

Abstract
The fungus Septoria tritici causes the disease septoria tritici blotch in wheat, one of the most economically devastating foliar diseases in this crop. To investigate signaling events and defense responses in the wheat–S. tritici interaction, we performed a time-course study of S. tritici infection in resistant and susceptible wheat using quantitative proteomics and phosphoproteomics, with special emphasis on the initial biotrophic phase of interactions. Our study revealed an accumulation of defense and stress-related proteins, suppression of photosynthesis, and changes in sugar metabolism during compatible and incompatible interactions. However, differential regulation of the phosphorylation status of signaling proteins, transcription and translation regulators, and membrane-associated proteins was observed between two interactions. The proteomic data were correlated with a more rapid or stronger accumulation of signal molecules, including calcium, H2O2, NO, and sugars, in the resistant than in the susceptible cultivar in response to the infection. Additionally, 31 proteins and 5 phosphoproteins from the pathogen were identified, including metabolic proteins and signaling proteins such as GTP-binding proteins, 14–3-3 proteins, and calcium-binding proteins. Quantitative PCR analysis showed the expression of fungal signaling genes and genes encoding a superoxide dismutase and cell-wall degrading enzymes. These results indicate roles of signaling, antioxidative stress mechanisms, and nutrient acquisition in facilitating the initial symptomless growth. Taken in its entirety, our dataset suggests interplay between the plant and S. tritici through complex signaling networks and downstream molecular events. Resistance is likely related to several rapidly and intensively triggered signal transduction cascades resulting in a multiple-level activation of transcription and translation processes of defense responses. Our sensitive approaches and model provide a comprehensive (phospho)proteomics resource for studying signaling from the point of view of both host and pathogen during a plant–pathogen interaction.
Plants mount diverse defense responses in order to survive during attacks by pathogens, and these responses are mediated by the recognition of characteristic non-self structures associated with invading microbes by plasma-membrane-localized receptors in the plant (1). The immediate downstream events of elicitor-receptor recognition include early signal transduction, such as ion fluxes across the plasma membrane, extracellular alkalization, transient increases in nitric oxide and cytosolic calcium concentration, the activation of mitogen-activated protein kinases (MAPKs)1 and calcium-dependent protein kinases (CDPKs) through protein phosphorylation, the production of reactive oxygen species (ROS), the biosynthesis of ethylene and jasmonic acid, and changes in sugar levels (1–3). Eventually, transcriptional cascades and the expression of defense responses, such as the expression of pathogenesis-related (PR) proteins, are activated (1).
Protein phosphorylation, as a rapid, reversible post-translational modification (PTM), achieves a fine-tuned regulation of protein function in a wide array of cellular processes including cellular development, differentiation, proliferation, apoptosis, and defense responses, from signaling cascades to gene expression (4). It appears to be the most predominant PTM in plants in response to pathogens (5). Many signaling components, such as protein kinases, phosphatases, and transcription factors, have been implied in relation to changes of phosphorylation status during plant–pathogen interactions (5). Therefore, studying phosphorylation dynamics is a useful strategy for unraveling the comprehensive signaling events and defense mechanisms in plants in response to pathogens. Previously, phosphoproteomics has been applied to plants treated with elicitors and abiotic stress (1, 6, 7). However, plant responses to living organisms (pathogens), as opposed to elicitor molecules or abiotic stresses, involve networks of molecular mechanisms that most likely vary depending on the nature of the agent or the stress signal. So far the only published phosphoproteomic report on a plant challenged with a microbe pathogen is about the A. thaliana–bacteria Pseudomonas syringae pv. tomato interaction, identifying the phosphorylation of five proteins, dehydrin, putative p23 co-chaperone, heat shock protein 81, plastid-associated protein/fibrillin, and the large subunit of rubisco, as part of the plant's basal defense (8). More recently, a phosphoproteomic study of grape attacked by the insect pest Lobesia botrana revealed the changes in phosphorylation of several kinases, phosphatases, and aquaporins, as well as eight significant phosphorylation motifs, including up-regulation of the X-G-S-X and S-X-X-D motifs and down-regulation of the R-X-X-S and S-D-X-E motifs (9).
In the present study, we applied high-throughput phosphoproteomics and proteomics to a model plant–microbe interaction between wheat and Septoria tritici in order to gain some insights into signaling networks and downstream defense responses. Septoria tritici (teleomorph: Mycosphaerella graminicola) is a well-characterized, fully sequenced, hemibiotrophic fungal pathogen (10). This fungus causes one of the most economically devastating foliar diseases of wheat, namely, septoria tritici blotch (11). The fungus initially infects the host via hyphal growth through stomatal openings (12, 13). Subsequently, it grows slowly into the mesophyll with hyphae extending in the intercellular spaces between the mesophyll cells. Approximately 10 days after inoculation (dai), the fungus rapidly switches to necrotrophic growth associated with disease lesions on the leaf surface (13). In contrast to many other fungal pathogens, S. tritici has not been reported to breach host cell walls or membranes or to develop any specialized penetration or feeding structures (12, 13). It remains in the apoplastic space to obtain nutrients throughout the entire infection cycle (14). So far, the transcriptome profiles of S. tritici in planta have been studied using microarray and expressed sequence tag library sequencing, focusing mainly on the necrotrophic phase because of the low fungal biomass at the biotrophic phase (10, 15–17). These studies revealed the expression of genes encoding cell-wall degrading enzymes (CDWEs) and genes involved in metabolism, transport, and signal transduction, such as MAPKs. However, host responses to fungal infection have never been deeply investigated at the proteome level, and they remain completely enigmatic with respect to post-translational responses. A few studies employing biochemical and qRT-PCR approaches revealed rapid expression of host PR genes and protein disulfide isomerase genes, the accumulation of ROS, changes in proteolytic activity, regulation of MAPK pathways, and programmed cell death in response to S. tritici infection (13, 18–22). Overall, the fundamental and abrupt lifestyle shift, the availability of the full genome sequence, the economical significance of S. tritici, and the lack of a systematic study of host defense responses and fungal proteome profiling during the initial biotrophic phase of interaction strongly encouraged us to investigate this biological system, which will serve as a model for molecular research in the field of plant–microbe interactions.
Quantitative proteomics and phosphoproteomics offer powerful and indispensable technologies for studying plant–pathogen interactions. By setting up a combined approach, we were able to identify plant signaling and defense-related proteins, as well as fungal metabolic and signaling proteins, in both susceptible and resistant wheat following perception of S. tritici during the biotrophic symptomless phase of the infection. In addition, several plant signal molecules and defense-related proteins were characterized. Our data, for the first time, shed light on the crosstalk among signaling networks, translational and post-translational responses in wheat during S. tritici infection, and the fungal proteome likely crucial for the initial pathogenicity.
EXPERIMENTAL PROCEDURES
Plant Growth and Inoculation
The susceptible wheat cv. Sevin and the resistant cv. Stakado were used in the experiments. The growth of plants, preparation of inoculum of S. tritici isolate IPO323 spores, and inoculation were performed as described previously (13). Control plants were mock-inoculated with water. At each time point and for each treatment, ∼40 leaves were collected from three separate pots, serving as one biological replicate. Three biological replicates were harvested at 3, 7, and 11 dai and immediately frozen in liquid nitrogen. The leaf samples were ground in liquid nitrogen and stored at −80 °C until use.
Fungal Biomass Determination
Total DNA was isolated from leaf powder using the DNeasy Plant Mini Kit (Qiagen, Venlo, The Netherlands). Fungal DNA was determined via qPCR using primers for S. tritici mating type gene 1–1 (23) as described previously (24). Statistical analysis of the data was performed via analysis of variance, assuming a normal distribution, using PC-SAS (release 9.3, SAS Institute, Cary, NC).
H2O2, Ca2+, NO, and Sugar Production
H2O2 was extracted from plant powder with cold 0.05 m Na3PO4 buffer, pH 7.4, and the content was determined using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen). Ca2+ was determined via o-cresolphthalein complexone assay (Sigma). NO was extracted by 1 m K3PO4, pH 6.4, containing 10 μm DAF-FM diacetate (Invitrogen) and measured using a FluoroStar instrument (GE Health Care) with 485-nm excitation and 520-nm emission. Glucose, fructose, and sucrose contents in samples were measured as described previously (20). Statistical analysis of data in infected versus control samples was performed as described above.
Preparation of Cell Wall Polysaccharides and Immunodot Assay
The preparation of cell wall polysaccharides was performed as described previously (25). The concentration of polysaccharides was measured via a phenol–sulfuric acid assay (26). For the immunodot assay, 10 pmol of each sample was applied to nitrocellulose membranes (Whatman) and allowed to air dry. Membranes were blocked with 1× PBS buffer containing 1.5% Tween 20 for 1 h prior to 2 h of incubation with rat monoclonal antibodies against wheat arabinoxylan (LM1) and β-1,3(4)-glucan (kindly provided by Professor William Willats, University of Copenhagen). After extensive washes by PBS buffer containing 1.5% Tween 20, the membranes were incubated with anti-mouse secondary antibodies conjugated to horseradish peroxidase (1:2000, Dako Glostrup, Denmark). Membranes were extensively washed prior to detection using the Immun-Star HRP Substrate Kit (Bio-Rad).
Enzyme Assays and Western Blotting
Proteins were extracted from plant powder in 25 mm sodium acetate, pH 4.5, at 4 °C. The protein concentration in the extracts was determined using the Bio-Rad Protein Assay (Bio-Rad) with bovine serum albumin as the standard. An azurine-crosslinked polysaccharide assay was performed as described elsewhere (27), with azurine-crosslinked casein (Megazymes Wicklow, Ireland) as the substrate for measuring protease activity. An in-gel protease activity assay was performed using 12% Zymogram gels embedded with casein (Bio-Rad). Protein bands were excised and subjected to in-gel trypsin digestion as described elsewhere (24). The extracted peptides were analyzed using an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher), and analysis was followed by protein identification through database searches against the TaGI wheat gene index, release 12.0 (released on April, 18, 2010; tentative consensus sequences, 93,508; expressed sequence tags, 128166; expressed transcript sequences, 251) using Proteome Discoverer (Thermo Fisher) as described below.
For Western blotting, proteins were separated on CriterionTM XT Precast Gels (12% Bis-Tris, Bio-Rad), and separation was followed by blotting to nitrocellulose membranes. Rabbit antibodies against barley PR-1, PR-2 (β-1,3-glucanase), and PR-3 (chitinase) (kindly provided by Professor David B. Collinge, University of Copenhagen) were used for Western blotting. Western blotting was carried out as described for the immunodot assay.
Protein Preparation for Proteome and Phosphoproteome Analysis
Leaf powder (200 mg) was suspended in 0.6 ml of phenol extraction buffer containing 0.7 m sucrose, 0.1 m KCl, 0.5 m Tris-HCl, pH 7.5, 50 mm EDTA, 2% (v/v) β-mercaptoethanol, “Complete” protease inhibitor mixture (Roche), “PhosSTOP” phosphatase inhibitor mixture (Roche), and 100 μm pervanadate. Phenol extraction was performed as described elsewhere (28). The protein pellet was dissolved in 100 μl of buffer (6 m urea, 2 m thiourea, and 50 mm Triethylammonium bicarbonate), treated with 10 mm DTT for 30 min at room temperature and 20 mm iodoacetamide for 30 min in the dark prior to trypsin (2%, w/w) digestion overnight at 37 °C. The resulting peptides were desalted on Poros Oligo R3 micro-columns packed in p200 pipette tips, and peptides were eluted with 60% acetonitrile and 0.1% TFA as described elsewhere (29–31). The eluate was lyophilized via vacuum centrifugation. The dried peptides were dissolved in 100 μl of 50 mm TEAB buffer prior to amino acid analysis for accurate protein quantification using a Biochrom 30+ Amino Acid Analyzer (Biochrom Cambridge, UK) following the manufacturer's instructions. One hundred micrograms of protein from each biological replicate of infected and control samples at 3, 7, and 11 days was labeled with TMT six-plex® (Thermo Scientific) according to the manufacturer's protocol (TMT-126 for the control at 3 dai, TMT-127 for the infected sample at 3 dai, TMT-128 for the control at 7 dai, TMT-129 for the infected sample at 7 dai, TMT-130 for the control at 11 dai, and TMT-131 for the infected sample at 11 dai).
Phosphopeptide Enrichment
Phosphopeptides were purified using TiO2 chromatography in a batch-mode format as described elsewhere (31). Briefly, labeled peptides were resuspended in a highly selective, low-pH loading buffer (80% acetonitrile, 5% TFA, and 1 m glycolic acid) and incubated with TiO2 beads with constant shaking for 30 min. The beads were washed with a buffer containing 80% acetonitrile and 2% TFA and then with a buffer containing 20% acetonitrile and 0.1% TFA. Phosphopeptides were eluted at high pH using ammonia water. Enriched phosphopeptides were desalted on a Poros Oligo R3 micro-column as described above, and the eluted phosphopeptides were lyophilized prior to hydrophilic interaction liquid chromatography (HILIC) fractionation and LC-MS/MS analysis. In order to characterize the protein expression (the total peptides for proteome analysis), the titanium dioxide flow-through was collected and desalted on a Sep-Pak C18 column (Waters Milford, MA) before being subjected to HILIC fractionation and LC-MS/MS analysis.
HILIC Fractionation
Isobaric labeled peptides (phosphorylated and total peptides) obtained from biological replicate 1 were fractionated on a TSKGel Amide 80 HILIC-HPLC column using the Agilent 1200 microHPLC instrument as previously described (32). Briefly, samples were suspended in solvent B (90% acetonitrile and 0.1% TFA) and peptides were eluted at 6 μl/min by decreasing the solvent B concentration (100%–60%) over 26 min. Fractions were collected in a 96-well plate, lyophilized, and stored at −20 °C until MS analysis.
Mass Spectrometry Analysis
Phosphopeptides and total peptides from biological replicate 1 were analyzed using an Easy-LC nanoHPLC (Thermo Fisher Scientific, Proxeon, Odense, Denmark) coupled with an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific). Samples were resuspended in 0.1% TFA and separated via reversed-phase liquid chromatography on a Reprosil-Pur C18 (3 μC; Dr. Maisch GmbH, Ammerbuch, Germany) column (22 cm × 100 μm inner diameter, in-house packed). The chromatographic gradient was 0%–34% solvent B (90% acetonitrile and 0.1% formic acid) for 90 min at a flow rate of 300 nl/min. The mass spectrometer was operated in a data-dependent mode, automatically switching between MS and MS/MS. A survey MS scan (400–1800 m/z) was acquired in the Orbitrap analyzer with a resolution of 30,000 at 400 m/z. The top three most intense ions with a threshold of 5000 were selected for both low-resolution collision-induced dissociation (total peptides) or mobility shift assay (phosphopeptides) in the ion trap at a normalized collision energy of 35 and high-resolution high-energy collisional activated dissociation (HCD) in the Orbitrap analyzer (normalized collision energy = 55; 7500 resolution at 400 m/z). Phosphopeptides and total peptides from biological replicates 2 and 3 without HILIC fractionation were performed using the same LC configuration with a 120-min gradient, but coupled to an LTQ-Orbitrap Velos. Following a survey MS scan at a resolution of 30,000 at 400 m/z, the top seven most intense ions were selected for high-resolution HCD-MS/MS (normalized collision energy = 48; 7500 resolution at 400 m/z). These two biological replicates were run twice using this setup. Raw data were viewed in Xcalibur v2.0.7 (Thermo Fisher Scientific).
Database Search and Bioinformatic Analysis
Raw MS/MS spectra were processed and quantified using Proteome Discoverer (version 1.2, Thermo Fisher) software. Quantification was set up as ratios of TMT-127/126 (infected/control at 3 dai), TMT-129/128 (infected/control at 7 dai), and TMT-131/130 (infected/control at 11 dai). Peptide identification was performed with MASCOT (v2.2, Matrix Science Ltd., London, UK) and SEQUEST (33) algorithms, searching against a target and decoy TaGI wheat gene index (release 12.0; released April, 18, 2010; TC sequences, 93,508; expressed sequence tags, 128,166; ETs, 251) and the DOE Joint Genome Institute gene index for Septoria tritici (released on September, 10, 2008; 10,933 genes). The following search criteria were set: trypsin as proteolytic enzyme with two missed cleavages, S-carbamidomethyl-cysteine as a fixed modification, oxidation (M), deamidation (N and Q), TMT® reagents (protein N-terminus and Lys side-chain), and phosphorylation (S, T, and Y for the modified fraction). The data were searched with a peptide ion mass tolerance of 10 ppm and a fragment ion mass tolerance of ±0.5 Da (±0.05 Da for high-resolution HCD spectra). False discovery rates were obtained using Percolator (34), selecting identifications with a q-value equal to or less than 0.01. Only peptide sequences with a Mascot ion score ≥ 23, Xcorr value > 2.2, rank 1, and phosphopeptides with a Mascot Delta score ≥ 9 (collision-induced dissociation–mobility shift assay activation) or ≥ 13 (HCD fragmentation) (35) were considered for further analysis. Quantification data were normalized using the log2-transformed median. Data from all biological and technical replicates were merged and exported to an Excel sheet using R. A threshold of quantification in at least two biological replicates and average ratios ≥ 1.5 or ≤ 0.7 at least at one time point were used to define the regulated peptides. The online motif-x algorithm was used to identify sequence motifs for phosphorylation. Profiling of the ratios of total peptides and phosphopeptides at three time points was performed using the GProX program (36). Fungal proteins were assessed for signal peptides using SignalP. All raw LC-MS/MS files and annotated MS/MS spectra containing information about identified (phospho)peptide sequences, activation type, modification sites, and ion score are available for downloading at the Proteome Commons Tranche data repository under the project name “Proteomics and phosphoproteomics analysis of wheat-Septoria tritici interaction.”
qRT-PCR Analysis
Primer design, RNA extraction, gDNA removal, cDNA synthesis, and qRT-PCR were performed as described elsewhere (37, 38). The Septoria tritici elongation factor was used as a reference gene. Primers are shown in supplemental Table S1. All relative expression values of genes were reported as means ± S.D. An analysis of significant differences in gene expression in infected Stakado versus Sevin at each time point was performed by means of Student's t test.
RESULTS
Monitoring Infection and Activation of Plant Defenses
S. tritici colonizes the host leaves biotrophically without causing symptoms in the susceptible cv. Sevin, typically in up to 9 to 11 days. Subsequently, the tissue turns necrotic and the pathogen starts sporulating. No symptom appears in the resistant cv. Stakado (13). In the present study, we focused on the entire biotrophic phase and thus harvested the samples after up to 11 days. In order to determine the level of fungal infection precisely during this period, fungal DNA was quantified. The concentration of fungal DNA increased from 3 to 7 dai and then remained constant in Stakado, whereas a sharp increase of fungal DNA was observed in Sevin from 3 to 11 dai (Fig. 1A). No fungal DNA could be detected in the control samples. In order to examine defense responses and biochemical changes triggered by S. tritici in the host, three interacting signal molecules (Ca2+, H2O2, and NO), sugar levels, protease activity, PR protein expression, and plant cell wall polysaccharides were characterized. High levels of Ca2+ and H2O2 were produced at 3 to 7 dai in infected Stakado, whereas their accumulation lasted to 11 dai in Sevin (Fig. 1B). Additionally, higher levels of Ca2+ and H2O2 were observed in infected Stakado than in infected Sevin at 3 and 7 dai, respectively. In Sevin, no significant changes in NO levels between infected samples and controls were observed, whereas in infected Stakado, levels of NO were higher than in the control at 7 dai and lower at 11 dai (Fig. 1B). In addition to their essential roles as substrates in energy metabolism and polymer biosynthesis, sugars have important hormone-like functions as primary messengers in signal transduction (3). Thus, the production of glucose, fructose, and sucrose was determined, and higher levels were found in infected Stakado at 7 and 11 dai and in infected Sevin at 11 dai than in the respective controls (Fig. 1C). Sugar production was also found to accelerate in S. tritici-infected wheat during the necrotrophic phase when visible symptoms appeared (10, 20). Because of previous observations of extracellular serine protease activity decreasing in a susceptible wheat cultivar and increasing in a resistant wheat cultivar inoculated with S. tritici from 4 to 14 dai (18), protease activity in protein extracts of the infected leaves was measured here. The darker blue rings in infected susceptible and resistant plants indicated higher protease activity from 3 to 11 dai (Fig. 2A). Subsequently, proteases were separated on gels, and the gel bands were excised and subjected to LC-MS/MS. However, no wheat protease could be identified (data not shown). In the Western blotting analysis, PR proteins, the markers of plant defense responses, were more abundant in both infected cultivars at 3, 7, and 11 dai, with the exception of PR-3 in Stakado (Fig. 2B). To confirm the finding that the plant cell wall is not breached by S. tritici (12), plant cell wall polysaccharides LM1 and β-1,3(4)-glucan were analyzed via immunodot assay, and no significant changes between infected and control samples at up to 11 dai were noted (Fig. 2C).
Fig. 1.
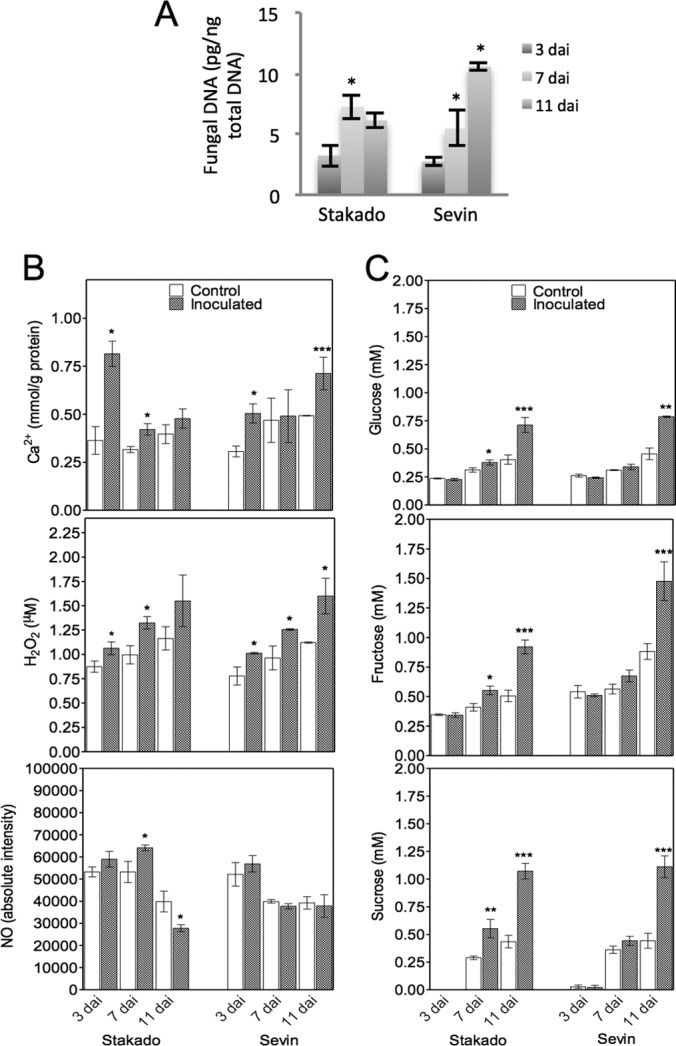
Presence of S. tritici infection in leaves of wheat cvs. Stakado and Sevin at 3, 7, and 11 dai. Significant differences between inoculated and control samples or in levels of fungal DNA are indicated by asterisks (***, p < 0.001; **, p < 0.01; *, p < 0.05). A, fungal biomass expressed as amount of S. tritici DNA in the infected samples. B, quantification of the signal molecules Ca2+, H2O2, and NO. C, quantification of glucose, fructose, and sucrose contents.
Fig. 2.
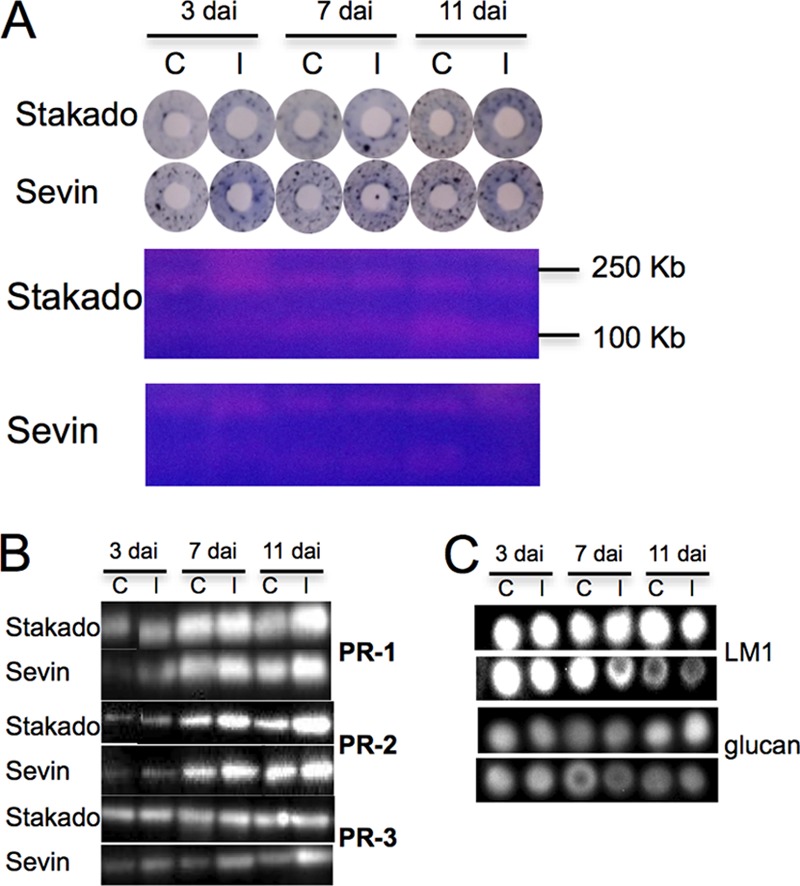
Detection of enzymes and cell wall polysaccharides in leaves of wheat cvs. Stakado and Sevin infected by S. tritici at 3, 7, and 11 dai. Results from one representative of three biological replicates are shown. C, control; I, infected sample. A, protease assay by azurine-crosslinked plates (upper panel) and Zymogram gels (lower panel) with casein as the substrate. B, Western blotting using antibodies recognizing plant PR-1, PR-2, and PR-3. C, immunodot assay using antibodies recognizing wheat LM1 polysaccharide and β-1,3(4)-glucan.
Detection and Quantification of Wheat Proteome and Phosphoproteome during Infection
The present MS-based approach, combining both proteomics and phosphoproteomics in one workflow, was designed to reduce the complexity of sample preparation and experimental procedures (supplemental Fig. S1). Peptides and phosphopeptides extracted from the first biological replicate were fractionated by means of offline HILIC prior to reverse-phase LC-MS/MS analysis, whereas peptides and phosphopeptides extracted from the other two biological replicates were analyzed twice using reverse-phase LC-MS/MS with a two-hour gradient. This resulted in the identification of 1600 proteins and 686 phosphopeptides in Stakado and 1610 proteins and 612 phosphopeptides in Sevin (Fig. 3A). Among the identifications, 695 proteins and 356 phosphopeptides were exclusively detected in Stakado, whereas 705 proteins and 282 phosphopeptides were uniquely identified in Sevin (Fig. 3A). To obtain reliable quantification, we defined the criteria for quantified proteins such that proteins had to be identified in at least two biological replicates with quantitative ratios. This led to the successful quantification of 454 proteins and 196 phosphopeptides in Stakado and 515 proteins and 104 phosphopeptides in Sevin, with 276 common proteins and 74 common phosphopeptides (Fig. 3A). Functional classification of quantified proteins and phosphopeptides showed that the majority of proteins were involved in primary metabolism, translation, and stress responses, whereas phosphopeptides were mainly involved in primary metabolism, signaling transduction, transport, and regulation of biological processes (Fig. 3B). In addition, similar patterns of protein functional classification were observed in the two cultivars (Fig. 3B). The common and exclusive quantified proteins and phosphopeptides in the two cultivars had diverse functions. To further elucidate the global patterns of host protein expression and phosphorylation during infection, we profiled average ratios of protein abundance and phosphorylation between infected and control samples at 3, 7, and 11 dai (Fig. 3C, supplemental Fig. S2). The ratio patterns revealed fewer changes in protein expression than in phosphorylation, which might indicate that plant cellular responses to fungal infection resulting from PTMs (i.e. phosphorylation) are independent of protein translation. Interestingly, major changes in protein expression were observed in Stakado at 3 dai (Fig. 3C) in association with a higher level of Ca2+ in Stakado than in Sevin at 3 dai, suggesting that rapid and intensive molecular responses might contribute to resistance against fungal infection. Detailed lists of all identified and quantified proteins and phosphopeptides with their respective phosphosites, along with the ratios in each biological and technical replicate, are provided in supplemental Tables S2 and S3.
Fig. 3.

Global view of identified and quantified wheat proteins and phosphopeptides in leaves of wheat cvs. Stakado and Sevin infected by S. tritici at 3, 7, and 11 dai. Each phosphopeptide contains unique position(s) of the phosphorylation site(s). A, Venn diagram depicting the comparison of the total number of proteins and phosphopeptides identified and quantified between the two cultivars. B, biological function classification of quantified proteins and phosphopeptides. C, distribution of the average normalized ratio (log2-scale) of proteins and phosphopeptides between infected and control samples.
Regulation of Wheat Proteome and Phosphoproteome by S. tritici
The selection criteria allowed us to identify 46 proteins and 70 phosphopeptides in Stakado and 47 proteins and 60 phosphopeptides in Sevin in response to S. tritici infection (supplemental Tables S4 and S5). The regulated proteins and phosphopeptides in the two cultivars had similar functions, although no overlap of protein accessions was detected. Proteins with known functions that were differentially expressed or phosphorylated in the two cultivars were involved in metabolic processes, translation, proteolysis, protein folding, stress responses, transport, and biological regulation (supplemental Tables S4 and S5). In particular, a substantial number of regulated phosphopeptides in both cultivars were involved in signal transduction, transport, regulation of transcription and translation, and cell component organization, of including receptor-like kinase, other protein kinases, transcription factors, ATPases, sugar transporters, and nuclear proteins (supplemental Table S5). We used the motif-x algorithm to identify sequence motifs for the phosphorylation sites in the regulated phosphopeptides, with the A. thaliana database as background and with a six-amino-acid residue sequence window surrounding the phosphorylated S. Two phosphorylation motifs (pS-X-X-X-D and pS-P) were significantly enriched (supplemental Fig. S3), and they could be the targets for MAPKs and casein kinase, respectively (39).
Identification of S. tritici Proteins and Phosphoproteins
Obtaining a large number of fungal identifications in a plant–fungus interaction system is challenging because of the dominance of host plant proteins, given the high ratio of plant to fungal biomass. Furthermore, it is difficult to distinguish whether a peptide originates from the plant, the fungus, or both when it can be identified in both host and pathogen databases, although it is more likely that the peptide is of plant origin. To obtain more fungal identifications with high confidence, both MASCOT and SEQUEST algorithms were used. Here, we show the proteins with peptides or phosphopeptides identified from fungal databases only. The 31 proteins and 5 phosphoproteins identified from either infected cultivar at 3, 7, and 11 dai were mainly related to basic cellular machinery and signaling (supplemental Table S6). These proteins included actin, ATPases, elongation factors, histones, ribosomal proteins, GTP-binding proteins, Ras GTPases, calcium-binding proteins, and 14–3-3 proteins. Notably, phosphoproteins, including a cell division/GTP-binding protein, a tetratrico peptide repeat, an ATPase, a glycoside hydrolase, and a hypothetical protein, could be identified only from infected Sevin, which might mean that protein phosphorylation contributes to fungal-specific infection strategies in the compatible interaction.
S. tritici Gene Expression
In order to investigate signaling networks activated in the fungus during its symptomless phase, we examined the expression of genes encoding fungal protein kinases (Slt2, Fus3, Hog1, and Tpk2), G protein (Gα3), and an ABC transporter (Atr4) in the infected hosts. It has been reported that S. tritici attempts to adapt to oxidative stress during the necrotrophic stage of infection through the high expression of genes implicated in stress tolerance (13, 17), and a series of genes encoding CDWEs are uniquely expressed in planta compared with in vitro (15). Therefore, genes encoding an ROS-scavenging enzyme Cu2+/Zn2+ superoxide dismutase and three CDWEs (arabinofuranosidase, xylanase, and glucosidase) were selected for gene expression analysis. All the transcripts were detectable in both cultivars with different profiles (supplemental Fig. S4). In both cultivars, the expression of signaling- and transport-related genes varied between 3 and 7 dai, with a more constant expression from 7 to 11 dai, except for an up-regulation of Tpk2 in Stakado at 7 to 11 dai. Superoxide dismutase was up-regulated from 3 to 11 dai in Sevin and from 3 to 7 dai in Stakado. The three CDWE genes were stably expressed or down-regulated from 3 to 7 dai, with variable expression profiles from 7 to 11 dai. Although the overall expression profiles of most genes were similar in the two cultivars, seven of the genes showed significantly different expression levels in the two cultivars at least at one time point (indicated by asterisks in supplemental Fig. S4).
DISCUSSION
Because no systematic study at the proteome level has been conducted on the interaction between wheat and the fungal pathogen S. tritici, we aimed at gaining a comprehensive picture of signaling networks and defense responses, as well as of fungal proteome profiling, by using large-scale quantitative proteomics and phosphoproteomics in combination with biochemical and gene expression approaches. However, the lack of a wheat genome sequence can hamper the protein identification and the phosphosite localization, as only partial information can be gathered from expressed sequence tag databases, homology searches, and de novo sequencing. The interpretation of the MS/MS spectra of phosphopeptides can be ambiguous because of the poor fragmentation of phosphopeptides or the loss of the phosphate group in the gas phase upon collision-induced dissociation and HCD. Different fragmentation techniques and localization algorithms should be introduced to improve the presence of site-determining ions and statistically estimate their likelihood. Careful manual and computational evaluation of the phosphopeptide MS/MS spectra is also required. In the present study, we applied both mobility shift assay and HCD fragmentation and the Mascot Delta score threshold to confidently localize the phosphorylation sites. Furthermore, large-scale phosphoproteomic analysis of eukaryotic and prokaryotic species has shown conservation of phosphorylation sites between closely evolutionarily related species. The verification of phosphorylation site assignments should take into account evolutionarily conserved phosphorylation sites in the different datasets or proteins from species that are closely related and have conserved function when an organism without a fully sequenced genome is analyzed.
Development of Biomarkers for the Symptomless Phase of Infection
The key features of S. tritici that distinguish it from most current fungal pathogen models are the facts that it remains strictly extracellular during a long biotrophic growth phase and does not cause any visible symptoms before a sudden appearance of disease symptoms associated with necrotrophic growth (12). The symptom expression in the compatible interaction has been suggested to be associated with the regulation of MAPK pathways and features of programmed cell death in the host, including DNA laddering, translocation of cytochrome c from mitochondria to the cytosol, electrolyte leakage, and degradation of RNA (22), and eventually with tissue degradation and a loss of cellular functions (13, 19, 20). However, it is currently unclear which effectors the fungus relies on to facilitate the initial symptomless growth phase and which disease resistance proteins can recognize these effectors to trigger plant immunity. In attempting to gain information on the potential fungal effectors and host defense-related mechanisms, we were particularly interested in the initial biotrophic stage of both compatible and incompatible interactions, and thus we harvested samples until the appearance of the first disease symptoms, before fungal-induced host necrosis occurred at the late necrotrophic stage. Although no macroscopic symptoms appeared, host defense responses were triggered by the pathogen because of higher levels of defense-related signal molecules H2O2, Ca2+, NO, and sugars, protease activity, and PR protein expression in infected hosts. These characteristics can serve as biomarkers for S. tritici infection, in particular in the symptomless growth stage. The accumulation of H2O2 has already been demonstrated as a defense response that inhibits S. tritici colonization during its initial biotrophic phase (13, 20). Moreover, it is noteworthy that the accumulation of PR-1 and PR-2 was observed in both cultivars, whereas PR-3 was accumulated only in Sevin, despite the fact that PR-2 and PR-3 function in the degradation of fungal cell walls (40). This indicates the stronger involvement of PR-2 than PR-3 in defense responses to S. tritici and that released β-1,3-glucan probably acts as the elicitor of further defense reactions during both compatible and incompatible interactions, as shown previously (19).
S. tritici Regulates Photosynthesis, Sugar Metabolism, and Translation Processes in the Hosts
As a consequence of S. tritici infection, a large number of proteins, including chloroplast chlorophyll a-b binding proteins, ribulose bisphosphate carboxylase, fructose-bisphosphate aldolase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate mutase, sucrose-phosphate synthase, UDP-N-acetylglucosamine pyrophosphorylase, and phosphoglucomutase, decreased in abundance and/or changed in phosphorylation status in one or both cultivars from 3 to 11 dai, suggesting the suppression of photosynthesis and changes in sugar metabolism. This result was correlated with the observation of significant increases in sugar contents and changes in phosphorylation levels of sugar transporters in response to S. tritici. Down-regulation of photosynthesis due to a decrease in chlorophyll a and an increase in sugar content were previously reported in the S. tritici-infected wheat cv. Sevin at the necrotrophic stage (20). It has been suggested that high sugar status reduces photosynthetic gene expression and enhances mobilization and sugar transport (3) and that fungal pathogens can target sugar transporters and induce the expression of the corresponding genes for nutritional gain (41). Our data indicate links among photosynthesis, sugar transport, and energy metabolism in response to S. tritici. Proteins involved in translation were shown to be up-regulated in Stakado and down-regulated in Sevin at expression level, whereas the proteins involved in amino acid transport and biosynthesis (i.e. amino acid selective channel protein, 2-oxoglutarate/malate translocator, and aspartate aminotransferase) were up-regulated in both cultivars, indicating that resistance to infection is probably related to the efficiency of protein translation, rather than the enhanced transport and synthesis of building blocks.
Activation of Defense Responses and Antioxidative Stress Mechanism during Two Interactions
Another major group of proteins changing in abundance and/or phosphorylation status in response to fungal infection are defense and stress related. As expected, several PR proteins (i.e. PR-2 and peroxidases (PR-9)) were up-regulated in both cultivars, whereas the increased abundance of PR-1 and PR-3 was observed only in Sevin, these findings were also confirmed via Western blotting analysis of PR-2 and PR-3. Shetty et al. (19) observed that the corresponding transcripts of PR-2 are up-regulated in infected Sevin (50-fold) and Stakado (3-fold) at 9 and 11 dai, whereas the expression of the distinct PR-3 gene is up-regulated in both cultivars at 9 dai but down-regulated in Stakado at 11 dai. Up-regulation of the ROS-scavenging enzymes catalase and peroxiredoxin in Sevin, associated with the accumulation of H2O2 and the expression of a fungal superoxide dismutase gene at up to 11 dai, strongly suggests that both pathogen and host attempted to overcome the increased oxidative stress during the compatible interaction. S. tritici is, in fact, able to tolerate ROS, but it does not thrive in the presence of H2O2 (20). Several heat shock proteins and chaperonins increased in abundance and were phosphorylated in the infected hosts. Heat shock proteins are known to play a critical role in plant innate immunity and defense or oxidative stress-related signaling by assisting in receptor processes and the stability of the defensome complex (42). We have also detected the accumulation of a wheat xylanase inhibitor and the expression of an S. tritici xylanase gene in two cultivars. Xylanase inhibitors were shown to be potential defense molecules in wheat that prevent cell wall degradation by fungal hydrolytic enzymes (43). Given that there were no significant changes in cell wall components between the infected and the control wheat, interplay between plant xylanase inhibitors and fungal xylanases might function in the inhibition of fungal degradation of host cell walls. Although one zinc-dependent protease was found to be up-regulated in Sevin, it seems unlikely this protein played a major role in the enhanced protease activity in the infected leaves because of its lower molecular weight (approximately 72 kDa) (Fig. 2A) relative to that observed from gel (>100 kDa).
Differential Regulation of Signaling Pathways, Transcription and Translation Processes, and Membrane Transport at Phosphorylation Levels between Compatible and Incompatible Interactions
Proteins with sites differentially phosphorylated in two cultivars included kinases involved in signaling, transcription, and translation factors, nuclear proteins, and membrane proteins. However, differential regulation of signaling pathways, transcription and translation processes, and membrane transport was found between two interactions, which might reflect resistance-related mechanisms. Receptor-like kinases play a major role in the perception and perpetuation of external environmental stress stimuli and downstream signal transduction (44). We identified a receptor-like kinase that was differentially phosphorylated in Stakado. A change in the cytoplasmic calcium concentration is one of the earliest responses to stress (45). CDPKs function as potential sensors that decode and translate the elevation of calcium into enhanced kinase activity and subsequent downstream signaling events (45). Here, CDPK 7 was phosphorylated in both cultivars, suggesting the importance of calcium and CDPK 7 in signaling in wheat in response to infection. MAPK cascades are involved in many aspects of plant growth and in the response of plants to stress, and in particular to pathogens (46). It was reported that a MAPK (TaMK3) was activated at the transcription, translation, and PTM levels during symptom development of S. tritici infection in susceptible wheat (22). We identified the differential phosphorylation of a mitogen-activated protein kinase kinase in Stakado and a DENN (AEX-3) domain-containing protein in Sevin, both of which are involved in the regulation of MAPK signaling pathways. A sucrose non-fermenting-related kinase (SNF), the master regulator of energy metabolism and one of the major components in sugar signaling (47), was found to be phosphorylated in Sevin at 3 dai, which was correlated with increased sugar levels in response to infection. SNF1 itself is activated by phosphorylation and can phosphorylate transcription repressors for the de-repression of genes involved in sugar metabolism (3). The alteration of the phosphorylation level of SNF1 has recently been identified in Arabidopsis in response to abscisic acid (6) and in Rhizobium-legume symbiosis (47). In addition, a protein kinase NPH1 and an RPT2-like protein showed altered phosphorylation levels in Sevin. NPH1 can regulate kinase activity in Arabidopsis in response to blue-light-induced redox changes (48), and PRT2 is a signal transducer involved in phototropic response and stomatal opening in Arabidopsis (49). This result can further substantiate the S. tritici-induced changes in redox and photosynthesis during the compatible interaction. We have also identified a phosphorylated pleckstrin homology domain and a dephosphorylated Src homology 3 domain in Stakado. Both domains have been found in proteins of signaling pathways regulating the cytoskeleton and related to Ras proteins, G protein, and protein kinase C (50, 51).
Several nuclear matrix proteins, a translation factor RNA-binding protein Rp120, and proteins involved in the regulation of transcription changed in phosphorylation level in one or both cultivars. These transcription regulators included transcription factors (acute myeloid leukemia protein 1-like, HMG-I/Y protein HMGa-like, methyl binding domain protein MBD109, alfalfa zinc finger protein-like, BRI1-KD interacting proteins, WRKY35-like protein, and ZF-HD homeobox protein) and post-transcription factors (alternative splicing regulator and RNA recognition motif family protein). Nuclear matrix proteins involved in nuclear remodeling, in conjunction with histone modification, chromatin remodelers, and other regulatory proteins, can result in transcription reprogramming in plants upon pathogen infection (52). WRKY transcription factors, participating in the control of defense-related genes and activated by MAPK-dependent phosphorylation, are the key players in the plant defense response (53). The alfalfa zinc finger protein has been reported as a transcription factor regulating gene expression due to abiotic stress (54). ZF-HD proteins have been implied in the activation of calmodulin expression in soybean in response to pathogens (55). Alternative splicing regulators can be influenced by biotic and abiotic stress in plants, subsequently altering the plant transcriptome post-transcriptionally in response to stress (56). It is noteworthy that major regulators were dephosphorylated in Sevin and phosphorylated in Stakado at 11 dai in response to infection, and this can be correlated with the observation that the abundance of proteins involved in translation were down-regulated in Sevin and up-regulated in Stakado, collectively indicating that the regulation of transcription and translation processes at both expression and phosphorylation levels might be critical for resistance to S. tritici.
In addition to sugar transporters, several proteins located in the membrane showed significant changes in phosphorylation levels, including plasma membrane (PM) ATPase and two PM intrinsic proteins (PIPs). PM ATPases are known to contribute to signaling events in plant–pathogen interactions (57). The activity of PM ATPases, often increasing during plant defense responses, can be positively and negatively influenced by phosphorylation, whereas transcription levels of these enzymes are unchanged (57). PIPs are water channel proteins and are regulated by phosphorylation (6, 58). Dephosphorylation of PIPs with a decrease in their activity has been found in Arabidopsis in response to abscisic acid (6) and NaCl (7). Increased phosphorylation levels of PIP1;3, PIP, and PM ATPase in Sevin and decreased phosphorylation level of PIP and PM ATPase in Stakado indicate that membrane transport may be targeted by S. tritici to increase plant susceptibility. Moreover, a vesicle-associated membrane protein involved in vesicle trafficking was phosphorylated in both cultivars. Vesicle trafficking has been shown to play an important role in the cellular processes of plant defense against pathogens and is possibly mediated by phosphorylation (47, 59).
A Role of Fungal Signaling Pathways in Facilitating Symptomless Growth
With respect to fungal proteins, we identified several basic cellular components and primary metabolic proteins. In addition, expression of ATPase and signaling proteins and the phosphorylation of a cell division/GTP-binding protein, a tetratrico peptide repeat, and an ATPase were detected during the initial host colonization. These signaling proteins included 14–3-3 proteins, several Ras GTPases involved in MAPK cascade, a GTP-binding protein, calcium-binding proteins, and a Sec7-like protein involved in ADP-ribosylation factor signal transduction for vesicular trafficking (60). Together with the expression of fungal genes encoding several MAPKs, a G protein α subunit, a protein kinase A, and an ABC transporter, which have been validated for pathogenicity (61), our results demonstrate that signal transduction might play an important role in the biotrophic growth of S. tritici and its initial pathogenicity.
CONCLUSION
In conclusion, based on the data obtained from the integrated approaches, we were able to track down several essential defense-related pathways and propose a model of crosstalk among signaling networks, as well as downstream defense responses and fungal symptomless growth, in the wheat–Septoria tritici interaction (Fig. 4). Recognition of the pathogen S. tritici by plant membrane receptors leads to several early signaling cascades, including of ROS, Ca2+, MAPKs, NO, and sugars, which subsequently activate SNF and several transcription and translation regulators such as WRKY transcription factor through phosphorylation controlling defense-related and carbohydrate metabolic gene expression. These defense responses are accompanied by suppressed photosynthesis and changes in sugar metabolism in the plant. Meanwhile, to overcome oxidative and starvation stress and to obtain nutrients from the host during the biotrophic growth, the transduction of signals such as MAPKs, G proteins, and Ca2+ and the expression of CDWEs and antioxidant enzymes are activated in the pathogen. Intriguingly, despite the molecular mechanisms of host responses shared by incompatible and compatible interactions, the differential regulation of various signaling pathways, membrane transport, and transcription and translation processes at protein expression and/or PTM levels associated with the distinct accumulation patterns of signal molecules could be observed in the two interactions. These observations constitute the first step toward elucidating the molecular processes occurring during the interactions between wheat and S. tritici and supply a reference for susceptibility or resistance to S. tritici. The earlier and/or more intense signaling involving ROS, Ca2+, NO, MAPKs, and sugars resulting in rapid and efficient transcription and translation processes of defense and stress-related genes might be essential for host resistance to S. tritici. The next challenge is to characterize the biological function of the identified proteins and their phosphorylation sites involved in this host–pathogen interaction, which will contribute to the development of novel wheat protection strategies against S. tritici. Overall, our well-established “-omics” strategy and model can be applied to other plant–pathogen interaction systems to identify a broad spectrum of proteins and protein modifications with potential roles in pathogenicity or host resistance from both plants and microbes.
Fig. 4.

Proposed model of crosstalk among signaling networks, as well as defense responses and fungal symptomless growth, in the wheat–S. tritici interaction. The pathogen S. tritici is perceived by receptors such as receptor-like kinase on the plasma membrane, which may be stabilized by Hsp70 and Hsp90. Perception triggers early signal transduction, including increases in NO and cytosolic Ca2+ concentration, the production of ROS, the activation of MAPKs and CDPKs, and increases in sugar levels. MAPK cascades and CDPK activate transcription factors such as WRKY that control the expression of defense-related genes such as PR genes, catalase (CAT), and peroxiredoxin (Prx). SNF kinase activated by sugar signaling can phosphorylate transcription factors to regulate the expression of genes involved in sugar metabolism. Sugar signaling is coordinated with the depression of photosynthesis and changes in sugar metabolism. Several signaling pathways including MAPKs, G proteins, and Ca2+ and the expression of genes encoding CDWEs and superoxide dismutase are activated in the pathogen during biotrophic colonization. Solid black lines link interacting partners, and dashed lines indicate putative interactions.
Supplementary Material
Acknowledgments
We thank Lene Jakobsen for technical assistance in proteomics and Kristina Egede Budtz for performing amino acid analysis.
Footnotes
* This work was supported by a grant from the Danish Research Council for Technology and Production (11–105997) to Fen Yang. Martin Røssel Larsen was supported by the Lundbeck Foundation (M.R.L. Junior Group Leader Fellowship).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are::
- CDPK
- calcium-dependent protein kinase
- CDWE
- cell-wall degrading enzyme
- dai
- days after inoculation
- HCD
- high-energy collisional activated dissociation
- HILIC
- hydrophilic interaction liquid chromatography
- MAPK
- mitogen-activated protein kinase
- PIP
- plasma membrane intrinsic protein
- PM
- plasma membrane
- PR
- pathogenesis-related
- PTM
- post-translational modification
- ROS
- reactive oxygen species
- SNF
- sucrose non-fermenting-related kinase.
REFERENCES
- 1. Benschop J. J., Mohammed S., O'Flaherty M., Heck A. J., Slijper M., Menke F. L. (2007) Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteomics 6, 1198–1214 [DOI] [PubMed] [Google Scholar]
- 2. Wendehenne D., Gould K., Lamotte O., Durner J., Vandelle E., Lecourieux D., Courtois C., Barnavon L., Bentéjac M., Pugin A. (2005) NO signaling functions in the biotic and abiotic stress responses. BMC Plant Biol. 5, s1–s3515927007 [Google Scholar]
- 3. Rolland F., Moore B., Sheen J. (2002) Sugar sensing and signaling in plants. Plant Cell 14, s185–s205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hunter T. (2000) Signaling—2000 and beyond. Cell 100, 113–127 [DOI] [PubMed] [Google Scholar]
- 5. Thurston G., Regan S., Rampitsch C., Xing T. (2005) Proteomic and phosphoproteomic approaches to understand plant–pathogen interactions. Physiol. Mol. Plant Pathol. 66, 3–11 [Google Scholar]
- 6. Kline K. G., Barrett-Wilt G. A., Sussman M. R. (2010) In planta changes in protein phosphorylation induced by the plant hormone abscisic acid. Proc. Natl. Acad. Sci. U.S.A. 107, 15986–15991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsu J. L., Wang L. Y., Wang S. Y., Lin C. H., Ho K. C., Shi F. K., Chang I. F. (2009) Functional phosphoproteomic profiling of phosphorylation sites in membrane fractions of salt-stressed Arabidopsis thaliana. Proteome Sci. 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones A. M., Bennett M. H., Mansfield J. W., Grant M. (2006) Analysis of the defence phosphoproteome of Arabidopsis thaliana using differential mass tagging. Proteomics 6, 4155–4165 [DOI] [PubMed] [Google Scholar]
- 9. Melo-Braga M. N., Verano-Braga T., León I. R., Antonacci D., Nogueira F. C., Thelen J. J., Larsen M. R., Palmisano G. (2012) Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol. Cell. Proteomics 11, 945–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keon J., Antoniw J., Carzaniga R., Deller S., Ward J. L., Baker J. M., Beale M. H., Hammond-Kosack K., Rudd J. J. (2007) Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Mol. Plant Microbe Interact. 20, 178–193 [DOI] [PubMed] [Google Scholar]
- 11. Eyal Z. (1999) The Septoria tritici and Stagonospora nodorum blotch diseases of wheat. Eur. J. Plant Path. 105, 629–641 [Google Scholar]
- 12. Kema G. H. J., Yu D. Z., Rijkenberg F. H. J., Shaw M. W., Baayen R. P. (1996) Histology of the pathogenesis of Mycosphaerella graminicola in wheat. Phytopathology 86, 777–786 [Google Scholar]
- 13. Shetty N. P., Kristensen B. K., Newman M. A., Møller K., Gregersen P. L., Jørgensen H. J. L. (2003) Association of hydrogen peroxide with restriction of Septoria tritici in resistant wheat. Physiol. Mol. Plant Pathol. 62, 333–346 [Google Scholar]
- 14. Rohel E. A., Payne A. C., Fraaije B. A., Hollomon D. W. (2001) Exploring infection of wheat and carbohydrate metabolism in Mycosphaerella graminicola transformants with differentially regulated green fluorescent protein expression. Mol. Plant Microbe Interact. 14, 156–163 [DOI] [PubMed] [Google Scholar]
- 15. Kema G. H., van der Lee T. A., Mendes O., Verstappen E. C., Lankhorst R. K., Sandbrink H., van der Burgt A., Zwiers L. H., Csukai M., Waalwijk C. (2008) Large-scale gene discovery in the septoria tritici blotch fungus Mycosphaerella graminicola with a focus on in planta expression. Mol. Plant Microbe Interact. 21, 1249–1260 [DOI] [PubMed] [Google Scholar]
- 16. Keon J., Antoniw J., Rudd J., Skinner W., Hargreaves J., Hammond-Kosack K. (2005) Analysis of expressed sequence tags from the wheat leaf blotch pathogen Mycosphaerella graminicola (anamorph Septoria tritici). Fungal Genet. Biol. 42, 376–389 [DOI] [PubMed] [Google Scholar]
- 17. Keon J., Rudd J., Antoniw J., Skinner W., Hargreaves J., Hammond-Kosack K. (2005) Metabolic and stress adaptation by Mycosphaerella graminicola during sporulation in its host revealed through microarray transcription profiling. Mol. Plant Pathol. 6, 527–540 [DOI] [PubMed] [Google Scholar]
- 18. Seggarra C. I., Casalongué C. A., Pinedo M. L., Cordo C. A., Conde R. D. (2002) Changes in wheat leaf extracellular proteolytic activity after infection with Septoria tritici. J. Phytopathol. 150, 105–111 [Google Scholar]
- 19. Shetty N. P., Jensen J. D., Knudsen A., Finnies C., Geshi N., Blennow A., Collinge D. B., Jørgensen H. J. L. (2009) Effects of β-1,3-glucan from Septoria tritici on structural defence responses in wheat. J. Exp. Bot. 60, 4287–4300 [DOI] [PubMed] [Google Scholar]
- 20. Shetty N. P., Mehrabi R., Lutken H., Haldrup A., Kema G. H., Collinge D. B., Jørgensen H. J. L. (2007) Role of hydrogen peroxide during the interaction between the hemibiotrophic fungal pathogen Septoria tritici and wheat. New Phytol. 174, 637–647 [DOI] [PubMed] [Google Scholar]
- 21. Ray S., Anderson J. M., Urmeev F. I., Goodwin S. B. (2003) Rapid induction of a protein disulfide isomerase and defense-related genes in wheat in response to the hemibiotrophic fungal pathogen Mycosphaerella graminicola. Plant Mol. Biol. 53, 741–754 [DOI] [PubMed] [Google Scholar]
- 22. Rudd J. J., Keon J., Hammond-Kosack K. E. (2008) The Wheat mitogen-activated protein kinases TaMPK3 and TaMPK6 are differentially regulated at multiple levels during compatible disease interactions with Mycosphaerella graminicola. Plant Physiol. 147, 802–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waalwijk C., Mendes O., Verstappen E. C., de Waard M. A., Kema G. H. (2002) Isolation and characterization of the mating-type idiomorphs from the wheat septoria leaf blotch fungus Mycosphaerella graminicola. Fungal Genet. Biol. 35, 277–286 [DOI] [PubMed] [Google Scholar]
- 24. Yang F., Jensen J. D., Spliid N. H., Svensson B., Jacobsen S., Jørgensen L. N., Jørgensen H. J., Collinge D. B., Finnie C. (2010) Investigation of the effect of nitrogen on severity of Fusarium head blight in barley. J. Proteomics 73, 743–752 [DOI] [PubMed] [Google Scholar]
- 25. Lionetti V., Raiola A., Camardella L., Giovane A., Obel N., Pauly M., Favaron F., Cervone F., Bellincampi D. (2007) Overexpression of pectin methylesterase inhibitors in Arabidopsis restricts fungal infection by Botrytis cinerea. Plant Physiol. 143, 1871–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dubois M., Gilles K. A., Hamilton J. K., Rebers P. A., Smith F. (1956) Colorimetric method for determination of sugars and related substances. Anal. Chem. 28, 350–356 [Google Scholar]
- 27. Schiøtt M., De Fine Licht H. H., Lange L., Boomsma J. J. (2008) Towards a molecular understanding of symbiont function: identification of a fungal gene for the degradation of xylan in the fungus gardens of leaf-cutting ants. BMC Microbiol. 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isaacson T., Damasceno C. M., Saravanan R. S., He Y., Catalá C., Saladié M., Rose J. K. (2006) Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 1, 769–774 [DOI] [PubMed] [Google Scholar]
- 29. Gobom J., Nordhoff E., Mirgorodskaya E., Ekman R., Roepstorff P. (1999) Sample purification and preparation technique based on nano-scale reversed-phase columns for the sensitive analysis of complex peptide mixtures by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 34, 105–116 [DOI] [PubMed] [Google Scholar]
- 30. Larsen M. R., Cordwell S. J., Roepstorff P. (2002) Graphite powder as an alternative or supplement to reversed-phase material for desalting and concentration of peptide mixtures prior to matrix-assisted laser desorption/ionization-mass spectrometry. Proteomics 2, 1277–1287 [DOI] [PubMed] [Google Scholar]
- 31. Larsen M. R., Thingholm T. E., Jensen O. N., Roepstorff P., Jørgensen T. J. (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886 [DOI] [PubMed] [Google Scholar]
- 32. Palmisano G., Jensen S. S., Le Bihan M. C., Lainé J., McGuire J. N., Pociot F., Larsen M. R. (2012) Characterization of membrane-shed microvesicles from cytokine-stimulated β-cells using proteomics strategies. Mol. Cell. Proteomics 11, 230–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 34. Käll L., Canterbury J. D., Weston J., Noble W. S., MacCoss M. J. (2007) Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 [DOI] [PubMed] [Google Scholar]
- 35. Savitski M. M., Lemeer S., Boesche M., Lang M., Mathieson T., Bantscheff M., Kuster B. (2011) Confident phosphorylation site localization using the mascot delta score. Mol. Cell. Proteomics 10, M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rigbolt K. T. G., Vanselow J. T., Blagoev B. (2011) GProX, a user-friendly platform for bioinformatics analysis and visualization of quantitative proteomics data. Mol. Cell. Proteomics 10, O110.007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang F., Jensen J. D., Svensson B., Jørgensen H. J., Collinge D. B., Finnie C. (2010) Analysis of early events in the interaction between Fusarium graminearum and the susceptible barley (Hordeum vulgare) cultivar Scarlett. Proteomics 10, 3748–3755 [DOI] [PubMed] [Google Scholar]
- 38. Yang F., Jensen J. D., Svensson B., Jørgensen H. J., Collinge D. B., Finnie C. (2012) Secretomics identifies Fusarium graminearum proteins involved in the interaction with barley and wheat. Mol. Plant Pathol. 13, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams J. A. (2001) Kinetic and catalytic mechanisms of protein kinases. Chem. Rev. 101, 2271–2290 [DOI] [PubMed] [Google Scholar]
- 40. Kim Y. J., Hwang B. K. (1997) Isolation of a basic 34 kDa Dam,3-glucanase with inhibitory activity against Phytophthora capsici from pepper stems. Physiol. Mol. Plant Pathol. 50, 103–115 [Google Scholar]
- 41. Chen L. Q., Hou B. H., Lalonde S., Takanaga H., Hartung M. L., Qu X. Q., Guo W. J., Kim J. G., Underwood W., Chaudhuri B., Chermak D., Antony G., White F. F., Somerville S. C., Mudgett M. B., Frommer W. B. (2010) Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L., Shimamoto K. (2011) Emerging roles of molecular chaperones in plant innate immunity. J. Gen. Plant Pathol. 77, 1–9 [Google Scholar]
- 43. Igawa T., Tokai T., Kudo T., Yamaguchi I., Kimura M. (2005) A wheat xylanase inhibitor gene, Xip-I, but not Taxi-I, is significantly induced by biotic and abiotic signals that trigger plant defense. Biosci. Biotechnol. Biochem. 69, 1058–1063 [DOI] [PubMed] [Google Scholar]
- 44. Haffani Y. Z., Silva D. R., Goring D. R. (2004) Receptor kinase signaling in plants. Can. J. Bot. 82, 1–15 [Google Scholar]
- 45. Harmon A. C., Gribskov M., Harper J. F. (2000) CDPKs—a kinase for every Ca2+ signal? Trends Plant Sci. 5, 154–159 [DOI] [PubMed] [Google Scholar]
- 46. Tena G., Asai T., Chiu W. L., Sheen J. (2001) Plant mitogen-activated protein kinase signaling cascades. Curr. Opin. Plant Biol. 4, 392–400 [DOI] [PubMed] [Google Scholar]
- 47. Rose C. M., Venkateshwaran M., Volkening J. D., Grimsrud P. A., Maeda J., Bailey D. J., Park K., Howes-Podoll M., den Os D., Yeun L. H., Westphall M. S., Sussman M. R., Ané J. M., Coon J. J. (2012) Rapid phosphoproteomic and transcriptomic changes in the Rhizobia-legume symbiosis. Mol. Cell. Proteomics 11, 724–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huala E., Oeller P. W., Liscum E., Han I. S., Larsen E., Briggs W. R. (1997) Arabidopsis NPH1: a protein kinase with a putative redox-sensing domain. Science 278, 2120–2123 [DOI] [PubMed] [Google Scholar]
- 49. Inada S., Ohgishi M., Mayama T., Okada K., Sakai T. (2004) RPT2 is a signal transducer involved in phototropic response and stomatal opening by association with phototropin 1 in Arabidopsis thaliana. Plant Cell 16, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harlan J. E., Hajduk P. J., Yoon H. S., Fesik S. W. (1994) Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature 371, 168–170 [DOI] [PubMed] [Google Scholar]
- 51. Mayer B. J., Baltimore D. (1993) Signalling through SH2 and SH3 domains. Trends Cell. Biol. 3, 8–13 [DOI] [PubMed] [Google Scholar]
- 52. Ma K. W., Flores C., Ma W. (2011) Chromatin configuration as battlefield in plant-bacteria interactions. Plant Physiol. 157, 535–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishihama N., Yoshioka H. (2012) Post-translational regulation of WRKY transcription factors in plant immunity. Curr. Opin. Plant Biol. 15, 431–437 [DOI] [PubMed] [Google Scholar]
- 54. Li Y., Sun Y., Yang Q., Kang J., Zhang T., Gruber M. Y., Fang F. (2012) Cloning and function analysis of an alfalfa (Medicago sativa L.) zinc finger protein promoter MsZPP. Mol. Biol. Rep. 39, 8559–8569 [DOI] [PubMed] [Google Scholar]
- 55. Park H. C., Kim M. L., Lee S. M., Bahk J. D., Yun D. J., Lim C. O., Hong J. C., Lee S. Y., Cho M. J., Chung W. S. (2007) Pathogen-induced binding of the soybean zinc finger homeodomain proteins GmZF-HD1 and GmZF-HD2 to two repeats of ATTA homeodomain binding site in the calmodulin isoform 4 (GmCaM4) promoter. Nucleic Acids Res. 35, 3612–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reddy A. S. (2007) Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 58, 267–294 [DOI] [PubMed] [Google Scholar]
- 57. Elmore J. M., Coaker G. (2011) The role of the plasma membrane H+-ATPase in plant–microbe interactions. Mol. Plant 4, 416–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Katsuhara M., Akiyama Y., Koshio K., Shibasaka M., Kasamo K. (2002) Functional analysis of water channels in barley roots. Plant Cell. Physiol. 43, 885–893 [DOI] [PubMed] [Google Scholar]
- 59. Baluška F., Mancuso S. (2009) Signaling in Plants, Signaling and Communication in Plants, Springer-Verlag, Berlin/Heidelberg, 287–301 [Google Scholar]
- 60. Morinaga N., Tsai S. C., Moss J., Vaughan M. (1996) Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc. Natl. Acad. Sci. U.S.A. 93, 12856–12860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mehrabi R. (2006) Signaling Pathways Involved in Pathogenicity and Development of the Fungal Wheat Pathogen Mycosphaerella graminicola. Ph.D. thesis, Wageningen University, The Netherlands [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.