
To demonstrate the feasibility of our strategy, we first investigated the mass isotopomers of 13C-UDP-HexNAc (UDP-GlcNAc and UDP-GalNAc) in a hepatoma cell line (Hepa1–6) in a time-dependent manner. The isotopomers of UDP-HexNAc had complex mass signals (Fig. 2A), as reported previously (15), because UDP-GlcNAc is synthesized from various precursors including glucose, UTP and acetyl-CoA, all of which contain structural elements ultimately derived from glucose. In order to reduce the data complexity because of the labeling at pyrimidine bases with 13C2-aspartate, we limited the labeling period to 6 h or less. Compositions of each isotopic ion of UDP-HexNAc were assigned by MS/MS analysis (supplemental Fig. S2).
An unlabeled [M-H]− ion of UDP-HexNAc was detected at m/z = 606.1 (Fig. 2A, 0 h). After labeling for 0.5 h, isotopic ions corresponding to six and eight mass unit increases appeared at m/z = 612.1 and 614.1, and these ions had increased at 2 h, indicating that UDP-HexNAc was labeled with 13C6-hexose and a 13C2-acetyl residue (supplemental Fig. 2). The labeling with a 13C5-ribose was delayed compared with that of 13C6-hexose and the 13C2-acetyl residue (Fig. 2A, 2 h). To quantify the replacement ratios of various structural elements, we calculated the replacement ratios of each motif in UDP-HexNAc using a similar approach reported previously (15) (Fig. 2C).
The replacement ratios of 13C6-hexose, 13C5-ribose and 13C2-acetyl motifs at 2 h were 42.9%, 22.2%, and 26.6%, and increased to 49.2%, 36.6%, and 39.6% at 6 h, respectively. The data were not consistent with the previous results that the 13C5-ribose motif was first labeled (15); however, this could be related to the specifics of the cell type in the UDP-GlcNAc synthetic pathway. Thus, the isotopomers of UDP-HexNAc should prove useful as an indicator to monitor the relevant contribution of the UDP-GlcNAc synthetic pathway, including the hexosamine pathway, pentose phosphate pathway, and glycolysis.
Tracing of UDP-GlcNAc Use Based on Mass Isotopomers of CMP-NeuAc
We next investigated the mass isotopomers of CMP-NeuAc (Fig. 2B, 0 h). Compositions of each isotopic ion of CMP-NeuAc were assigned by MS/MS analysis (supplemental Fig. S3). After labeling for 0.5 h, a characteristic isotopic ion corresponding to a three-unit mass increase appeared at m/z = 616.1, indicating that CMP-NeuAc was labeled with 13C3-pyruvate, a product of glycolysis. The labeled ion with 13C3-pyruvate was immediately evident compared with that of the 13C2-acetyl residue (Fig. 2B, 2 h), because the 13C2-acetyl residue is derived from acetyl-CoA, a product downstream of glycolysis. Furthermore, the relative peak intensities of the isotopic ions in CMP-NeuAc, labeled with 13C6-glucose (m/z = 622.1) and the 13C2-acetyl residue (m/z = 624.1), compared with the unlabeled ion (m/z = 613.1) (Fig. 2B, 2 h), were weaker than those of the equivalent isotopic ions in UDP-HexNAc at m/z = 612.1 and m/z = 614.1 (Fig. 2A, 2 h). These data indicate that the labeling rates of 13C6-hexose and 13C2-acetyl motifs in CMP-NeuAc were lower than those in UDP-GlcNAc as expected; NeuAc lies downstream of UDP-GlcNAc.
Thus, the 13C6-hexose motif in CMP-NeuAc is incorporated only via the synthetic pathway (Fig. 2, upper), suggesting that the replacement ratio of the 13C6-hexose motif in CMP-NeuAc is a marker of tracing UDP-GlcNAc use in CMP-NeuAc de novo synthesis. The ratios of 13C6-hexose replacement in CMP-NeuAc was in the range of 23.1–28.3% at 2 h, and increased to 37–50% at 6 h (Fig. 2C).
Comparison of UDP-GlcNAc Metabolism Between Hepatoma and Insulinoma Cell Lines
Based on the replacement ratio of each motif of the nucleotide sugars from 12C to 13C isotopomers, we compared the metabolic flow between Hepa1–6 and pancreatic insulinoma (Min6) cell lines (29, 30). We previously found that the hepatic glucose transporter 2 (GLUT2) is highly sialylated (unpublished data). In contrast, pancreatic β-cell GLUT2 was rarely sialylated (31, 32). Glycomic profiling of the N-glycans in pancreatic β-cells also revealed that sialylated N-glycans are rarely expressed in the pancreas (33). Thus it was anticipated that Hepa1–6 and Min6 have different UDP-GlcNAc metabolisms with respect to glycosylation.
In Min6 after labeling for 6 h (Fig. 3A), the ratios of 13C6-hexose and 13C5-ribose in UDP-HexNAc isotopomers were slightly lower than those in Hepa1–6. Notably, the ratios of the 13C6-hexose motif in CMP-NeuAc was remarkably lower in Min6 (6.5–8.9%), compared with that in Hepa1–6 (37.2–51.0%) (Fig. 3B). Conversely, the unlabeled ions of CMP-NeuAc were the major ones detected in Min6 up to 24 h (Fig. 3C) where the ratio of unlabeled ion to total isotopomers was 57% (hatched line). In contrast, the ratio in Hepa1–6 rapidly decreased below 20% after 6 h and completely disappeared after 24 h (Fig. 3C, solid line). These data indicate that the flow of CMP-NeuAc de novo synthesis from UDP-GlcNAc (UDP-GlcNAc use) in the insulinoma cell line was much slower compared with that in the hepatoma cell line.
Fig. 3.
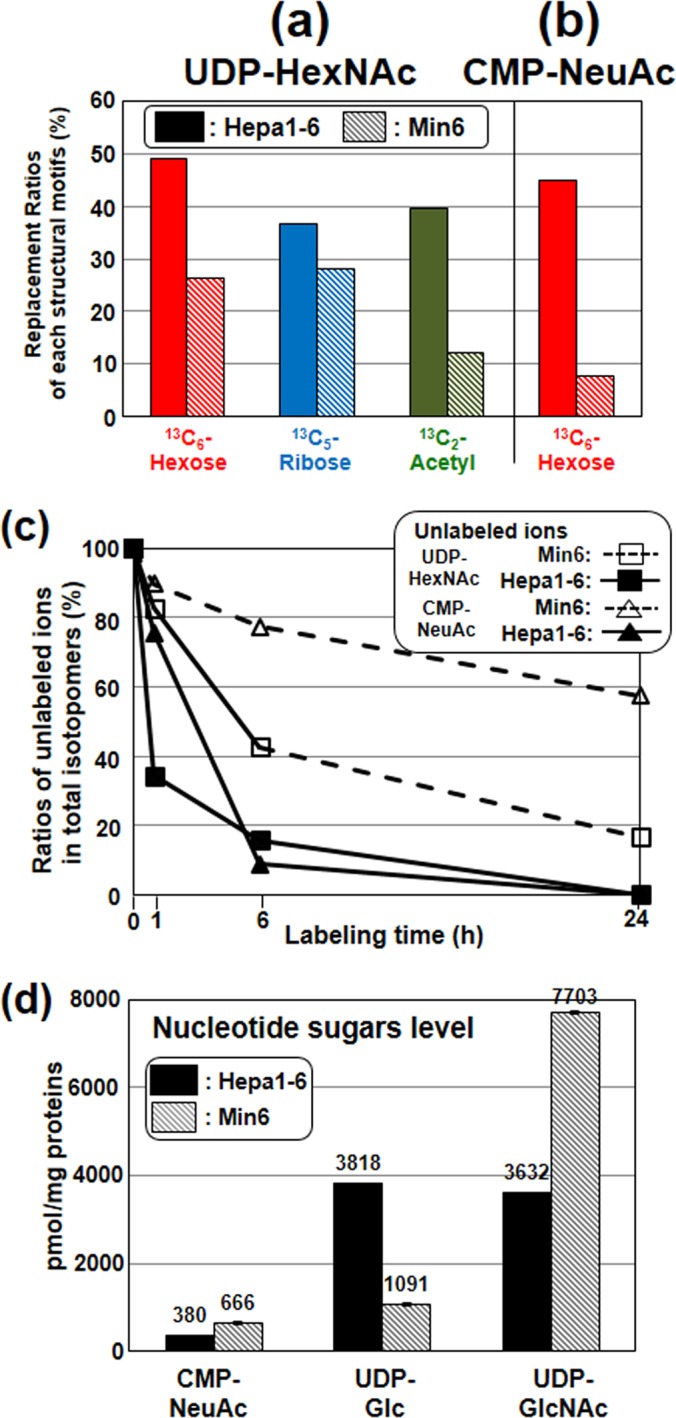
Comparison of UDP-GlcNAc metabolism between hepatoma (Hepa1–6) and pancreatic insulinoma cell lines (Min6). 13C-Nucleotide sugars in both cell lines were analyzed using the same conditions described in Fig. 2. (A and B): The replacement ratios of labeled motifs in UDP-HexNAc and CMP-NeuAc after labeling for 6 h. The values were calculated by the same procedure described in Fig. 2. C, Time course of the ratios of unlabeled ions in UDP-HexNAc and CMP-NeuAc isotopomers. Symbols are defined in the figure. D, CMP-NeuAc, UDP-Glc and UDP-GlcNAc in both cell lines were determined by HPLC with UV detection. Triplicate samples were analyzed and the data were normalized to units of pmol/mg protein.
As confirmed by the quantitative HPLC analysis of the nucleotide sugars as shown in Fig. 3D (18), Min6 had significantly higher UDP-GlcNAc and CMP-NeuAc levels than Hepa1–6. Gene expression levels of various related enzymes such as GNE (UDP-GlcNAc 2-epimerase/ManNAc kinase) and SLC35a1 (CMP-NeuAc transporter) were similar or higher in Min6 compared with Hepa1–6 (supplemental Fig. S4). These data imply that the slow CMP-NeuAc synthesis in the insulinoma cell line seems to be a result of the slow epimerization from UDP-GlcNAc by feedback inhibition of the activity of UDP-GlcNAc epimerase (10).
Tracing of UDP-GlcNAc and CMP-NeuAc Use in Glycan Synthesis Based on Mass Isotopomers of N- and O-Glycans
To dynamically trace the use of UDP-GlcNAc and CMP-NeuAc in glycan synthesis, we further established a second method based on the isotopomer analysis of N- and O-glycans obtained by sequential release from the cells metabolically labeled with 13C2-glucosamine for 24 h (Fig. 1B). These glycan pools were analyzed as their alditol forms by graphite carbon capillary LC-ESI-MS and ESI-MS/MS to separate isometric glycans. Fig. 4A and b show the base peak chromatograms of N- and O-glycans of Hepa1–6 before metabolic labeling. Glycan structures were identified according to previous reports (23). Table I lists common glycans found in both Hepa1–6 and Min6 cell lines. High-mannose type N-glycans [Hex]5–10[HexNAc]2 (peaks 1–6, Fig. 4A and Table I) were abundantly detected and hybrid type N-glycans (peaks 7–9) and core-fucosylated, or non-fucosylated complex type N-glycans with mono-, or di-sialic acid residues (peaks 10–16) were also detected. These isobaric structures of the NeuAc linkage, or fucose residues alone or in combination, were separated and identified as reported previously (23). In addition, sialyl core 1 and disialyl T (peaks 17–18, Fig. 4B and Table I) were detected in the O-glycan samples.
Fig. 4.

LC-MS profiles and the isotopomers of 13C-labeled glycan alditols. Hepa1–6 was cultured in a medium containing 13C2-glucosamine for 24 h. N- and O-glycan alditols were analyzed by LC-MS as described in the experimental procedures. (A and B): Base peak chromatograms of N- and O-glycan alditols. C–F Mass spectra of isotopomers of (C) peak 4 (Man8GlcNAc2), (D) peak 13 (nonfucosylated disialo-biantennary glycans), (E) peak 15 (core-fucosylated disialo-biantennary glycans) and (F) peak 17 (sialyl core 1). Labeling with 13C2-glucosamine led to increases in the mass signals because of the labeling with the motifs highlighted with bold asterisks. G, Time course of the ratios of unlabeled ions in each glycan. Symbols are defined in the figure.
Table I.
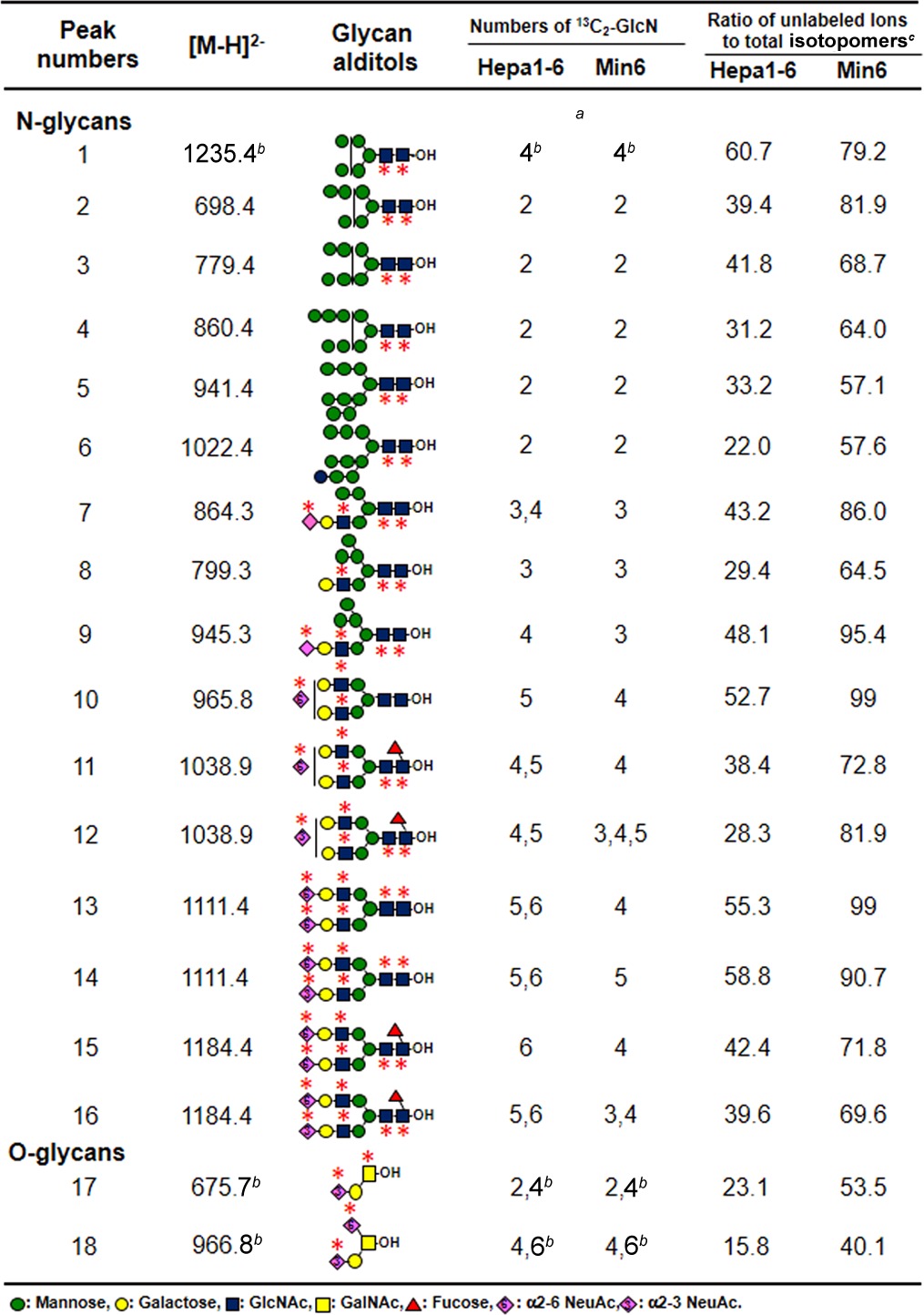
a The mass shifts of isotopomers represent the numbers of incorporated 13C2-GlcN in each glycan.
b The incorporations of one 13C2-GlcN in these glycans lead to the 2 mass shift for one 13C2-GlcN residue. since the glycans were detected as single charged ions.
c Ratio of unlabeled ions to total isotopomers were calculated by the procedure described in “Experimental Procedures”.
After labeling with 13C2-glucosamine, the incorporation of one 13C2-glucosamine residue in the N-glycans, except for peak 1, resulted in a mass shift of m/z = 1 in the isotopomers, because the glycans were detected as doubly charged ions (Fig. 4C–4E). O-Glycans and peak 1 were detected as single charged ions, and hence the incorporation of one 13C2-glucosamine residue resulted in a two-mass unit shift (Fig. 4F). Thus, the mass shifts of the isotopomers can be used to calculate the number of incorporated 13C2-glucosamine residues in each glycan structure (mass numbers increase when 13C2-glucosamine is incorporated into GlcNAc, GalNAc and NeuAc residues). For example, the m/z of the isotopomers of peak 4 (Man8GlcNAc2) increased by 2 m/z units owing to the labeling with two GlcNAc residues in the N-glycan cores as indicated with asterisks (Fig. 4C). The m/z of the isotopomers of peaks 13 and 15 (biantennary glycans with two α2–6 sialic acids) increased up to 6 m/z units owing to the labeling with four GlcNAc and two NeuAc residues (Fig. 4D and 4E). The mass shifts of other glycans are summarized in Table I.
Since 13C2-GlcN is converted to various metabolites via Fruc-6-P (16), we assumed that there are multiple ways by which 13C originating from 13C2-GlcN is incorporated into glycans. To examine this possibility, the compositions of the mass isotopomers of [Hex]9[HexNAc]2-ol (peak 5), which has a single expected structure (Man9GlcNAc2-ol), were evaluated by MS/MS analysis (supplemental Fig. S5). The isotopomers of ions expected from the number of HexNAc residues, detected at m/z = 943.32−, were observed at m/z = 862.2, corresponding to a loss of hexose (m/z = 81.12−). In addition, the isotopomers of the fragment ions of the isotopic ions 2 units above the expected mass, detected at m/z = 944.32−, showed fragment ions at m/z = 862.32−, which correspond to the loss of 13C2-hexose (m/z = 82.12−). These data indicate that a fraction of the glycan was also labeled with 13C2-mannose by the conversion of 13C2-GlcN to Fruc-6-P. In this study, for the comparison of labeling efficiencies, we calculated the ratios of unlabeled ions to total isotopomers in each glycan (Hepa1–6, Table I right).
Fig. 4G shows the time-dependent reduction of unlabeled ions in typical glycans (peaks 4, 13, 15 and 17). The ratio of unlabeled O-glycans (peak 17) was found to decrease rapidly below 23% after 24 h, whereas the ratios of N-glycans modestly decreased as follows: high mannose type (peak 4), 31%; core-fucosylated glycans (peak 15), 42%; and nonfucosylated glycans (peak 13), 55%. Among the high mannose type glycans, the unlabeled ions of Man8–9GlcNAc2 (peaks 4–5) and Glc1Man9GlcNAc2 (peak 6) rapidly decreased, compared with those representing the short high-mannose glycans (peaks 1–3), because Man8–9GlcNAc2 is the precursor of all the others. Core-fucosylation and sialylation also affected variations in labeling efficiencies as follows. The ratio of the unlabeled ion of monosialo-glycan (peak 9) showed high values when compared with the asialo glycan (peak 8); that of disialo-glycans (peak 15) also showed a high ratio when compared with the monosialo-glycans (peaks 11), because sialo N-glycans are synthesized later. On the other hand, core-fucosylated glycans (peaks 11 and 15) showed rather lower values than those of non-fucosylated glycans (peaks 10 and 13) for unknown reasons.
Importantly, most of UDP-HexNAc and CMP-NeuAc in Hepa1–6 were 13C-labeled after 24 h of 13C2-GlcN labeling (supplemental Fig. S6), indicating that the mass isotopomers in each glycan reflect the synthetic ratios of the glycans rather than the availability of labeled nucleotide sugars. Therefore, the mass isotopomers of glycans should prove useful as an indicator of UDP-GlcNAc metabolism in glycan synthesis.
Comparison of N-Glycan Synthesis Between Hepatoma and Insulinoma Cell Lines
Next we investigated whether the slow metabolic flow in CMP-NeuAc, as mentioned above, affects the labeling efficiencies of glycans in the insulinoma cell line, because CMP-NeuAc was barely labeled with 13C2-GlcN (supplemental Fig. S6). We compared the dynamic turnover of N-glycans in Hepa1–6 and Min6 cell lines (Fig. 5, supplemental Fig. S7). The isotopomers of peak 4 (Man8GlcNAc2) and peak 15 (core-fucosylated disialo-glycans) were taken as examples. The isotopomers of peak 4 were increased by 2 m/z units in both cell lines (Table I). The ratio of unlabeled ion remained high in the insulinoma cell line (Table I, right), suggesting that the overall turnover of N-glycans is slower in the Min6 cell line.
Fig. 5.
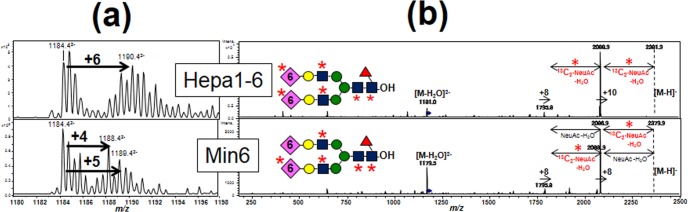
Comparison of the isotopomers of 13C-labeled glycan alditols. A, Mass spectra of isotopomers of peak 15 (core-fucosylated disialo-biantennary glycans) derived from Hepa1–6 and Min6 cells after labeling for 24 h, respectively. Labeling with 13C2-glucosamine led to increases of 4–6 mass units in peak 15. B, MS/MS spectra of peak 15 from Hepa1–6 and Min6 for the labeled parent ion at m/z = 1190.4 and m/z = 1188.4.
With regard to the peak 15, we observed that the isotopomers with a mass increase of 4 or 5 m/z units were major species in Min6, whereas those with a mass increase of 6 m/z units were found in Hepa1–6 (Fig. 5A).
To identify the labeled sugar residues derived from 13C2-glucosamine, we further performed MS/MS analysis of the captured major isotopic ions at m/z = 1190.42− and m/z = 1188.42− in each cell line (Fig. 5B). In both MS/MS spectra, desialylated fragment ions at m/z = 1795.8, corresponding to the loss of two 13C2-NeuAc motifs, were detected as a single charged ion. In addition, we observed the monosialylated fragment ion at m/z = 2088.9 in Hepa1–6 showing a 293 mass difference (13C2-NeuAc-H2O), whereas in Min6, we also observed a peak at m/z = 2086.9 with a 291 mass fragment (unlabeled NeuAc-H2O), compared with the desialylated fragment ion at m/z = 1795.8 (Fig. 5B). These data indicate that NeuAc residues were not completely labeled in the insulinoma cell line, which reflects the low efficiency of the cell line in de novo sialic acid biosynthesis compared with the hepatoma cell line.
Quantitative Analysis of N-Glycans and Secreted Hyaluronic Acids
To quantitatively evaluate the amounts of sialo N-glycans in each cell line, we quantified the total N-glycans by HPLC with fluorescent detection after pyridylamination (Fig. 6A) (25). Total N-glycans derived from both cell lines were separated by anion exchange chromatography according to their sialic acid content (27), and neutral N-glycans were further analyzed by reversed-phase mode (supplemental Fig. S8) (28). As shown in Fig. 6A, there was no significant difference in the total amounts of high mannose type N-glycans between these cell lines. Interestingly, Min6 had asialo complex type N-glycans in abundance, whereas the amounts of di, tri and tetra-sialo N-glycans were much smaller in Min6 compared with those in Hepa1–6 (Fig. 6A). These data, combined with the data we obtained from the stable isotope labeling of glycans, implied that the slow de novo synthesis of CMP-NeuAc resulted in the poor sialylation of N-glycans in insulinoma cells.
Fig. 6.

Quantitative analysis of N-glycans and secreted hyaluronan. (a) Cellular pyridylaminated N-glycans in both cell lines were quantified by HPLC with fluorescence detection. The data are represented as pmol per 1 × 107 cells. The amounts in the six categories (high mannose, asialo, monosialo, disialo, trisialo and tetrasialo types) of N-glycans in Hepa1–6 and Min6 cells were quantified based on the peak areas of the chromatograms. B, Secreted hyaluronic acids were quantified by ELISA, and the data represented as pmol per 1 × 107 cells. The amount of secreted hyaluronic acid in mouse fibroblasts that are known to highly express hyaluronic acid was also quantified as a control sample.
Because hyaluronic acid is a major type of glycoconjugate containing GlcNAc, we further quantified secreted hyaluronic acids by an ELISA assay. No significant differences were observed between the two cell lines (Fig. 6B). The amounts in the two cell lines were 26 times lower than those from a fibroblast line in which hyaluronic acid is highly expressed (34). O-GlcNAc modifications were also analyzed and no significant differences were observed by Western blot analysis between the two cell lines (supplemental Fig. S9). Taken together, hyaluronic acids and O-GlcNAc modifications may not compete with N- or O-glycosylation for the UDP-GlcNAc pool (i.e. they may not have a major impact on the use, or turnover of UDP-GlcNAc), at least in the cell lines we have studied.
DISCUSSION
We proposed a strategy for the dynamic tracing of cellular GlcNAc use (i.e. “GlcNAc cycle”) by a combination of two LC-MS methods based on mass isotopomer analysis. Our combination of isotopomer analyses of labeled nucleotide sugars and glycans is ideal for systematically assessing the UDP-GlcNAc synthetic pathways that modulate nucleotide sugar levels, and for further characterizing how nucleotide sugar changes affect the synthesis of glycans with specific structural motifs. However, we still do not have the equations to quantify the metabolic flow based on the replacement ratios in each structural motif. Further efforts that incorporate a systems biology-type approach are required.
Our approach using LC-MS has particular advantages over the methods proposed previously. 1) LC-MS analysis of 13C-labeled nucleotide sugars (first method) can simultaneously observe the isotopomers of other nucleotide sugars. Thus, the method can reveal not only the contribution of relevant pathways to UDP-GlcNAc metabolism but also provide an overview of the metabolic flow leading to the synthesis of other nucleotide sugars. For example, the reduction of unlabeled UDP-GlcA ions (supplemental Fig. S10, black cycle), which lie downstream of UDP-Glc in the biosynthetic pathway, was quite slow, compared with that of UDP-Glc. 2) Analysis of glycans labeled with 13C2-glucosamine (second method) can evaluate the variations of the labeling pattern in each structural motif because 13C2-glucosamine efficiently labels GlcNAc, GalNAc, and NeuAc residues in glycans. Indeed, glycans synthesized through a simple process (O-glycans or high mannose type N-glycans) showed high labeling efficiencies (Fig. 4G and Table I). We found higher labeling efficiencies of core-fucosylated N-glycans (Table I), even though fucosylated glycans undergo an additional modification, which is not derived from glucosamine. 3) Because LC-MS analysis can roughly separate complex cellular glycans, this method enabled us to evaluate the labeling efficiencies of each isometric glycan separated on a graphite carbon column. The directly infused approach that relies on permethylated glycans metabolically labeled with 15N-glutamate in the hexosamine pathway has been reported previously (17). However, tracing the dynamics of isometric glycans is not approachable by a directly infused approach. Indeed, we also found variations in the labeling efficiencies in positional isomers of α2–3- and α2–6-sialylated N-glycans (peaks 11 and 12, and peaks 15 and 16).
Importantly, 4) our approach can be also applied for monitoring the metabolic flow of other glycoconjugates. For example, the mass isotopomers of the glycans derived from gangliosides and secreted hyaluronic acids could be evaluated in a similar manner, as performed for N- and O-glycans (supplemental Table S2). Hepa1–6 abundantly expressed GM2 (supplemental Fig. S11), consistent with a previous report (35); however, the labeling efficiency was lower than those of O-glycans for unknown reasons. In contrast, the labeling efficiency of hyaluronic acids was much higher than other groups of glycans evaluated in this study, likely because hyaluronic acids are synthesized on the plasma membrane using the cytosolic UDP-GlcNAc pool. This is the first report of radioisotope-independent detection of glycan synthesis in cells. At present, we are analyzing the mechanism behind these glycan dynamics.
The limitation of our approach is that the metabolic flow to O-GlcNAcylation from UDP-GlcNAc is not effectively monitored because O-GlcNAcylation is not approachable by a glycomic approach (36). Currently, we are also developing a proteomic approach that identifies 13C-GlcNAc labeled glycoproteins including O-GlcNAc modified proteins to reveal glycan dynamics at each glycosylation site of glycoproteins.
By applying our approach, we observed slow CMP-NeuAc synthesis (by the first method, Fig. 3) and slow incorporation of labeled NeuAc in sialo N-glycans (by the second method, Fig. 5) in the Min6 insulinoma cell line. Although not mentioned so far, we also found a low labeling efficiency of the 13C2-acetyl residue in UDP-HexNAc (Fig. 3A; the replacement ratio of the 13C2-acetyl residue was 12.5% after 6 h), and the absence of CMP-NeuAc isotopomers labeled at the 13C3-pyruvate motif (supplemental Fig. S12) in Min6 cells. These data are consistent with previous reports that a rat insulinoma cell line has slower glycolysis flux before the tricarboxylic acid cycle than hepatocytes (37). These data imply that low sialylation (Fig. 6) in the insulinoma cell line is caused by slow CMP-NeuAc synthesis, which in turn is caused by slow glycolysis and/or slow epimerization of UDP-GlcNAc caused by feedback inhibition of the epimerase by high CMP-NeuAc levels (Fig. 7). Burleigh et al. reported that lack of glutamine in the medium coupled with reduced glycolysis causes a reduction in the sialylation and branching of N-glycans in recombinant glycoproteins (38). A significant difference between our finding and this finding is that the Min6 insulinoma cell line contained a high level of nucleotide sugars. It is known that cellular NeuAc is a regulator of sialylation (39). In our analysis, the replacement ratio of the 13C5-ribose motif in CMP-NeuAc (an indicator of the salvage pathway from the cellular NeuAc pool) is lower in Min6 compared with Hepa1–6 (supplemental Fig. S12), suggesting that the salvage pathway may be also slightly less active in the Min6 insulinoma cell line. Further studies will be required to explain the regulatory mechanism of glycosylation in the Min6 insulinoma cell line.
Fig. 7.

Model of metabolic flux regulation of sialylation in pancreatic β-cell insulinoma. (1) Slow CMP-NeuAc synthesis is caused by slow glycolysis and/or slow epimerization (hatched line) of UDP-GlcNAc by feedback inhibition of the activity of UDP-GlcNAc 2-epimerase by CMP-NeuAc. (2) low sialylation and (3) low amounts of sialo glycans and abundant asialo N-glycans (hatched line). No significant differences in hyaluronan synthesis and O-GlcNAcylation (solid line).
Nevertheless, our approach is useful in identifying a metabolic bottleneck that affects the turnover speed and patterns of cellular glycosylation. Our findings may be relevant to pancreatic β-cell glycosylation patterns and functions in vivo, as our recent work has indicated that highly branched N-glycans on glucose transporter 2 of the pancreatic β-cell surface have important roles in maintaining glucose transportation leading to insulin secretion (31, 32). Low sialylation of N-glycans on β-cells may help cell surface localization of the glucose transporter 2, because terminal sialylation prevents the interaction between glycans and galactose-binding lectins that maintains the cell surface localization of the transporters (40).
In conclusion, we have demonstrated that our approach is efficient and convenient for cellular GlcNAc tracing. Our method provides valuable information toward understanding dynamic glycosylation regulatory mechanisms, which should lead to new insights in the field of glycobiology.
Supplementary Material
Acknowledgments
We thank Dr. Kazuaki Kakehi and Dr. Mitsuhiro Kinoshita (Kinki University) for providing the protocol for the preparation of whole cellular proteins for glycan analysis, and Dr. Miyako Nakano (Hiroshima University) for helpful suggestions for glycan analysis. Dr. Yoshiki Yamaguchi (Riken) provided us with 13C6-glucose, and Dr. Gerald Hart and Dr. Natasha Zachara provided us the O-GlcNAc antibody (CTD110.6) and cell lysates for the control experiments. We also thank Dr. Takemichi Nakamura and Dr. Yayoi Hongo (Riken) for helping MS analysis of nucleotide sugars by ESI-IT-TOF MS. We also thank Ms. Kaori Tanaka for technical assistances.
Footnotes
* This work is supported by Grant-in-Aid for Scientific Research (A), 20249018 (to NT), Grant-in-Aid for Challenging Exploratory Research, 22659067, Grant-in-Aid for B, 24790301 (to KN), and by the global COE program (to NT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
 This article contains supplemental Figs. S1 to S12.
This article contains supplemental Figs. S1 to S12.
1 The abbreviations used are::
- GlcNAc
- N-acetyl glucosamine
- CMP-NeuAc
- cytidine monophospho-N-acetyl-D-neuraminic acid
- Fruc-6-P
- fructose-6-phosphate
- GlcN
- glucosamine
- ManNAc
- N-acetyl mannosamine
- UDP-Glc
- uridine diphosphate glucose
- UDP-GlcA
- uridine diphosphate glucuronic acid
- UDP-GlcNAc
- uridine diphosphate N-acetyl glucosamine
- UDP-GalNAc
- uridine diphosphate N-acetyl galactosamine
- UTP
- uridine triphosphate
REFERENCES
- 1. Ohtsubo K., Marth J. D. (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 [DOI] [PubMed] [Google Scholar]
- 2. Hakomori S. (2002) Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. U.S.A. 99, 10231–10233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taniguchi N. (2009) From the gamma-glutamyl cycle to the glycan cycle: a road with many turns and pleasant surprises. J. Biol. Chem. 284, 34469–34478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shirato K., Nakajima K., Korekane H., Takamatsu S., Gao C., Angata T., Ohtsubo K., Taniguchi N. (2011) Hypoxic regulation of glycosylation via the N-acetylglucosamine cycle. J. Clin. Biochem. Nutr. 48, 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Slawson C., Copeland R. J., Hart G. W. (2010) O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem. Sci. 35, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oguri S., Minowa M. T., Ihara Y., Taniguchi N., Ikenaga H., Takeuchi M. (1997) Purification and characterization of UDP-N-acetylglucosamine: alpha1,3-D-mannoside beta1,4-N-acetylglucosaminyltransferase (N-acetylglucosaminyltransferase-IV) from bovine small intestine. J. Biol. Chem. 272, 22721–22727 [DOI] [PubMed] [Google Scholar]
- 7. Sasai K., Ikeda Y., Fujii T., Tsuda T., Taniguchi N. (2002) UDP-GlcNAc concentration is an important factor in the biosynthesis of beta1,6-branched oligosaccharides: regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 12, 119–127 [DOI] [PubMed] [Google Scholar]
- 8. Grigorian A., Lee S. U., Tian W., Chen I. J., Gao G., Mendelsohn R., Dennis J. W., Demetriou M. (2007) Control of T Cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J. Biol. Chem. 282, 20027–20035 [DOI] [PubMed] [Google Scholar]
- 9. Keppler O. T., Hinderlich S., Langner J., Schwartz-Albiez R., Reutter W., Pawlita M. (1999) UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science 284, 1372–1376 [DOI] [PubMed] [Google Scholar]
- 10. Sommar K. M., Ellis D. B. (1972) Uridine diphosphate N-acetyl-D-glucosamine-2-epimerase from rat liver. I. Catalytic and regulatory properties. Biochim. Biophys. Acta 268, 581–589 [DOI] [PubMed] [Google Scholar]
- 11. Zamboni N., Fendt S. M., Rühl M., Sauer U. (2009) (13)C-based metabolic flux analysis. Nat. Protoc. 4, 878–892 [DOI] [PubMed] [Google Scholar]
- 12. Toya Y., Ishii N., Hirasawa T., Naba M., Hirai K., Sugawara K., Igarashi S., Shimizu K., Tomita M., Soga T. (2007) Direct measurement of isotopomer of intracellular metabolites using capillary electrophoresis time-of-flight mass spectrometry for efficient metabolic flux analysis. J. Chromatogr. A. 1159, 134–141 [DOI] [PubMed] [Google Scholar]
- 13. Gallinger A., Biet T., Pellerin L., Peters T. (2011) Insights into neuronal cell metabolism using NMR Spectroscopy: Uridyl diphosphate N-acetyl-glucosamine as a unique metabolic marker. Angew. Chem. Int. Ed. 50, 1–4 [DOI] [PubMed] [Google Scholar]
- 14. Wellen K. E., Lu C., Mancuso A., Lemons J. M., Ryczko M., Dennis J. W., Rabinowitz J. D., Coller H. A., Thompson C. B. (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 24, 2784–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moseley H. N., Lane A. N., Belshoff A. C., Higashi R. M., Fan T. W. (2011) A novel deconvolution method for modeling UDP-N-acetyl-D-glucosamine biosynthetic pathways based on 13C mass isotopologue profiles under non-steady-state conditions. BMC Biol. 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz. S., Varki. A. (2009) Metabolic radiolabeling of animal cell glycoconjugates. Curr. Protoc. Protein Sci. 12, 1–55 [DOI] [PubMed] [Google Scholar]
- 17. Orlando R., Lim J. M., Atwood J. A, 3rd, Angel P. M., Fang M., Aoki K., Alvarez-Manilla G., Moremen K. W., York W. S., Tiemeyer M., Pierce M., Dalton S., Wells L. (2009) IDAWG: Metabolic incorporation of stable isotope labels for quantitative glycomics of cultured cells. J. Proteome Res. 8, 3816–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakajima K., Kitazume S., Angata T., Fujinawa R., Ohtsubo K., Miyoshi E., Taniguchi N. (2010) Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology 20, 865–871 [DOI] [PubMed] [Google Scholar]
- 19. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 20. Turnock D. C., Ferguson M. A. (2007) Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot. Cell 8, 1450–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamada K., Kinoshita M., Hayakawa T., Nakaya S., Kakehi K. (2009) Comparative studies on the structural features of O-Glycans between leukemia and epithelial cell lines. J. Proteome Res. 8, 521–537 [DOI] [PubMed] [Google Scholar]
- 22. Wilson N. L., Schulz B. L., Karlsson N. G., Packer N. H. (2002) Sequential analysis of N- and O-linked glycosylation of 2D-PAGE separated glycoproteins. J. Proteome Res. 1, 521–529 [DOI] [PubMed] [Google Scholar]
- 23. Nakano M., Saldanha R., Gobel A., Kavallaris M., Packer N. H. (2011) Identification of glycan structure alterations on cell membrane proteins in desoxyepothilone B resistant leukemia cells. Mol. Cell. Proteomics 10, 11, M111.009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E. (2009) Essentials of Glycobiology 2nd Ed, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 25. Hase S., Ikenaka T., Matsushima Y. (1978) Structure analyses of oligosaccharides by tagging of the reducing end sugars with a fluorescent compound. Biochem. Biophys. Res. Commun., 85, 257–263 [DOI] [PubMed] [Google Scholar]
- 26. Tokugawa K., Oguri S., Takeuchi M. (1996) Large scale preparation of PA-oligosaccharides from glycoproteins using an improved extraction method. Glycoconj. J. 13, 53–56 [PubMed] [Google Scholar]
- 27. Hase S. (1994) High-performance liquid chromatography of pyridylaminated saccharides. Methods Enzymol. 230, 225–237 [DOI] [PubMed] [Google Scholar]
- 28. Korekane H., Korekane A., Yamaguchi Y., Kato M., Miyamoto Y., Matsumoto A., Hasegawa T., Suzuki K., Taniguchi N., Ookawara T. (2011) N-Glycosylation profiling of recombinant mouse extracellular superoxide dismutase produced in Chinese hamster ovary cells. Glycoconj. J. 28, 183–196 [DOI] [PubMed] [Google Scholar]
- 29. Maier K., Hofmann U., Reuss M., Mauch K. (2010) Dynamics and control of the central carbon metabolism in hepatoma cells. BMC Syst. Biol. 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fernandez C., Fransson U., Hallgard E., Spégel P., Holm C., Krogh M., Wårell K., James P., Mulder H. (2008) Metabolomic and proteomic analysis of a clonal insulin-producing beta-cell line (INS-1 832/13). J. Proteome Res. 7, 400–411 [DOI] [PubMed] [Google Scholar]
- 31. Ohtsubo K., Takamatsu S., Minowa M. T., Yoshida A., Takeuchi M., Marth J. D. (2005) Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell 123, 1307–1321 [DOI] [PubMed] [Google Scholar]
- 32. Ohtsubo K., Chen M. Z., Olefsky J. M., Marth J. D. (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med. 17, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takamatsu S., Antonopoulos A., Ohtsubo K., Ditto D., Chiba Y., Le D. T., Morris H. R., Haslam S. M., Dell A., Marth J. D., Taniguchi N. (2010) Glycobiology 20, 4, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tammi R., Tammi M. (1991) Correlations between hyaluronan and epidermal proliferation as studied by [3H]glucosamine and [3H]thymidine incorporations and staining of hyaluronan on mitotic keratinocytes. Exp. Cell Res. 195, 524–527 [DOI] [PubMed] [Google Scholar]
- 35. Suzuki A., Hashimoto Y., Abe M., Kiuchi Y., Yamakawa T. (1984) Adv. Exp. Med. Biol. 174, 263–272 [DOI] [PubMed] [Google Scholar]
- 36. Zachara N. E., Vosseller K., Hart G. W., Gao Y. (2001) Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr. Protoc. Protein Sci. 12, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berman H. K., Newgard C. B. (1998) Fundamental metabolic differences between hepatocytes and islet beta-cells revealed by glucokinase overexpression. Biochemistry 37, 4543–4552 [DOI] [PubMed] [Google Scholar]
- 38. Burleigh S. C., van de Laar T., Stroop C. J., van Grunsven W. M., O'Donoghue N., Rudd P. M., Davey G. P. (2011) Synergizing metabolic flux analysis and nucleotide sugar metabolism to understand the control of glycosylation of recombinant protein in CHO cells. BMC Biotechnol. 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bork K., Reutter W., Gerardy-Schahn R., Horstkorte R. (2005) The intracellular concentration of sialic acid regulates the polysialylation of the neural cell adhesion molecule. FEBS Lett. 579, 22, 5079–5083 [DOI] [PubMed] [Google Scholar]
- 40. Hirabayashi J., Hashidate T., Arata Y., Nishi N., Nakamura T., Hirashima M., Urashima T., Oka T., Futai M., Muller W. E., Yagi F., Kasai K. (2002) Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim. Biophys. Acta 1572, 232–254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.